
Abstract
Delayed neuronal death in the gerbil hippocampal CA1 sector occurs 48 to 72 hours after severe forebrain ischemia. DNA fragmentation is observed in the hippocampal CA1 neurons at around that time. We show here that an inhibitor of proteolytic process of apoptosis, N-tosyl-L-phenylalanyl chloromethyl ketone (TPCK), protected hippocampal neuronal damage by inhibition of the DNA fragmentation in a dose-dependent manner and that TPCK induced an apoptosis-regulating molecule, Bcl-2 protein, in the surviving neurons. These results suggest the prevention of apoptosis-related DNA fragmentation by TPCK may be an attractive therapeutic strategy for preserving hippocampal neurons from ischemic insult.
The specific DNA damage shown as internucleosomal DNA fragmentation is the most important evidence for apoptotic cell death, which is an active cell-deletion process with protein synthesis (McConkey et al., 1990; Wyllie et al., 1984). Significant neuronal damage in the hippocampus of the experimental model of severe forebrain ischemia is observed 48 to 72 hours after transient ischemia. The DNA fragmentation in the CA1 neurons of the hippocampus has been considered as a key phenomenon for delayed neuronal death, which has been investigated by terminal deoxynucleotidyl transferase (TDT)-mediated dUTP-biotin in situ nick-end labeling (TUNEL), gel electrophoresis, and electron microscope (Heron et al., 1993; Iwai et al., 1995; MacManus et al., 1993; Nitatori et al., 1995).
Cytoplasmic proteases play a functional role in the pathway leading to apoptotic cell death (Chow et al., 1995; Bruno et al., 1992; Lazebnik et al., 1994). Some protease inhibitors have been reported to block apoptosis-associated internucleosomal DNA fragmentation. Among them, a strong apoptosis-inhibitory effect has focused on a serine protease inhibitor, N-tosyl-L-phenylalanyl chloromethyl ketone (TPCK; Higuchi et al., 1995; Lotem and Sachs, 1996). This compound was reported to exert its proteinase inhibiting actions as an anti-inflammatory agent in vivo (Nakagawa et al., 1981; Hall et al., 1980).
In the current study, we evaluated the neuroprotective effect of TPCK, using intraperitoneal administration, on delayed neuronal death in the CA1 sector of the gerbil hippocampus after severe forebrain ischemia.
MATERIALS AND METHODS
Induction of ischemia and drug treatment
Male Mongolian gerbils (Meriones ungiculatus), weighing 65 to 75 g, were subjected to severe forebrain ischemia. Anesthesia was induced with halothane. The bilateral common carotid arteries were isolated through an anterior midcervical incision and occluded with microclips. After 5 minutes of forebrain ischemia, the clips were removed, restoration of blood flow was visually confirmed, and then the wound was closed. N-Tosyl-L-phenylalanyl chloromethyl ketone (Sigma Chemical Co., St. Louis, MO, U.S.A.) at a dose of 100 mg/kg in 0.05% polysorbate 80 was injected intraperitoneally 1 hour before or 1 or 24 hours after the ischemic insult. In some gerbils, another serine protease inhibitor, N-α-p-tosyl-L-lysinyl chloromethyl ketone (TLCK), at a dose of 100 mg/kg was also used in the same manner as TPCK. Rectal temperatures were maintained at 37° ± 0.3°C using a heating pad from the induction of anesthesia until 3 hours after ischemia. Sham-operated animals underwent the same surgical manipulation without occlusion of the bilateral common carotid arteries. In the ischemic control group and sham-operated group, intraperitoneal administration of vehicle (0.05% polysorbate 80) was performed. Six to eight animals per group were used for each experiment.
Histologic evaluation
Ninety-six hours after ischemic insult, animals were anesthetized with pentobarbital and perfused transcardially with saline and then with buffered 10% formalin. Brains were removed and processed for paraffin embedding. Five-micrometer coronal sections were cut at the level of the dorsal hippocampus and then used for hematoxylin and eosin staining, TUNEL, and Bcl-2 immunostaining. Neurons that had shrunken cell bodies were defined as damaged cells. Histologic analysis was performed by a blinded observer and the average of right and left damaged cell numbers in a single section of dorsal hippocampus was calculated for each gerbil as reported by Kirino et al. (1986). The degree of hippocampal CA1 damage was determined by measuring the number of damaged cells, as a proportion of the entire cell number of CA1.
In situ labeling of DNA fragmentation
TUNEL was performed as described previously (Hara et al., 1995a, 1995b) with some modification of the method of Gavrieli et al. (1992). After incubation with 20 μg/mL proteinase K (Sigma), the serial sections used for hematoxylin and eosin staining were immersed in TDT buffer (30 mmol/L Trizma base, pH 7.2, 140 mmol/L sodium cacodylate, 1 mmol/L cobalt chloride). Biotinylated dUTP (Boehringer Mannheim GmbH, Mannheim, Germany) and TDT (Boehringer Mannheim GmbH) were diluted in TDT buffer at a concentration of 0.8 nmol and 0.15 EU/μL, respectively. The solution was placed on the sections, and then incubated at 37°C for 60 minutes. The sections were covered with streptavidin peroxidase (Dako, Carpinteria, CA, U.S.A.), and stained with 3,3′-diaminobenzidine as a substrate for the peroxidase. Finally, counterstaining was done using Mayer's hematoxylin. The percentage of TUNEL-positive cells in hippocampal CA1 was determined by measuring the number of CA1 neurons possessing dark-brown staining nuclei, as a proportion of the entire cell number of CA1.
Immunostaining for Bcl-2 protein
The Bcl-2 immunostaining was performed as described previously (Hara et al., 1996a, 1996b). Briefly, the deparaffinized sections, which were cut serially to those used for hematoxylin and eosin staining, were heated and boiled for 1 minute by microwaving in citrate buffer. Anti-Bcl-2 antibody (N-19) was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, U.S.A.). Anti-Bcl-2 antibody, used at a dilution of 1:1000, was added to the slides and incubated overnight at 4°C. Expression of Bcl-2 protein was shown by the labeled streptavidin biotin method using the LSAB kit (DAKO) containing blocking reagent, biotinylated link antibody, and peroxidase-labeled streptavidin reagents. The peroxidase binding sites were detected by staining with 3,3′-diaminobenzidine. Finally, counterstaining was performed using Mayer's hematoxylin.
RESULTS
Administration of TPCK (100 mg/kg) produced a marked neuroprotective effect when given 1 hour before or 1 hour after transient forebrain ischemia. Moreover, TPCK was effective in preventing neuronal damage after transient forebrain ischemia even when administered up to 24 hours after the ischemic insult (Fig. 1a). CA1 neuronal damage after transient forebrain ischemia was assessed to determine the dose-response curve of TPCK administration 1 hour after ischemic insult at the doses of 25, 50, and 100 mg/kg (Figure 1c). More than 50 mg/kg of TPCK prevented neuronal damage after transient forebrain ischemia, whereas almost no neuronal preservation was observed at a dose of 25 mg/kg. Administration of 100 mg/kg TPCK was not fatal to the gerbil. Paralytic ileus was recognized in some gerbils at 96 hours after ischemic insult.
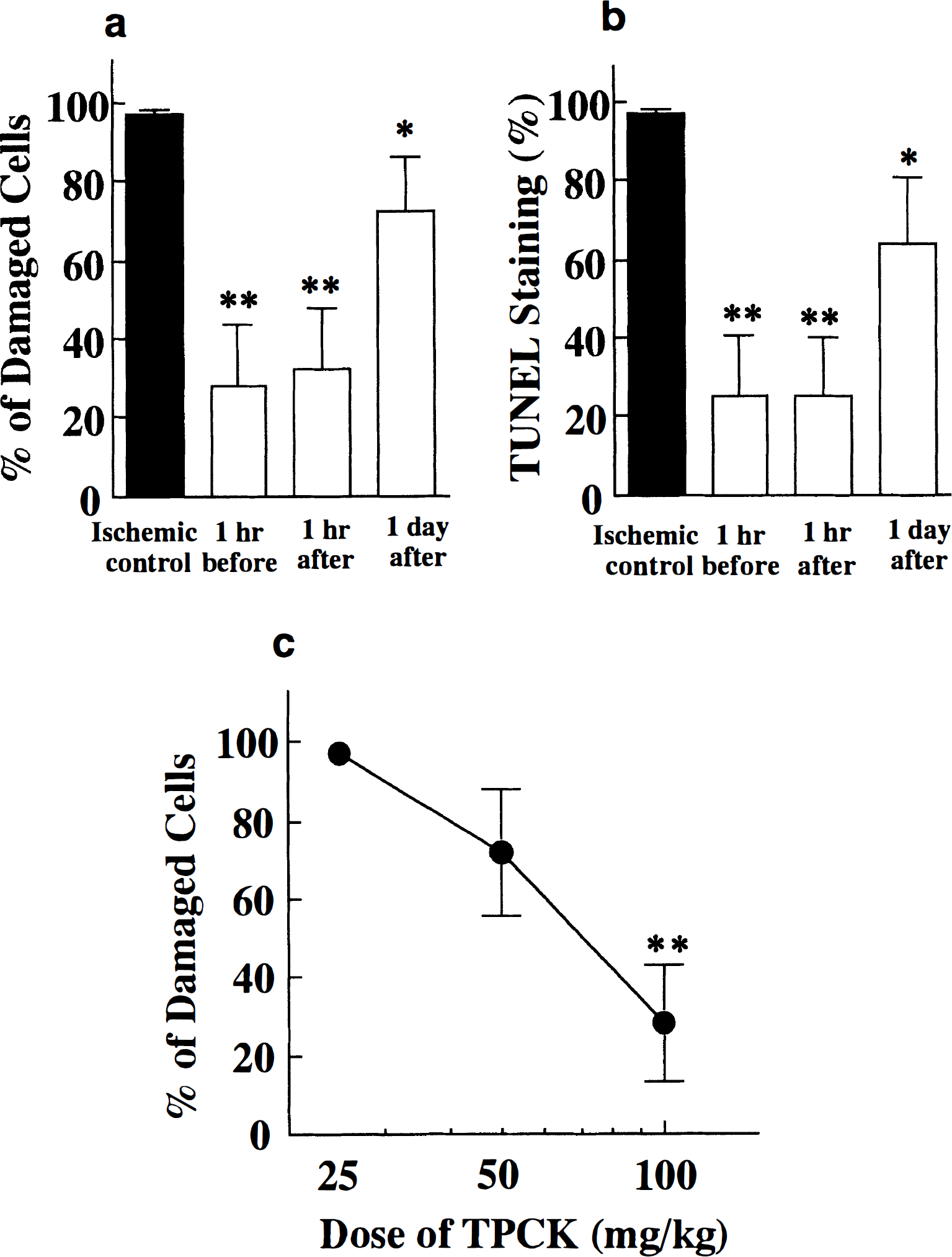
Effect of TPCK on the neuronal damage, histologically evaluated
In the ischemic control group (vehicle administration), TUNEL exhibited a strong positive reaction (Fig. 2, a and c), indicating DNA fragmentation occurred exclusively in damaged CA1 neurons. Administration of TPCK (100 mg/kg) 1 hour before or 1 hour after ischemic insult lead to a remarkable decrease of the number of TUNEL-positive CA1 neurons (Fig. 2, b and d). Even in gerbils treated with TPCK (100 mg/kg) 24 hours after ischemia, significantly fewer TUNEL-positive cells were observed in the hippocampal CA1 region than in that of vehicle-treated ischemic controls (Fig. 1b). The neuroprotection by TPCK was always observed laterally in the CA1 regions. Another serine protease inhibitor, TLCK (100 mg/kg, intraperitoneally) had no protective effect on CA1 neurons after transient forebrain ischemia.
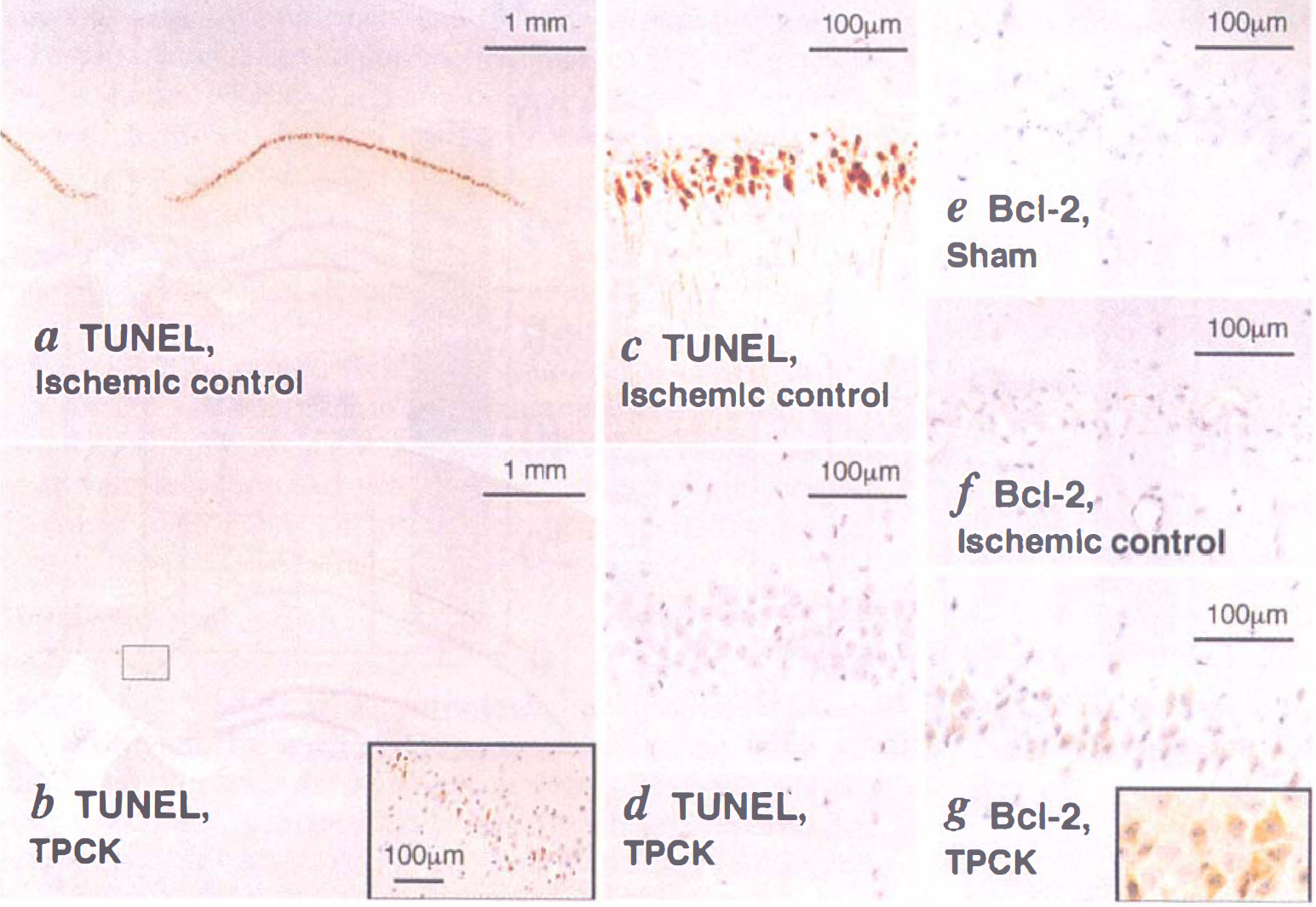
TUNEL staining
To assess the contribution of Bcl-2 protein to TPCK-induced protection against ischemic insult, expression of Bcl-2 protein in preserved CA1 neurons after ischemic insult was investigated. No immunoreactivity of Bcl-2 protein was detected in either CA1 neurons of sham-operated gerbils (Fig. 2e) or damaged CA1 neurons of ischemic control animals (Fig. 2f). Positive immunoreactivity of Bcl-2 was detected in many CA1 neurons that were well-protected by TPCK treatment from ischemic insult (Fig. 2g). Interestingly, all the Bcl-2-positive CA1 neurons were observed in CA1 layers that showed 0% to 10% TUNEL-positive rate, but not when the rate was above 10%. The Bcl-2-positive cells were observed in the preserved neurons by TPCK treatment, which were located laterally in the CA1 regions.
DISCUSSION
In the present study, we demonstrated a protective effect of TPCK, which inhibits the proteolytic process of apoptosis, against postischemic neuronal damage in the gerbil hippocampus. It is possible that some neuroprotective agents are effective because of their hypothermic effect during or after ischemia. Intraischemic hypothermia prevents delayed neuronal death in the gerbil hippocampus (Welsh et al., 1990; Iwai et al., 1993). However, the postischemic hypothermia is effective only if initiated within 3 hours after ischemic insult (Carroll and Beek, 1992). Therefore, we prevented hypothermia during ischemia and for 3 hours after ischemia. Moreover, TPCK was effective in preventing neuronal damage after transient forebrain ischemia even when administered up to 24 hours after the ischemic insult. For these reasons, we considered that the protective mechanism of TPCK was not related to hypothermia.
Data from various experimental system support a role for proteolysis in apoptotic cell death. Interleukin-1β-converting enzyme (ICE)-like proteases have been described to have a central role in proteolysis during apoptosis in HL-60 cells treated with cycloheximide for induction of apoptotic cell death (Lu and Mellgren, 1996). Overexpression of ICE gene by cDNA transfection causes apoptosis in mammalian fibroblasts (Miura et al., 1993). Although ICE possesses a cysteine protease activity, it is different from TPCK-sensitive serine protease. In addition, TPCK does not inhibit the proteolytic activity of ICE-like proteases (Xue et al., 1996). It has been suggested that apoptosis is associated with a multistep cascade of activated proteases that may act in a common pathway leading to apoptotic cell death (Lotem and Sachs, 1996). In this proteolytic pathway, TPCK-sensitive serine protease is considered to be associated with the final step for induction of internucleosomal DNA fragmentation (Yoshida et al., 1996). Our results revealed that another serine protease inhibitor, TLCK, has no inhibitory effect on apoptosis-related DNA fragmentation. Both TPCK and TLCK inhibit serine and cysteine proteases (Lotem and Sachs, 1996). Chymotrypsin-like proteases are targets of TPCK, whereas trypsinlike proteases are targets of TLCK (Higuchi et al., 1995). It is suggested that TPCK-sensitive proteases are associated with delayed neuronal death in gerbil hippocampal CA1.
Recent studies demonstrate that very low levels of Bcl-2 protein, which has been shown to inhibit programmed cell death and promote neuronal survival (Chen et al., 1997), are found in rat (Krajewski et al., 1995) and gerbil (Hara et al., 1996b; Shimazaki et al., 1994) CA1 neurons. Brief ischemia, which causes no neuronal damage, induces tolerance to subsequent ischemia and increases levels of Bcl-2 protein (Shimazaki et al., 1994). In the present study, increased expression of Bcl-2 protein was detected in many CA1 neurons that were protected from ischemic insult by TPCK administration. The precise mechanistic relationship between cell survival and expression of Bcl-2 protein in TPCK-protected CA1 neurons is currently unknown. However, the expression of Bcl-2 protein in CA1 neurons is considered closely related to the acquisition of resistance to ischemia-induced neuronal death, although the expression of Bcl-2 protein is not recognized in all the cells.
Significant morphologic damage and TUNEL-positive DNA fragmentation in gerbil CA1 neurons was observed only after 72 hours (Iwai et al., 1995). In the present study, TPCK was effective in preventing neuronal death after transient forebrain ischemia in the gerbil when administered up to 24 hours after ischemic insult. The first 24 hours after ischemic insult is considered to be a window of opportunity in which the biomolecular process leading to apoptotic cell death may be reversible. In conclusion, our results suggest that the prevention of apoptosis-related DNA fragmentation by TPCK, an inhibitor of the proteolytic process of apoptosis, may be an attractive therapeutic strategy in the prevention of neurodegeneration after stroke or cardiac arrest.