
Abstract
Programmed cell death (apoptosis) signaling pathways have been implicated in seizure-induced neuronal death and the pathogenesis of human temporal lobe epilepsy (TLE). End-stage DNA fragmentation during cell death may be mediated by nucleases including caspase-activated DNase (CAD), apoptosis-inducing factor (AIF) and endonuclease G. In the present study, we investigated the subcellular localization of these nucleases in resected hippocampus from TLE patients and autopsy controls. Subcellular fractionation determined levels of CAD were significantly higher in the nuclear fraction of TLE samples compared with controls, and semiquantitative immunohistochemistry revealed cleaved caspase-3 positive cells in TLE sections but not controls. While mitochondrial levels of AIF and endonuclease G were higher in TLE samples than controls, nuclear localization of AIF was limited and restricted to cells that were negative for cleaved caspase-3. Nuclear accumulation of endonuclease G was not found in TLE samples. These data support ongoing caspase-dependent apoptosis signaling in human TLE and suggest that interventions targeting such pathways may have potential as adjunctive neuroprotective therapy in epilepsy.
Introduction
Temporal lobe epilepsy (TLE) is the most common form of epilepsy in adults and the most intractable. Neuroimaging studies suggest that damage may be ongoing in patients who continue to experience seizures and temporal lobe pathology involving neuronal loss in the hippocampus is a common feature (Pitkanen and Sutula, 2002). Experimental seizure models have confirmed that seizures, particularly when prolonged, are capable of causing hippocampal damage in a pattern that closely resembles clinical observations (Pitkanen and Sutula, 2002).
Neurochemical analysis of seizure-damaged hippocampus has implicated the molecular machinery of apoptosis (Henshall and Simon, 2005). Apoptosis is a physiologic process for removing unwanted cells that is essential for normal organism development and homeostasis. Apoptosis is coordinated by several families of genes including the caspases; cysteine proteases that are responsible for cleaving key structural and functional proteins within cells leading to cell death. Activation of caspases has been reported after evoked seizures in rats and their pharmacological inhibition confers varying degrees of protection on the hippocampus (Henshall and Simon, 2005). Moreover, increased expression and processing of caspase-3 is found in resected neocortex from TLE patients (Henshall et al, 2000).
Deoxyribose nucleic acid (DNA) fragmentation is a hallmark of the later stages of apoptosis, which can be mediated by several nucleases (Nagata et al, 2003). The caspase-activated DNase (CAD, also called DNA fragmentation factor 40) was identified as the nuclease responsible for cleaving DNA into the characteristic apoptotic ladder pattern of ~200 bp (Enari et al, 1998). Caspase-activated DNase is expressed as a heterodimer with its inhibitor ICAD; ICAD is cleaved by caspase-3 leading to CAD activation (Nagata et al, 2003). At least two other nucleases may be involved in apoptotic DNA fragmentation. Apoptosis-inducing factor (AIF) is a mitochondrial flavoprotein that translocates to the nucleus following apoptotic stimuli, where it induces chromatin condensation and large-scale fragmentation of DNA (Susin et al, 1999). Endonuclease G is also mitochondrially localized and is capable of oligonucleosomal DNA fragmentation when redistributed to the nucleus (Li et al, 2001).
Because DNA fragmentation is a consistent finding after experimentally evoked seizures, and apoptosis signaling pathways are activated in human TLE (Henshall et al, 2000, 2004; Henshall and Simon, 2005), we investigated the localization of CAD in human TLE samples and compared findings to those for AIF and endonuclease G.
Materials and methods
Human Brain Samples
This study was approved by the Legacy Health System Institutional Review Board and informed consent was obtained from all patients. Clinical data for patients have previously been published (Henshall et al, 2004). All patients (n = 10) were referred for surgical resection of the temporal lobe by an epileptologist after neurologic assessment, video-EEG recording and MRI/neuroimaging. Each patient was determined to have medically intractable epilepsy with a history of recurring seizures, but no patient had experienced status epilepticus during the year in which their surgery was performed. All patients were taking anticonvulsant medication before surgery. Seizure frequency for each patient was typically in the range of 1 to 2 per week (range 2 to 30 per month). All patients underwent left or right temporal lobe resection and the hippocampus and in some cases the adjacent temporal cortex were obtained. Specimens were immediately frozen in liquid nitrogen and stored at −70°C until use. The hippocampus was separated from the adjacent temporal cortex and was analyzed in its entirety without further microdissection of subfields. Specimens were first sectioned for immunohistochemistry and then coronal slabs of ~1 mm thickness prepared from the remaining sample for biochemical analysis. Human control hippocampi (n = 6) were obtained from the Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, MD, USA. These specimens were similar fresh frozen, en bloc hippocampi from people who died of causes not related to known neurologic diseases. Subcellular fractionation was performed on a selection of age- and gender-matched control (C1, C2 and C3) and epilepsy specimens (E2, E3 and E4) as previously described (Henshall et al, 2004).
Subcellular Fractionation
Subcellular fractionation to isolate the cytoplasm, mitochondria, microsomes (endoplasmic reticulum and Golgi body) and nucleus was performed as previously described with modifications (Henshall et al, 2004). Briefly, brain samples were homogenized in a mannitol/sucrose buffer, mitochondria separated from the homogenate by centrifugation (10,000g) and the crude cytosol fraction centrifuged (100,000g) to obtain the cytosol and microsomal fraction (pellet). The crude nuclear fraction was further purified by centrifugation in sucrose buffer (10 mmol/L Tris, pH 7.5, 300 mmol/L sucrose and 1 mmol/L EDTA with 0.1% NP-40). All steps were performed at 4°C. Subcellular fractionation markers cytochrome IV oxidase (CoxIV, mitochondria), protein disulfide isomerase (PDI, microsomes) and lamin A/C (nucleus) were used to verify fraction purity.
Western Blotting
Western blotting was performed as previously described (Henshall et al, 2004). In total, 50 μg protein samples were boiled in gel-loading buffer and separated on 12% to 15% SDS-PAGE gels. Proteins were transferred onto polyvinylidene difluoride membranes (BioRad, Hercules, USA) and then incubated with antibodies against the following: AIF and CAD (Santa Cruz Biotechnology, Santa Cruz, USA), CoxIV (Molecular Probes, Eugene, USA), PDI and endonuclease G (Abcam, Cambridge, USA), and lamin A/C (Cell Signaling Technology, Beverley, USA). Membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies (1:2000 dilution) followed by chemiluminescence detection (NEN Life Science Products, Boston, USA), and then exposed to Kodak Biomax film (Kodak, Rochester, USA).
Immunohistochemistry
Hippocampal sections (12 μm) were postfixed in 10% formalin and processed for immunohistochemistry as previously described (Henshall et al, 2004). Briefly, sections were blocked in 5% goat serum and then incubated overnight at 4°C with antibodies against AIF, CAD, cleaved caspase-3 (Cell Signaling Technology) or NeuN (Chemicon, Temecula, USA). Nonspecific staining was assessed by omitting the primary antibody. After washing, sections were incubated for 2 h at room temperature in a 1:500 dilution of goat anti-rabbit or anti-mouse Cy3 or fluorescein isothiocyanate (FITC) (Jackson Immunoresearch, Plymouth, USA). Sections were counterstained with 4',6 diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, USA) to assess nuclear morphology. Images were visualized using a Hamamatsu Orca 285 camera attached to a Nikon 2000s epifluorescence microscope under Ex/Em wavelengths of 330 to 380/420 nm (blue), 472/520 nm (green) and 540 to 580/600 to 660 nm (red).
Data Analysis
Western blot protein levels were determined using gel-scanning integrated optical density software (Bioquant, Nashville, USA). The proportion of CAD- and AIF-expressing cells was assessed from 10 randomly selected ×60 lens fields. Counts of cleaved caspase-3 immunoreactive cells were the mean sum of 10 randomly selected fields studied under ×40 lens magnification. Data are presented as mean ± s.d. Data were analyzed using the Mann–Whitney U-test (StatView software, SAS Institute Inc., Cary, USA). Significance was accepted at P < 0.05.
Results
Nuclear Accumulation of Caspase-activated DNase in Human Temporal Lobe Epilepsy Brain
Figure 1A shows representative Western blots verifying the presence of CoxIV in mitochondrial, PDI in microsomal and lamin A/C in nuclear fractions that were used in these studies. We first examined CAD expression in subcellular fractions of human hippocampi. Caspase-activated DNase was detected at low levels at its predicted molecular weight of ~40 kDa in cytoplasmic fraction samples from control brain (n = 3) (Figure 1B). In contrast, CAD was largely undetectable in the cytoplasmic fraction of TLE samples (n = 3) (Figure 1B). Small amounts of CAD were also detectable in the mitochondrial and microsomal fractions, but no significant differences were found between control and TLE samples (Figure 1B). Significantly higher levels of CAD were detected in the nuclear fraction of TLE samples compared to controls (Figures 1B and 1C).
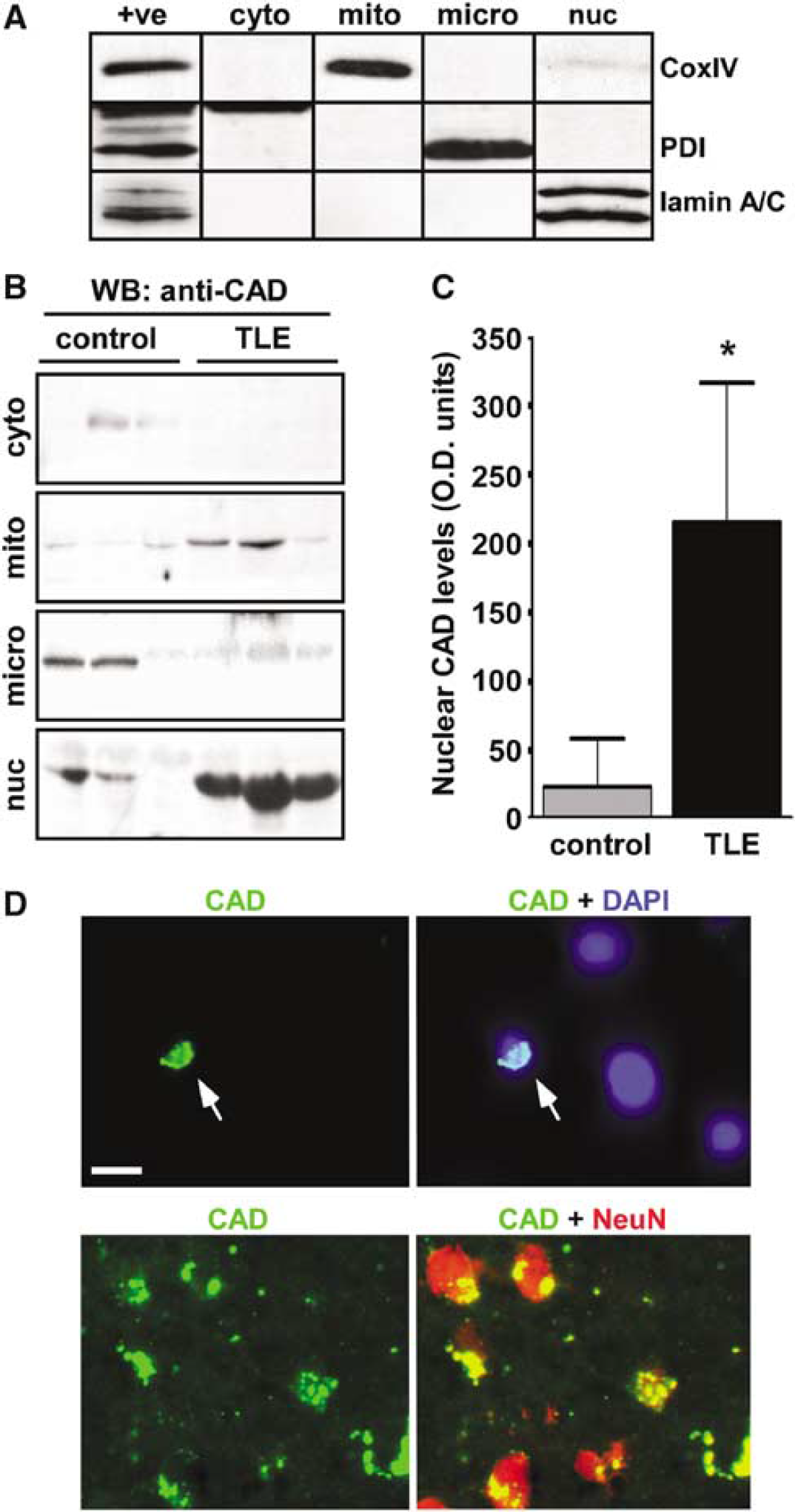
Nuclear accumulation of CAD and cleaved caspase-3 in human TLE brain. (
Next, we used fluorescence microscopy to visualize CAD in TLE hippocampal sections. Caspase-activated DNase immunoreactivity was detected at low levels in 22% (range 6% to 68%, from n= = 3 TLE patient sections) of cells and appeared in both perinuclear (data not shown) and nuclear (Figure 1D) distributions. Counter-staining sections with antibodies against the neuronal phenotype marker NeuN revealed CAD was expressed by neurons (Figure 1D), but CAD was also detectable in non-neuronal cells, mostly with a microvascular distribution and appearance (data not shown).
Caspase-3 Cleavage in Human Temporal Lobe Epilepsy Brain
Since caspase-3 may be an upstream promoter of nuclear CAD accumulation, we next undertook fluorescence microscopy to visualize activated caspase-3 in human control and TLE hippocampi (n = 5 to 6 per group). Cleaved caspase-3 positive cells were not detected in control sections (Figure 2A). In contrast, small numbers of cleaved caspase-3 positive cells were detected in TLE brain sections (Figures 2A and 2B). Cleaved caspase-3 distribution appeared largely nuclear and colocalized with CAD (Figure 2C), although some process staining was also occasionally detected (Figure 2B).
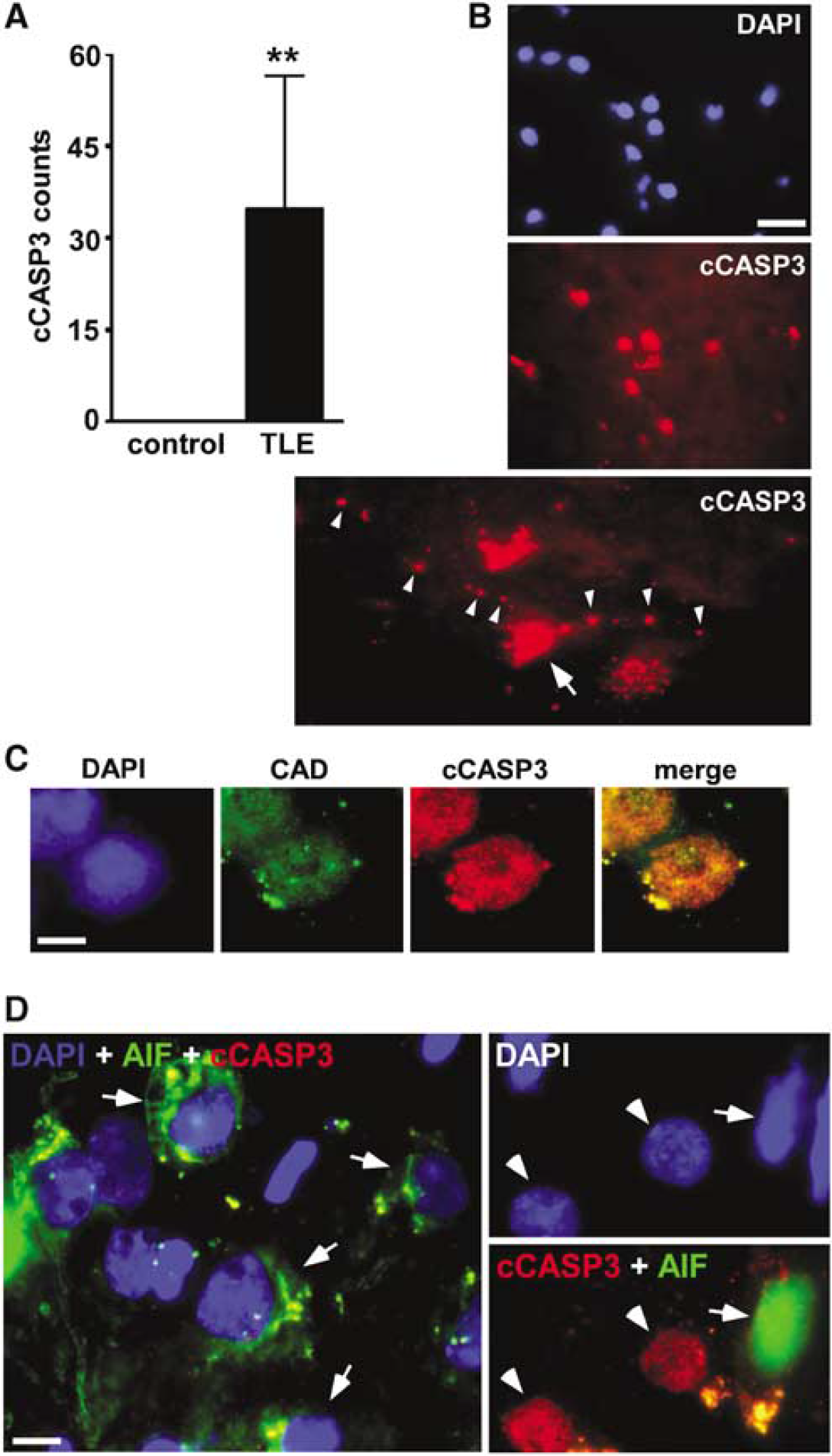
Cleaved caspase-3, CAD and AIF immunohistochemistry in human TLE brain. (
Mitochondrial Overexpression but Limited Nuclear Accumulation of Apoptosis-inducing Factor and Endonuclease G in Human Temporal Lobe Epilepsy Brain
We next studied the localization of AIF in human hippocampus. In controls, AIF was expressed within the mitochondrial fraction, mostly at ~67 kDa corresponding to its preprotein form, but also as a lower weight fragment at ~62 kDa corresponding to its mature form (Otera et al, 2005) (Figure 3A). Apoptosis-inducing factor and its processed form could also be detected in the cytoplasmic fraction and nucleus of control samples (Figure 3A). Mitochondrial levels of AIF and its processed form (data not shown) were significantly higher in TLE samples than controls (Figures 3A and 3B). While some nuclear AIF could be detected in human TLE samples, this was the unprocessed form and levels were significantly lower in TLE samples compared with controls (Figures 3A and 3C). Only very low levels of processed AIF were detected in the cytoplasmic, microsomal and nuclear fractions of human control and TLE brain samples and levels did not significantly differ.
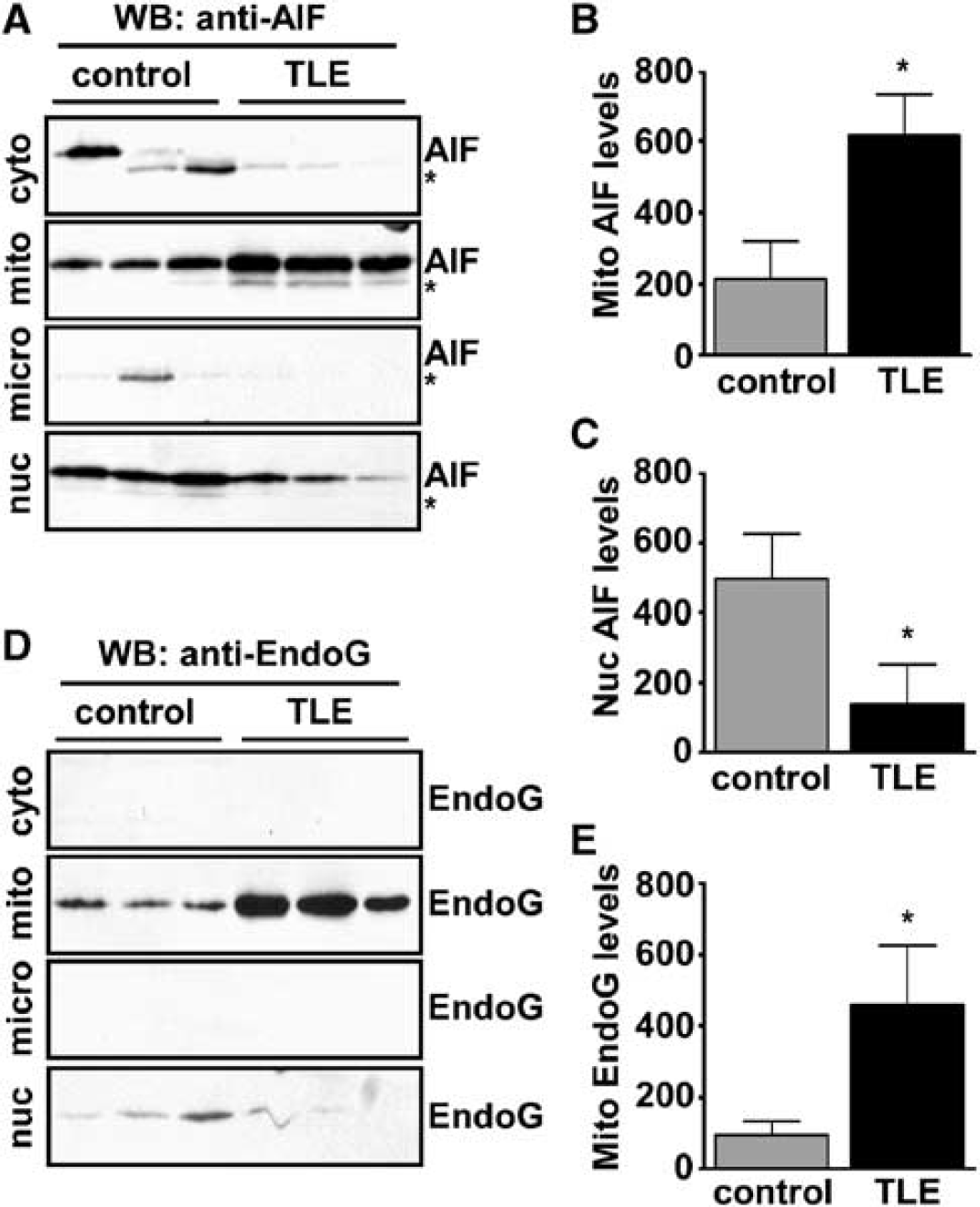
AIF and endonuclease G subcellular localization in human TLE. (
Endonuclease G, another mitochondrially localized nuclease proposed to mediate apoptotic DNA fragmentation, was detected at ~35 kDa almost exclusively in the mitochondrial fraction of control and TLE samples. Mitochondrial endonuclease G levels were significantly higher in TLE brain compared with controls (Figures 3D and 3E). Endonuclease G was not detected in the cytoplasmic or microsomal fractions and endonuclease G was largely undetectable in the nucleus of control or TLE samples (Figure 3D).
To determine whether the presence of nuclear AIF in controls was an artifact we simulated an 8 h autopsy delay, which corresponded to the mean delay in the human controls, using mouse hippocampus. These experiments revealed that some AIF but not CAD or endonuclease G appears in the nucleus as a result of post-mortem changes (Supplementary Figure 1 and data not shown). Accordingly, AIF localization in TLE brain was further investigated. Using fluorescence microscopy, AIF immunoreactivity was observed in 26% (range 7% to 47%, from n = 3 TLE patient sections) of cells examined and appeared almost exclusively in a perinuclear (mitochondrial) distribution (Figure 2D). Staining of AIF within the nucleus was seldom observed (six of 130 cells examined) and in double-labeled sections <1% of cleaved caspase-3 positive cells expressed nuclear AIF (Figure 2D and data not shown).
Discussion
Our data suggest ongoing apoptosis signaling in human TLE brain, as evidenced by the presence of cleaved caspase-3 and nuclear accumulation of CAD. Moreover, they suggest a selective involvement of apoptotic nucleases since endonuclease G and to a lesser extent AIF remained in the mitochondrial fraction. These findings add to the emerging evidence of apoptosis signaling in the pathogenesis of TLE and suggest that interventions targeting such pathways may have potential as adjunctive neuroprotective therapy in epilepsy.
While the molecular mechanisms underlying seizure-induced neuronal death and the pathogenesis of hippocampal sclerosis in TLE remain incompletely understood, evidence is accumulating for the involvement of apoptosis signaling pathways (Henshall and Simon, 2005). Altered expression of Bcl-2 family proteins, caspases and other apoptosis-regulatory proteins is seen in neocortex and hippocampal resection material from TLE patients with intractable seizures (Henshall et al, 2000, 2004; Henshall and Simon, 2005). Deoxyribose nucleic acid fragmentation is a commonly reported biochemical feature of seizure-induced neuronal death (Henshall and Simon, 2005), but the enzyme(s) responsible are not known. Caspase-activated DNase, a caspase-dependent DNase (Enari et al, 1998), has been implicated in mediating DNA fragmentation during ischemic brain injury (Cao et al, 2001) and also during seizure-induced DNA fragmentation based on the observation that seizures in rats trigger degradation of ICAD (Kondratyev et al, 2002), and cell death is delayed after kainic acid treatment in DFF45 (CAD deficient) mutant mice (Zhang et al, 2001). Our subcellular fractionation analyses build on these experimental data, suggesting that CAD accumulates in the nucleus in TLE brain. In line with reports on CAD expression in the human (Mukae et al, 1998) and rat (Cao et al, 2001), we found CAD was only expressed at low levels in the brain, but this included a neuronal and nuclear distribution. Caspase-3 is thought to be the major enzyme responsible for proteolyzing ICAD (Enari et al, 1998), and our observation of cleaved caspase-3 in human TLE sections provides a mechanistic explanation for increased nuclear CAD. The mostly nuclear localization of cleaved caspase-3 fits well with other reports on the movement of activated caspase-3 during CAD activation in vivo (Cao et al, 2001). In light of caspase-7 sharing substrate preference with caspase-3, we cannot rule out other caspases as being involved in CAD activation, and whether caspase-3 processing occurs downstream of death receptor (extrinsic) or mitochondrial/Bcl-2 family-regulated (intrinsic) pathways will require further investigation.
While CAD is considered responsible for caspase-mediated DNA fragmentation, other nucleases are capable of DNA fragmentation during apoptosis (Nagata et al, 2003). Apoptosis-inducing factor, a mitochondrial flavoprotein that contributes to oxidative phosphorylation, is released during apoptosis to promote chromatin condensation and large-scale DNA fragmentation (Susin et al, 1999; Vahsen et al, 2004). Studies on AIF's apoptotic functions support a caspase-independent mechanism, which may involve poly(ADP-ribose) polymerase-1 and/or calpain I (Polster et al, 2005; Susin et al, 1999; Yu et al, 2002), although AIF release in some models can be blocked by caspase inhibitors (Arnoult et al, 2003). Apoptosis-inducing factor has been implicated in promoting seizure-induced DNA fragmentation on the basis that mice hypomorphic for AIF are seizure-damage resistant (Cheung et al, 2005). Our results show nuclear AIF localization is limited in human TLE. Interestingly, we also found that cells expressing cleaved caspase-3 did not express nuclear AIF, supporting separate signaling pathways. These data suggest that AIF may be more important during apoptosis signaling after acute prolonged seizures (Cheung et al, 2005).
We also investigated a second mitochondrially localized nuclease, endonuclease G (Li et al, 2001), that like AIF has recently been implicated in mediating seizure-induced neuronal death on the basis of seizure-damage resistance in endonuclease G heterozygous mice (Wu et al, 2004). However, the retention of endonuclease G in the mitochondrial fraction was near complete in human TLE samples, suggesting that endonuclease G is probably not an important apoptotic nuclease in TLE. Like AIF, endonuclease G has been reported to function in caspase-independent apoptosis pathways (Nagata et al, 2003), but its release has also been shown to be blocked by caspase inhibitors (Arnoult et al, 2003). Interestingly, reports have brought into question the significance of endonuclease G as a mediator of apoptotic DNA fragmentation in vivo (Nagata et al, 2003).
Certain limitations should be considered in the interpretation of the present findings. While well balanced in terms of age and gender, differences naturally exist between our control and TLE material including possible effects of medication and sampling (autopsy) delay. Indeed, post-mortem effects might explain the presence of nuclear AIF in controls (supplementary data, Figure 1). Second, since some CAD is normally present in the nucleus (Cao etal, 2001), the observed nuclear accumulation of CAD in TLE brain falls short of functional evidence of its activation. Studies to assess whether CAD transcription, translation or turnover is altered in TLE might also provide further insight. An apparent paradox left by the present data is why so few cells exhibit DNA fragmentation ('TUNEL (terminal desoxynucleotidyl transferase-mediated Terminal deoxynucleotidyl transfease-mediated dUTP nick end labeling) positivity') in human TLE specimens (Henshall et al, 2004) despite caspase-3 cleavage and an apparent abundance of nuclear CAD. Certainly, the absence of serial time-point sampling with clinical material may underestimate cells exhibiting DNA fragmentation. However, antiapoptotic signaling intermediates may be actively engaged in suppressing the effects of the caspase-3/CAD system in TLE brain. Indeed, a block in caspase activity might explain our AIF and endonuclease G data since their release can be prevented using caspase inhibitors (Arnoult et al, 2003). Several antiapoptotic proteins are upregulated in human TLE samples (Henshall et al, 2000; Henshall and Simon, 2005) and specific CAD inhibitors have also been identified, including a nuclear phosphatidylinositol 3,4,5-triphosphate receptor, nucleophosmin (NPM)/B23 (Ahn et al, 2005). Finally, DNA fragmentation is not essential for (apoptotic) cell death and CAD deficient cells can still be killed by apoptotic stimuli, albeit with an often delayed time scale (Nagata et al, 2003).
Taken together, our data point to CAD as the apoptotic nuclease(s) involved in human TLE. Despite detecting cleaved caspase-3 and largely nonnuclear AIF and endonuclease G, the context-specific requirement for caspases in AIF/endonuclease G activation (Arnoult et al, 2003) precludes our ruling out caspase-independent apoptosis signaling pathway involvement in human TLE. Further studies, perhaps using experimental models, may expand our understanding of the functional significance of these nucleases in seizure-regulated apoptosis signaling pathways and human TLE. In conclusion, our data represent the first profile of nuclease distribution in human TLE and suggest that caspase-dependent pathways may be the prominent apoptosis signaling mechanism in the pathogenesis of TLE.
Footnotes
Acknowledgements
The authors thank Dr Heiko Duessmann, Carmen Bellver-Estelles and Jing-Quan Lan for technical support, and the Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, MD, USA.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.