
Abstract
Using in situ hybridization, Northern blot analysis, Western blot analysis, and immunocytochemistry, mRNA and protein expression of the novel DNA damage-inducible gene GADD45 was examined in the rat brain at 0.5, 2, 4, 8, 16, 24, 48, and 72 hours after 15 minutes of transient global ischemia. Transient ischemia produced by the four-vessel occlusion method resulted in DNA double-strand breaks and delayed neuronal cell death in vulnerable neurons of the hippocampal CA1 sector, the hilus, dorsal caudate-putamen, and thalamus, as shown by in situ DNA nick end-labeling and histologic staining. GADD45 mRNA was transiently increased in less-vulnerable regions such as the parietal cortex (up to 8 hours after ischemia) and dentate granule cells (up to 24 hours after ischemia) but was persistently increased in vulnerable neurons such as CA1 pyramidal neurons (up to 48 hours). GADD45 immunoreactivity was increased in both vulnerable and less-vulnerable regions at earlier reperfusion periods (4 to 16 hours), but thereafter immunoreactivity was decreased below control levels in most vulnerable regions before delayed cell death and DNA double-strand breaks. At 72 hours after transient ischemia, a moderate increase in GADD45 immunoreactivity was still detectable in some CA3 neurons and in a few surviving neurons in the CA1 region. Double staining performed at 16 to 72 hours after ischemia revealed that GADD45 immunoreactivity was persistently increased in neurons that did not develop DNA damage. Because GADD45 protein may participate in the DNA excision repair process and because it has been shown that this protein is also overexpressed in neurons that survive focal ischemia and kainate-induced epileptic seizures, the results reported here support the hypothesis that GADD45 could have a protective role in neuronal injury.
Active oxygen-derived free radicals (AOR) such as superoxide and hydrogen peroxide are common byproducts of normal cellular metabolism, being produced along the mitochondrial electron transport chain during aerobic respiration and by a variety of enzymes during the metabolism of endogenous substances. To prevent the attack by AOR, cells rely on an elaborate antioxidant defense system dedicated to reducing the levels of oxidants (Halliwell, 1989). This antioxidant system is composed of a variety of enzymatic and nonenzymatic factors that can scavenge most but not all AOR produced. Thus, DNA and other cellular macromolecules constantly receive attacks by AOR which have escaped from the antioxidant system (Adelman et al., 1988; Halliwell and Aruoma, 1991; Ames et al., 1993a; 1993b). The second line of defense that cells rely on is an efficient repair system composed of a number of enzymes and related proteins that can quickly repair oxidative DNA lesions (Sancar and Sancar, 1988). Because of the higher rate of oxygen consumption, a higher content of oxidizable lipids, and lower levels of some antioxidant, brain cells generate higher levels of AOR and, as a result, encounter a higher steady state level of oxidative DNA damage as compared to other tissues (Adelman et al., 1988). However, the neuron is able to maintain its DNA integrity and normal cellular functions because of the action of both antioxidant and DNA repair systems (Ames et al., 1993b).
Under various pathologic conditions, high levels of AOR are generated in the cell and cause oxidative stress and excessive DNA damage (Janssen et al., 1993). It has been suggested that cells are able to respond to such stress by activating the expression of a variety of genes that enhance endogenous defensive mechanisms (Ames et al., 1993a; Janssen et al., 1993). Among the induced gene products are antioxidant enzymes (Holley et al., 1992; Janssen et al., 1992) and DNA repair proteins (Schraufstatter et al., 1986; Fornace et al., 1989b; Fritz et al., 1991; Fornace 1992). Failure of this response may result in the accumulation of oxidative DNA lesions which subsequently activate certain signal transduction pathways and result in apoptotic cell death (Janssen et al., 1993; Payne et al., 1995). Thus, the inducible expression of these genes may affect the fate of injured cells under certain circumstances. Recently, a group of novel DNA damage- and repair-related genes, termed growth arrest and DNA damage inducible (GADD) genes, has been identified. These genes were originally isolated in Chinese hamster ovarian cells on the basis of rapid induction by ultraviolet radiation (Fornace et al., 1988). Subsequently, it was found that they were induced in a variety of mammalian cells by many other stimuli that produced DNA base damage (Fornace et al., 1989a). Among the five GADD genes, GADD45 is the only one that has been found to be induced in the brain neurons in vivo by a DNA-damaging agent (Yoshida et al., 1994) or by the N-methyl-D-aspartate agonist, quinolinic acid (Hughes et al., 1996). Functionally, GADD45 protein can stimulate DNA excision repair in vitro by interacting with proliferating cell nuclear antigen (PCNA) (Smith et al., 1994). Thus, GADD45 may be a DNA repair gene. Kastan et al recently have found that ataxia-telangiectasia cells fail to express GADD45, and suggested that this may be in part responsible for the hypersensitivity of AT cells to DNA-damaging stimuli (Kastan et al., 1992).
The recent observation that the GADD45 protein is overexpressed in neurons that survive ischemic infarction suggests that this gene may have a protective role in injured brain (Jin et al., 1996). To further investigate the role of GADD45 in neuronal DNA damage and ischemic neuronal injury, we examined the regional distribution and time profile of the expression of GADD45 mRNA and protein in a rat model of transient global ischemia. Delayed and selective neuronal death after transient ischemia is a relatively slow process involving glutamatetriggered oxidative stress (Chan et al., 1993; Simonson et al., 1993; Globus et al., 1995a; 1995b; Lancelot et al., 1995; Suzuki et al., 1995). A number of recent studies suggest that neuronal death in this setting of ischemic injury has the characteristics of apoptosis and the DNA damage occurs in vulnerable neurons before or associated with cell death (MacManus et al., 1993; Kihara et al., 1994; Nitatori et al., 1995; Tobita et al., 1995). The molecular events associated with DNA damage and cell death or survival after transient global ischemia, however, have not been explored.
METHOD
Animal preparation
Experiments were performed using 70 male Sprague-Dawley rats each weighing 300 to 350 g. Transient global ischemia was induced in isoflurane-anesthetized rats by the method of Pulsinelli (Pulsinelli et al., 1982), with slight modifications (Chen et al., 1996). In brief, anesthesia was induced with 3.5% isoflurane through a face mask, rats were then intubated, and ventilated with 1% isoflurane in a mixture of 25% O2 and 74% N2O. Blood pressure, blood gases, and blood glucose concentration were maintained in the normal range throughout the experiments. Rectal temperature was continuously monitored and kept at 37 to 37.5°C using a heating pad and a heating lamp throughout the experiment. Brain temperature was monitored by a 29-Ga thermocouple implanted in the left striatum and allowed to decrease spontaneously from 36.2±0.1°C to 34.5±0.5°C during ischemia. Within 5 to 8 minutes of reperfusion, brain temperature returned to pre-ischemia range in all animals. Animals were placed in a Kopf steriotaxic frame, and vertebral arteries were coagulated and transected at the level of the junction of first and second cervical vertebrae. Then, their common carotid arteries were exposed, bilateral external carotid arteries were ligated to block the potential collateral flow from the vertebral artery system, and the anesthesia was discontinued. Three minutes later, both common carotid arteries were occluded with microvascular clips during continuous monitoring of the EEG. The EEG became isoelectric within 10 seconds after carotid artery occlusion in all animals. At various time points after the completion of ischemia, the animals were killed according to the experimental protocols. Sham-operation was performed in additional animals using the same anesthesia and surgical exposure procedures except the arteries were not occluded; the animals were killed at 24 hours after surgery and their brains were used as nonischemic controls.
In situ hybridization
Rats used for in situ hybridization were anesthetized with 8% chloral hydrate and decapitated at 0.5, 2, 4, 8, 16, 24, 48, or 72 hours after 15 minutes of global ischemia (n = 3 to 4 per time point) or 24 hours after sham-operation (n = 3). Their brains were rapidly removed, frozen in 2-methylbutane at −30°C, covered with mounting medium and stored at −80°C. Coronal sections (20-μm thick) were cut on a cryostat at −20°C, collected on precleaned Probe-On-Slides (Fisher Scientific, Pittsburgh, PA), and processed for in situ hybridization using the method previously described (Chen et al., 1995). Briefly, an antisense oligodeoxynucleotide probe (5'-GGA TGT TGA TGT CGT TCT CGC AGC AGA ACG CCT GGA TGA GG-3') corresponding to codons 246–286 of the rat GADD45 gene sequence (Yoshida et al., 1994) was synthesized. A GenBank search revealed that this sequence is nearly identical to the GADD45 gene sequence of human and Chinese hamster origins but is not homologous with any other known gene sequences. We previously confirmed the specificity of this oligo probe by performing duplicate Northern blots using both oligo and cDNA probes in a rat model of focal ischemia (Jin et al., 1996). The oligonucleotide probe was 5'-end labeled with 35S-dATP using terminal deoxynucleotidyltransferase (Gibsol BRL, Gaithersburg, MD) and hybridized (1 times 107 cpm/mL) at 42°C for 18 hours with the sections. After hybridization, the sections were rinsed in 1 × SSC (150 mmol/L sodium chloride, 15 mmol/L sodium citrate, pH 7.4) at 56°C for 60 minutes with four changes in 1 × SSC, rinsed again in 1 × SSC, at room temperature, and dehydrated. All slides were then exposed to the same sheet of Kodak SB-5 film for 3 weeks and developed according to the manufacturer's instructions. Relative changes in mRNA expression were then semiquantified by determining the optical density (OD) radio of the specified regions (SR) in ischemic brains versus control brains using an image analysis system (micro computer imaging device [MCID], St. Catharine's, Ontario, Canada). OD of nonspecific radioactive background was also measured in each section and the values were subtracted from that of SR. For OD measurement in the hippocampal formation (CA1-3 and dentate, at the level of anterior-posterior [AP] = − 0.3 mm from the bregma), hippocampal white matter was chosen for background measurement because this region contains a relatively small amount of cells and OD varies proportionally to the background of the entire section. For OD measurement in the cortex and caudate putamen (at the level of AP = + 0.2 mm from the bregma), OD in the corpus callosum and cingulum was measured as background levels. Fold (s) changes of OD in SR after ischemia were then calculated using the following equation: [OD of a SR (ischemia)—background OD of the same section]/[OD of an SR (nonischemia control)—background OD of the same section]. Cellular localization of the labeled mRNA was evaluated by coating slides with Kodak NTB-2 emulsion at 4°C for 5 weeks and counterstaining with cresyl violet.
Northern blot analysis
Northern blot analysis was performed to confirm the expression of GADD45 mRNA in ischemic brains. Hippocampal total RNA was extracted from sham-operated brains and from brains subjected to 15 minutes of ischemia followed by 4 hours or 24 hours of reperfusion (n = 3 per time point). Northern blot analysis was performed by electrophoresis through a 1% agarose-2.2-mol/L formaldehyde gel and subsequent transfer to nylon filters. The transfer efficiency was examined by staining the filters with methylene blue (0.04% in 0.5 mol/L sodium acetate, pH 5.2). The GADD45 oligodeoxynucleotides were 3' end-labeled with 40 μCi 32P-dATP (specific activity > 106 Ci/mol) using terminal deoxynucleotidyltransferase (GIBCO BRL). Unincorporated nucleotides were separated using P-60-sephorose columns. The labeled probe was denatured by heat and hybridized to the filters at 42°C overnight in a buffer containing 50% formamide, 25% dextran sulfate, 0.3 mol/L NaCl, 12.5 mmol/L sodium phosphate buffer, 125 mmol/L Tris, 10 mmol/L dithiothreitol, 0.8 mg/mL tRNA, 0.4 μg poly(A), 1 nmol/L S-riboATP, and 50 μg/mL S-RNA. The washing procedure was performed under high stringency conditions in 2 × SSC/0.1% sodium dodecyl sulfate (SDS) (45 minutes for three times at 52°C). Membranes were then exposed to Kodak X-omat AR films (Eastman Kodak Company, Rochester, NY) using intensifier screens at −80°C for 72 hours. Autoradiogram signals on the films were semiquantified (OD × area) by a gel densitometric scanning program using the MCID image analysis system. To control for variation in the amount of total RNA in different samples, the original probe was stripped off in a solution containing 0.1 × SSC and 0.5% SDS at 100°C for 15 minutes; all blots were rehybridized with an oligonucleotide probe (5'-ACGGTATCTGATCGTCTTCGAACC-3') corresponding to 18 S-rRNA. All densitometric values for GADD45 were normalized to values for 18 S-rRNA obtained on the same lane.
Immunocytochemistry
Rats were given intraperitoneal injection of 100 U/kg heparin and anesthetized with 8% chloral hydrate at 4, 8, 16, 24, or 72 hours after 15 minutes of global ischemia (n = 3 to 4 per time point) or 24 hours after sham-operation (n = 3). Five minutes later, they were perfused with 4% paraformaldehyde in 0.1-mol/L phosphate-buffered saline (PBS) solution. Their brains were removed, postfixed for at least 4 hours, sectioned 50-μm thick on a vibratome, and processed for immunocytochemical staining. The sections were reacted for the expression of GADD45 protein using a mouse monoclonal antibody against human GADD45 recombinant protein (Sata Cluz Biochemical, San Diego, CA). This antibody lacks cross-reactivity with other GADD45-related proteins such as GADD153 and GADD34. Immunocytochemistry was performed using the avidin-biotin-horseradish peroxidase method (Elite Vecstatin, Burlingame, CA). Sections were placed in PBS containing 2% horse serum, 0.2% Triton X-100, and 0.1% bovine serum albumin (BSA) for 2 hours at room temperature. This was followed by a 72-hour incubation at 4°C in the GADD45 antibody diluted 1:1000 in PBS-horse serum, and then in a second antibody (biotinylated horse anti-mouse immunoglobulin G) for 2 hours at room temperature. They were placed in an avidin-horseradish peroxidase solution for 1 hour, washed twice with PBS, and reacted for horseradish peroxidase using 0.015% diaminobenzidine and 0.001% hydrogen peroxide. Sections were then washed for 1 hour in PBS and mounted on gelatinized slides. As negative controls, alternative sections were incubated in the absence of primary antibody.
Western blot analysis
Western blot analysis was performed to verify the reaction specificity of the anti-GADD45 antibody referred above against rat brain tissues. Rats were killed at 2, 4, 8, 16, 24, or 72 hours after 15 minutes of global ischemia (n = 3 per time point) or 24 hours after sham-operation. Their hippocampi were dissected, homogenized, and lysed in a buffer containing 0.1 mol/L NaCl, 0.01 mol/L Tris-Cl, NaCl (pH 7.6), 0.001 mol/L edetic acid (pH 8.0), 1 μg/mL aprotinin, with 100 μ/mL polymethylsulfonylfluoride. Lysates were cleared by centrifugation at 14,000 g for 30 minutes at 4°C and boiled at 100°C in SDS-gel loading buffer (100 mmol/L Tris-Cl, 200 mm dithiothreitol, 4% SDS, 0.2% bromophenol blue and 20% glycerol) for 6 minutes. Forty micrograms of protein samples from each brain were loaded on to a 12% SDS-polyacrylamide gel. Western blots were performed using standard techniques (Sambrook et al., 1989). The transferred polyvinylidene difluoride membrane was incubated in GADD45 monoclonal antibody in the dilution of 1:500 at 4°C overnight. This was followed by three washes in buffer and then incubation in the AP-conjugated goat antimouse secondary antibodies at room temperature for 60 minutes. A chemiluminescent substrate (Clontech, Palo Alto, CA) was applied to the side of the membrane containing the blotted proteins. The blot was wrapped in plastic wrap and exposed to Kodak X-OMAT film; the film was developed according to the manufacturer's instructions. Immunoreactivity for GADD45 on each individual lane of the blots was quantified by a gel densitometric scanning program using the MCID image analysis system.
DNA strand break detection and histology
A procedure using the large (Klenow) fragment of DNA polymerase I was performed to detect DNA strand breaks in cells on cryostat brain sections obtained from rats killed at 16, 24, 48, or 72 hours after 15 minutes of ischemia (n = 3 per time point) or 24 hours after sham-operation (n = 3). This detection method is based on the specific binding of the Klenow fragment of polymerase I to the exposed DNA template followed by the incorporation of biotin-labeled deoxynucleotides. The sections were fixed with 4% paraformaldehyde in PBS for 15 minutes followed by three washes in PBS (pH 7.4). After the sections were permeabilized with 1% triton X-100 in PBS for 15 minutes and quenched with 2% hydrogen peroxide for 15 minutes followed by three PBS washes, they were incubated in a moist-air chamber at 37°C for 45 minutes in a reaction mixture containing: 5 mmol/L MgCl2; 5 mmol/L 2-mercaptoethanol; 20 μg/mL BSA; 10 μmol/L each of dGTP, dCTP, dTTP, biotinylated dATP; and 30 U/mL of Klenow fragment of DNA polymerase I (Sigma Biochem. Co) in PBS (pH 7.4). The reaction was terminated by washing the sections twice in PBS for total 20 minutes. The slides were then incubated with streptavidin-horseradish-peroxidase in PBS-BSA (VECTASTAIN Elite ABC, Vector Co., Burlingame, CA) for 45 minutes at room temperature. Detection of the biotin-streptavidin-peroxidase complex was performed using a VIP substrate kit which produces an intense, violet-colored precipitate in positively stained cells (Vector Co., Burlingame, CA). To determine nonspecific labeling, alternative slides were incubated in the reaction mixture without Klenow fragment of DNA polymerase I.
To confirm the delayed neuronal cell death in this model, brain sections obtained 24 hours (n = 6) and 72 hours (n = 6) after 15 minutes of global ischemia were stained with cresyl violet.
Double staining
To determine if GADD45 protein is expressed in the same cells that contain DNA strand breaks after ischemia, double staining for GADD45 immunoreactivity and DNA strand break labeling was performed in brain sections obtained at 16, 24, and 72 hours after ischemia (n = 3 each time point). Brain sections were first processed for GADD45 immunostaining using the method described above. After the sections were washed in PBS for 1 hour, an avidin/biotin blocking kit (Vector Co., Burlingame, CA) was used to block the biotin or avidin binding sites potentially resulting from the immunostaining. The sections were then incubated in the reaction mixture containing the Klenow fragment of DNA polymerase I for DNA strand break detection as described above. The incorporated biotinylated dATP in damaged DNA was detected using Rhodamine-avidin D (Cell sorting grade, Vector Co., Burlingame, CA) which selectively binds to biotin and produces a red color under fluorescent microscope using a fluorescent filter for Rhodamine (maximum absorption at 550 nm; maximum emission at 575 nm).
To determine if GADD45 protein is expressed in cell populations other than neurons after ischemia, double immunostaining of GADD45 and glial fibrillary acidic protein (for astrocytes) and of GADD45 and factor VIII-associated antigen (for endothelial cells and blood vessels) were performed on brain sections obtained at 16, 24, and 72 hours after ischemia or 24 hours after sham-operation (n = 3 each time point). In brief, GADD45 immunostaining was first performed using the procedure described above except that the biotinylated secondary antibody (horse antimouse immunoglobulin G) was detected using the Rhodamine-avidin D method. This was followed by an avidin/biotin-blocking procedure. Then, the sections were incubated at room temperature over night in buffer containing either anti—glial fibrillary acidic protein or anti—factor VIII-related antigen antibodies, followed by an incubation in a biotinylated second antibody at room temperature for 2 hours. After the washing procedure, sections were placed in a fluorescein-avidin D solution (Cell sorting grade, Vector Co., Burlingame, CA) for 15 minutes, washed three times in PBS for 2 hours, coversliped, and examined under a fluorescent microscope using filters for Rhodamine and fluorescein, respectively.
RESULTS
Northern blot analysis
The GADD45 oligodeoxynucleotide probe hybridized in a single band at approximately 1.0 kb with both control and ischemic brain samples (Fig. 1). GADD45 mRNA is present at a very low level in the normal control brain, but the level increased 2.3-fold at 4 hours and 4.7-fold at 24 hours after transient global ischemia as determined by densitometric analysis of gels.
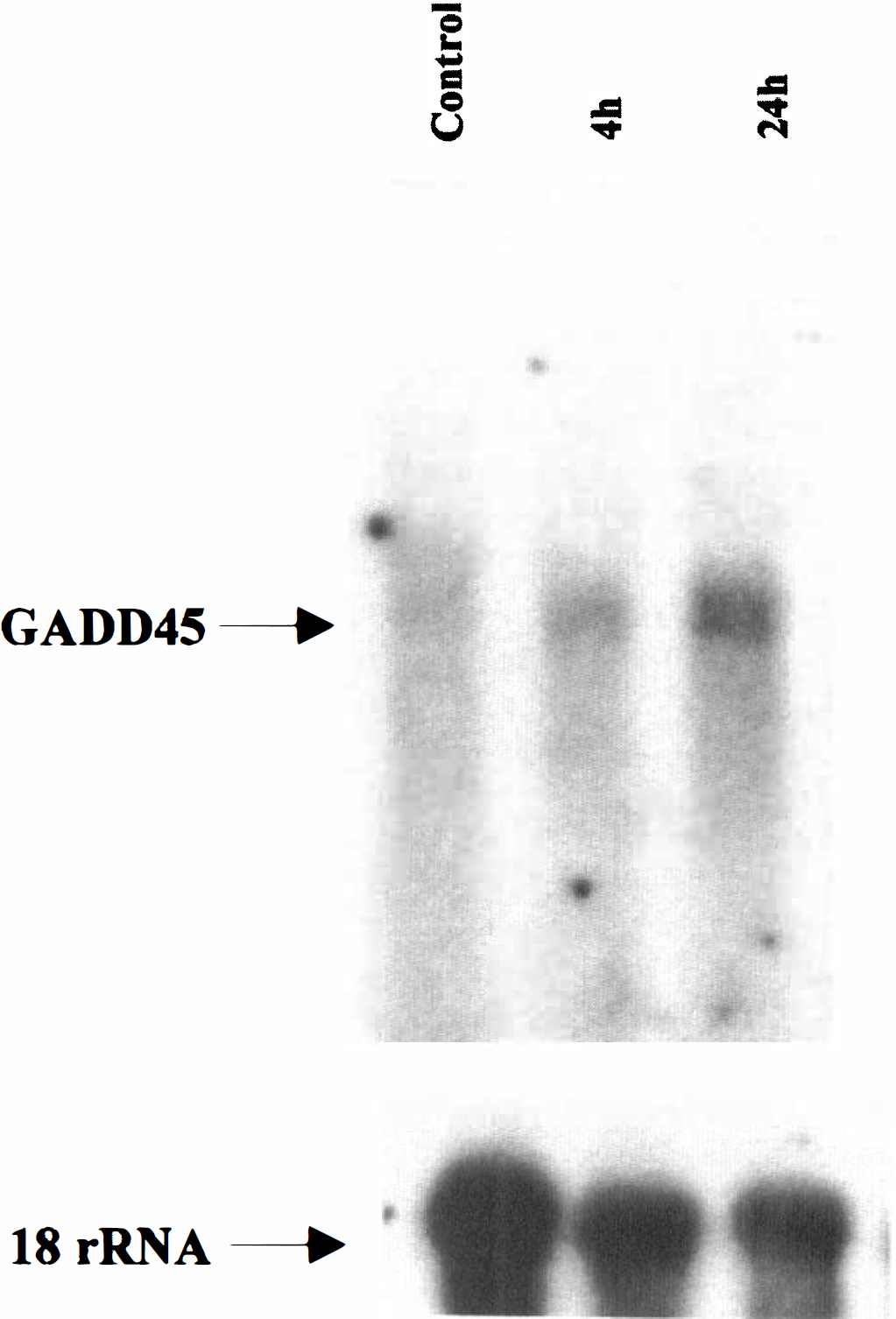
Northern blot analysis of GADD45 mRNA expression in the rat brain after transient global ischemia. Total RNA was isolated from rat hippocampi either 24 hours after sham-operation (control) or 4 hours and 24 hours after ischemia (n = 3 per time point), and electrophoresed through a 1% agarose formaldehyde gel (20 μg of RNA per lane). The only transcription species resulting from hybridizing with the GADD45 oligonucleotide probe is approximately 1.0 kb, consistent with the predicted molecular size of rat GADD45 mRNA. Bottom: the same blot was hybridized with the 18S rRNA probe as a control for sample loading.
In situ hybridization
In control nonischemic rats, basal expression of GADD45 mRNA was detectable, but at an extremely low level, in a variety of cell populations in the brain including the hippocampal neurons, large and medium cortical neurons, and neurons in the striatum and thalamus. In the hippocampus, the basal mRNA level was very low in the pyramidal neurons but relatively high in dentate granule cells (Fig. 2B). Sections hybridized with the sense oligonucleotide probe showed a low-level nonspecific background signal that was homogeneous throughout the brain section (data not shown).
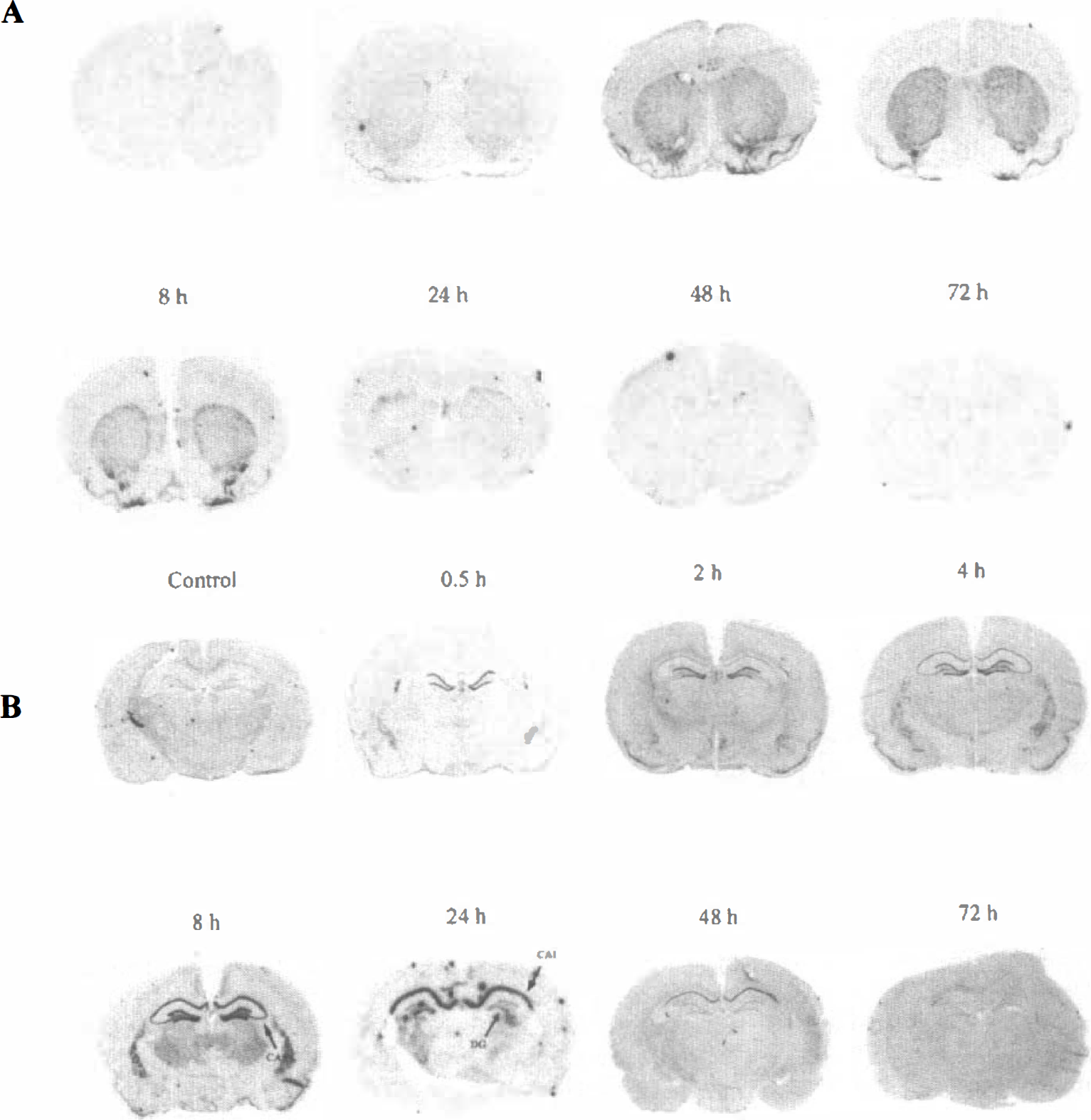
Representative in situ hybridization autoradiograms showing the regional distribution of GADD45 mRNA expression in the rat brain at various time points after transient global ischemia. (
Figures 2A and B show the effect of transient global ischemia on GADD45 mRNA expression in the brain. The relative changes in mRNA measured in the hippocampus, cortex, and caudate putamen are summarized in Fig. 3. Thirty minutes after ischemia, increased GADD45 mRNA signal was first detected in the dentate granule cells. Two hours after ischemia, the signal was further increased in the dentate and began to be increased in the CA1 region, piriform cortex and caudate putamen. At 4 hours after ischemia, the signal was further increased in CA1, the dentate, and caudate putamen, and began to be increased in CA3. At 8 hours after ischemia, the signal was maximally increased in the CA1 and CA3 regions, the dentate and thalamus, but subsided to near control levels in the cortex and caudate putamen. At 24 hours and 48 hours after ischemia, the mRNA signal was maintained at increased level only in the CA1 region, but subsided to control levels in other regions mentioned above. At 72 hours after ischemia, the signal was decreased to below control levels in CA1, consisting with extensive cell loss in this region.
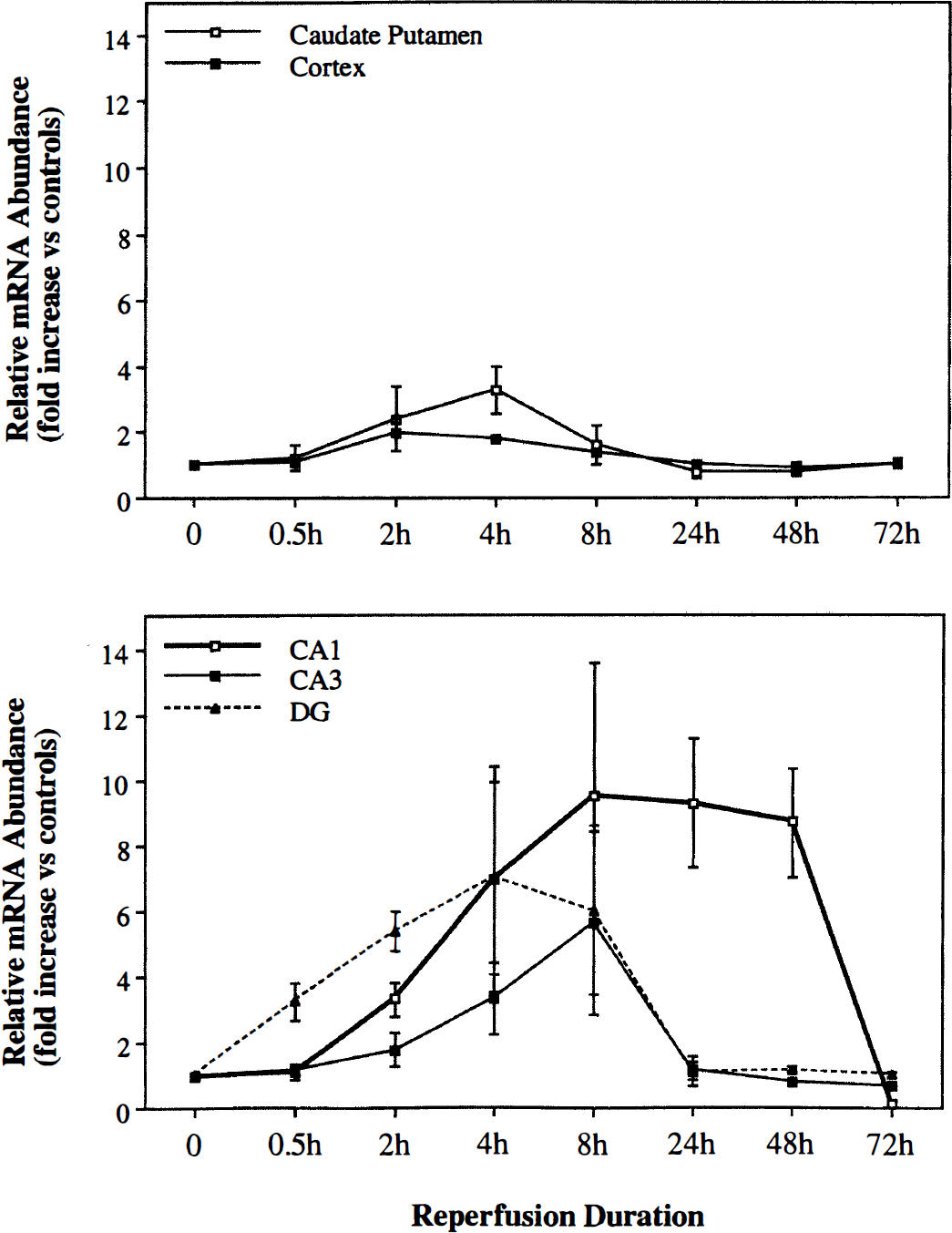
Semiquantitative results of relative levels of GADD45 mRNA expression in various brain regions at various time points after transient ischemia, as determined by measuring the regional optical density (ROD) on in situ hybridization autoradiograms. Data are mean±SD of fold(s) changes of ROD in regions of interest versus nonischemic control brains. Two sections per brain, 3 to 4 brains per time point were analyzed. CA1, the hippocampal CA1 region; CA3, the hippocampal CA3 region; DG, the dentate gyrus.
Examination of emulsion-coated slides revealed that the GADD45 mRNA in situ hybridization signals in various brain regions emanated mainly from neuronal elements (Fig. 4).
Representative micrograms taken from emulsion-coated in situ hybridization slides showing increased GADD45 mRNA signals (silver grains) in various brain regions at 4 hours after ischemia (
Western blot analysis
Western blot analysis detected a major band at approximate 21 kd, the predicted size for the rat GADD45 protein (Yoshida et al., 1994) in all samples tested. GADD45 protein was present at very low levels in control brain samples, but the levels were higher in ischemic samples (Fig. 5A). The quantitative results are summarized in Fig. 5B.
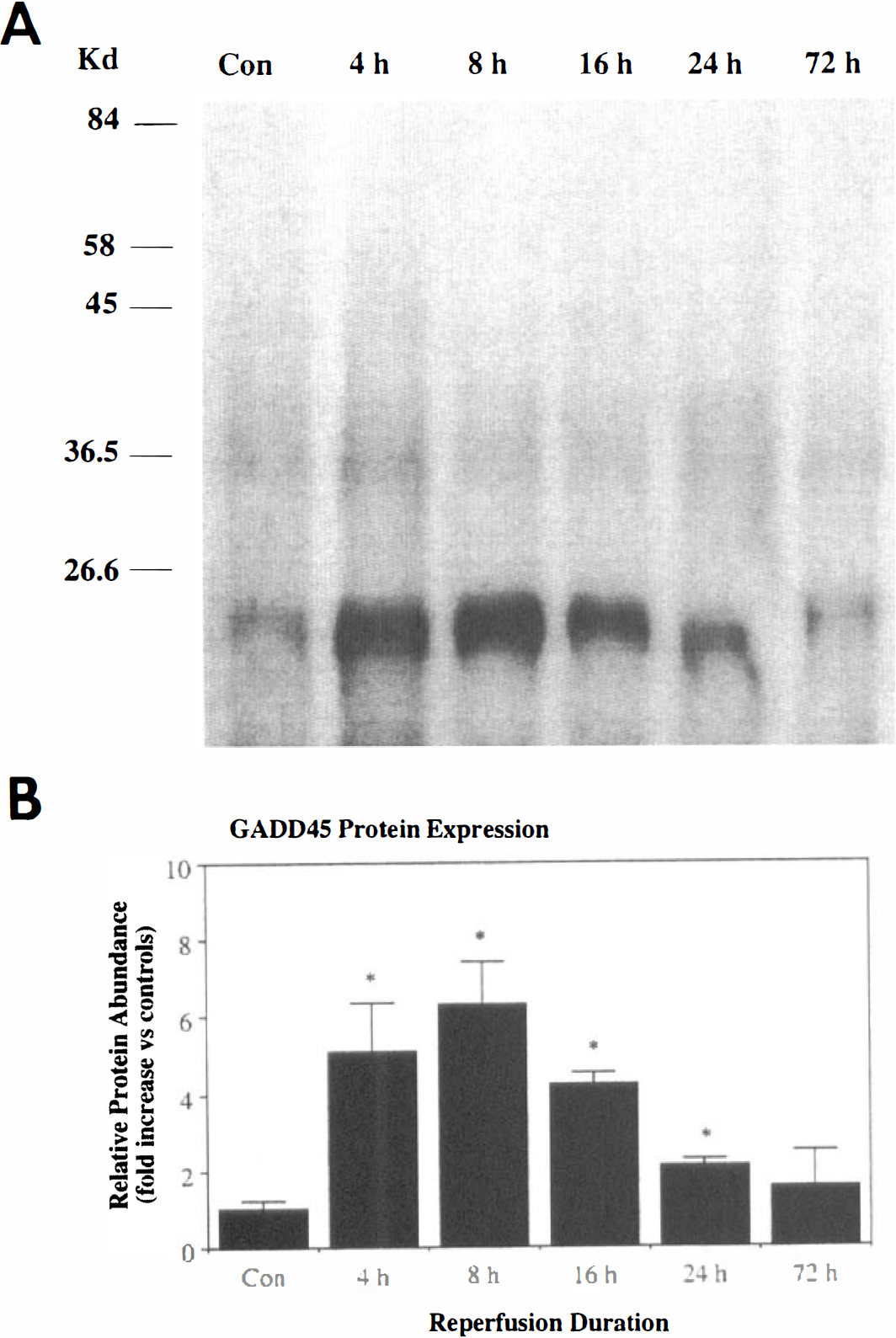
(
Immunocytochemistry
Low level basal GADD45 immunoreactivity was present in neurons of a variety of brain regions, including large and medium neurons in the cortex, and neurons in the caudate putamen and the thalamus. In the hippocampus, basal GADD45 immunoreactivity was detectable mainly in dentate granule cells and, to a lesser extent, in the CA1 pyramidal neurons. After ischemia, GADD45 immunoreactivity was increased in the brain (Fig. 6). Four hours after ischemia, increased GADD45 immunoreactivity was first detected in the hippocampus, including CA1–3 pyramidal neurons, neurons in the hilus, and the dentate granule cells. At 8 hours after ischemia, a remarkable increase in GADD45 immunoreactivity was detected throughout the hippocampus. Increased immunoreactivity was also present in large and medium neurons in layers II, III, V, and VI of the cortex, neurons in the caudate putamen and thalamus. At 16 hours after ischemia, GADD45 immunoreactivity remained at higher levels in the hippocampus and caudate putamen, but subsided to nearly control levels in the cortex and thalamus. At 24 hours, GADD45 immunoreactivity was present in moderately high levels in CA3 neurons and some dentate granule cells, but was decreased to below control levels in the majority of CA1 neurons. At 72 hours after ischemia, moderately increased GADD45 immunoreactivity was still detectable in some CA3 neurons and in a few scattered neurons in the CA1 region.
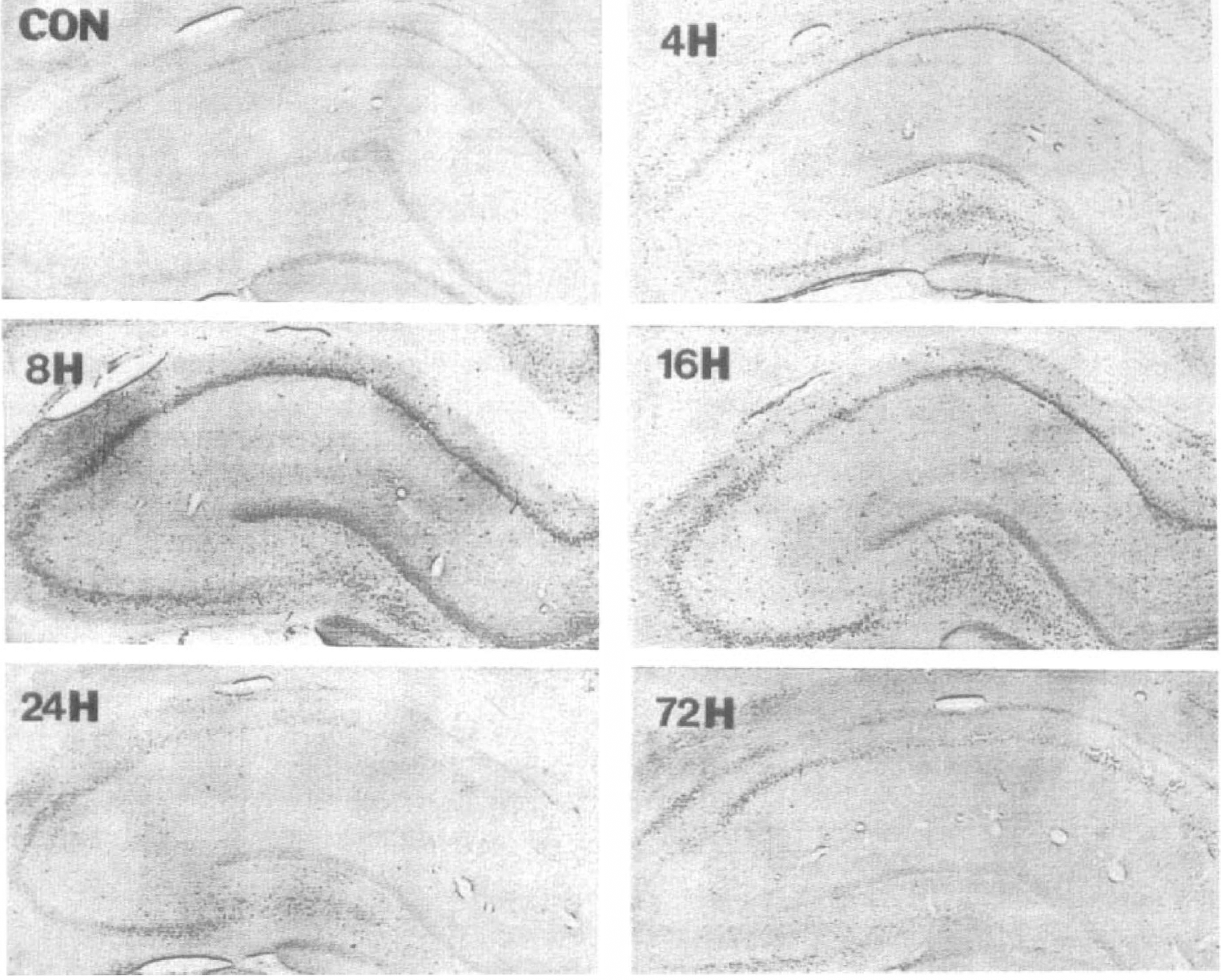
Representative micrograms show the basal GADD45 immunoreactivity in a control hippocampus (Con) and altered immunoreactivity at 4, 8, 16, 24, and 72 hours after ischemia. Immunoreactivity was increased throughout the hippocampus at 4 to 16 hours after ischemia, but was decreased in the CA1 regions at 24 to 72 hours after ischemia. Magnification = x40 for all photos.
DNA strand break labeling and histology
Cresyl violet staining revealed that the hippocampus remained intact at 24 hours after ischemia. At 72 hours after ischemia, however, the total number of neurons in the CA1 region decreased to less than 50% of that in control brains. The majority of the remaining CA1 neurons were pyknotic neurons. Cell death was also detected in several other regions, including the hilus of hippocampus, dorsal caudate-putamen, and dorsal and lateral nuclei of the thalamus.
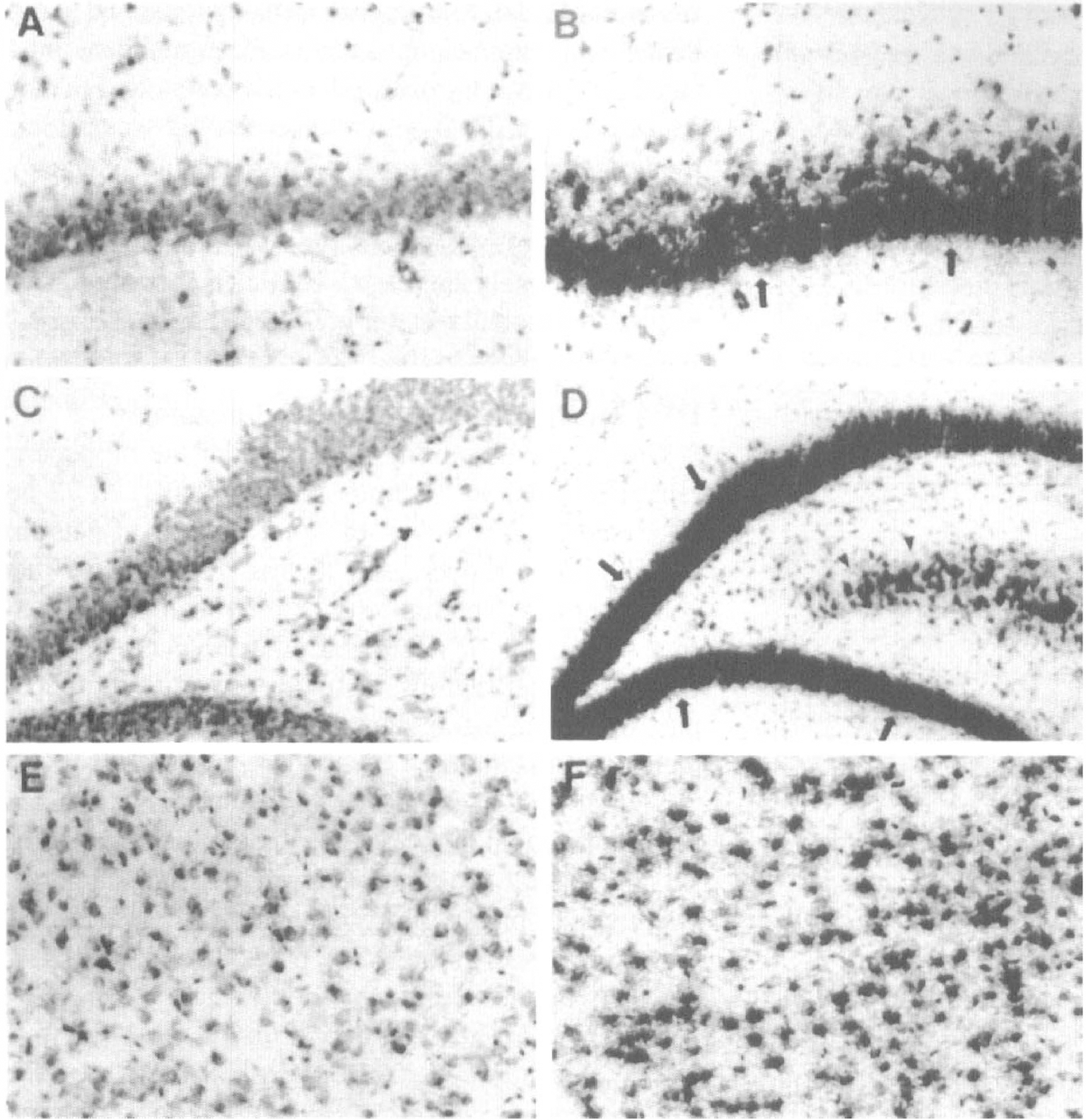
Sixteen to 24 hours after ischemia, many neurons in the hippocampal hilus, dorsal caudate-putamen, and dorsal and lateral thalamus showed positive labeling for DNA strand breaks. Forty-eight to 72 hours after ischemia, increased amounts of DNA-damaged cells were detected in these regions. In the hippocampus, DNA damage was exclusively present in the CA1 region (Fig. 7A–B) and in a few cells in the hilus. Positively labeled cells showed shrinkage and condensation of chromatin; some of them showed fragmented nucleus or the formation of two or more dense masses around the nucleus suggestive of apoptotic bodies (Fig. 7C–D). DNA strand break labeling performed in the absence of the Klenow fragment of DNA polymerase I resulted in negative staining in all sections tested (data not shown).
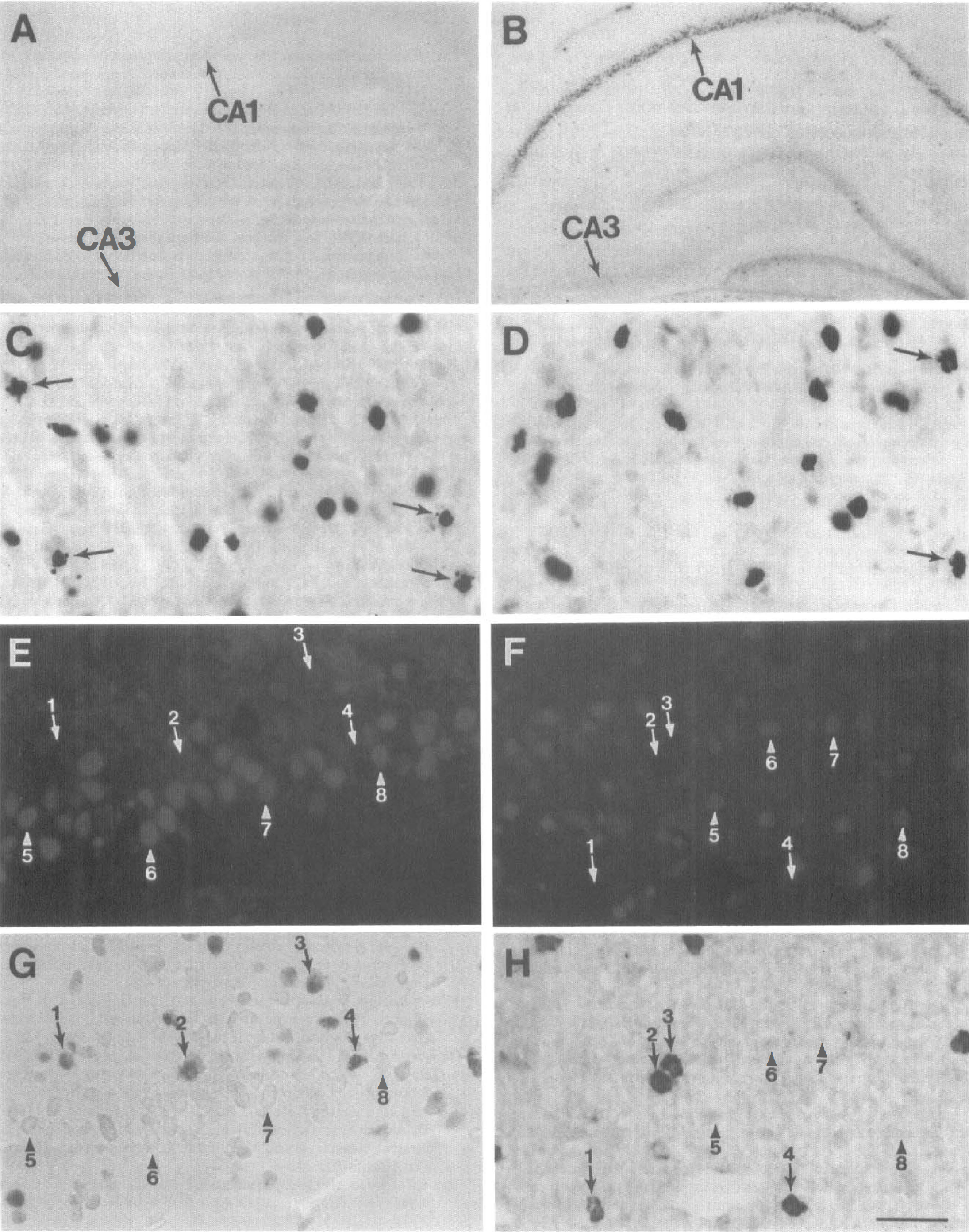
Representative micrograms showing DNA double-strand break labeling after transient ischemia (
Double staining
Positive double staining for DNA strand breaks and GADD45 immunoreactivity was obtained in the dorsal caudate-putamen and thalamus regions at 16 to 24 hours after ischemia, and also in the hippocampal CA1 region at 72 hours after ischemia. Increased GADD45 immunoreactivity was found in cells that did not show DNA strand breaks. Cells that contained DNA strand breaks showed low levels or absence of GADD45 immunoreactivity (Fig. 7E–H).
Double staining was also performed to colocalize GADD45 immunoreactivity to either astrocytes or endothelial cells and blood vessels. No evidence of increased GADD45 immunoreactivity in these cell populations was found in any brain sections tested (data not shown).
DISCUSSION
Similar to many non-neuronal systems, cellular responses to oxidative stress and DNA damage in the brain may involve the inducible expression of gene products that enhance defense against oxidative injury or accelerate recovery from cellular injury. The data presented here show that GADD45, a gene that may be involved in the DNA excision repair process, is induced in neurons after transient global ischemia and reperfusion. Consequently, these results extend the previous observations of GADD45 induction in brain neurons after DNA base damage (Yoshida et al., 1994), quinolinic acid lesioning, (Hughes et al., 1996), ischemic infarction (Jin et al., 1996), or kainate-induced epileptic seizures (Zhu et al., 1997) to another form of brain insult involving glutamate neurotoxicity and oxidative injury.
Consistent with previous findings, both GADD45 mRNA and protein are expressed at very low levels in the control nonischemic brains. During the early reperfusion periods (0.5 to 8 hours after ischemia), expression of GADD45 mRNA is markedly upregulated in neurons located within both vulnerable (e.g., hippocampal CA1 neurons) and less vulnerable regions (e.g., dentate granule cells, CA3 neurons, and the cortex). After longer periods of reperfusion (24 to 48 hours after ischemia), increased GADD45 mRNA is exclusively detected in selectively vulnerable regions, including CA1 pyramidal neurons, scattered neurons in dorsal caudate-putamen, and in the dorsal and lateral thalamus. Therefore, the distribution of persistent expression of GADD45 mRNA (but see below for protein expression) coincides with the distribution of DNA damage detected by in situ nick end-labeling and histologic cell death in this model.
The precise mechanism for GADD45 gene regulation after transient global ischemia is currently uncertain. A number of in vitro studies have shown that GADD45 mRNA can be induced by a variety of genotoxic stimuli that cause high (Fornace et al., 1989a; Fornace, 1992) or low levels of DNA base damage or DNA strand breaks (Papathanasiou et al., 1991; Zhan et al., 1994). In many, but not all, cases, the induction of GADD45 mRNA in cells depends on the normal function of the wild-type p53 tumor-suppressor protein but is not related to the activity of protein kinase C (Papathanasiou et al., 1991). Ionizing-radiation induction of GADD45 is absent in several human tumor cells lines lacking the wild-type p53 phenotype (Fornace et al., 1991). Further, p53 protein transcriptionally regulates GADD45 expression via a highly conserved p53-binding site (the consensus sequence: 5'-PuPuPuC(A/T)(T/A)GPyPyPy-3') in the third intron of the GADD45 gene (Kastan et al., 1992; Hollander et al., 1993; Zhan et al., 1994). Thus, GADD45, along with other genes that contain the p53-binding site consensus sequence such as WAF/p21, Bax, and proliferating cell nuclear antigen (PCNA), have been defined as p53-responsive genes (Kastan et al., 1992; el-Deiry et al., 1993; Miyashita and Reed, 1995). Because oxidative damage to nuclear DNA appears to be an early event in vulnerable brain regions after transient ischemia (Tobita et al., 1995; Liu et al., 1996), and because p53 protein was previously found to be overexpressed in vulnerable neurons after such a brain insult (Wieloch et al., 1995), DNA damage-mediated activation of the p53-dependent pathway could upregulate the expression of GADD45 mRNA after transient global ischemia. This mechanism may explain the persistent expression of GADD45 mRNA in hippocampal CA1 neurons found in the present study. However, induction of GADD45 mRNA was already detected in some regions as early as 0.5 hours (dentate gyrus) and 2 hours (CA1 and caudate putamen) after ischemia, before the induction of p53 in the brain (Wieloch et al., 1995) (Chen et al., unpublished data). Moreover, GADD45 was also induced in some resistant regions such as CA3 and the parietal cortex, where the induction of p53 was not observed. Thus, it appears unlikely that p53 is involved in upregulating the early expression of GADD45. This observation suggests that a p53-independent pathway for GADD45 gene expression may also be present in the brain.
The pattern of GADD45 immunoreactivity during the late reperfusion periods (24 to 72 hours) is distinct from that of GADD45 mRNA expression. After an early (4 to 16 hours after ischemia) protein induction in regions similar to those of mRNA expression, GADD45 immunoreactivity was decreased below control levels in vulnerable neurons although the mRNA was persistently induced in these cells. These changes were synchronous with or preceded histologic evidence of neuron degeneration and DNA fragmentation in these regions. The discrepancy between the mRNA and protein expression shown here suggests that a translational blockage of GADD45 gene expression occurs in dying neurons after transient global ischemia. This blockage of GADD45 protein translation may reflect the impairment of protein synthesis capacity in these severely injured cells. It has been well documented that general protein synthesis is largely compromised in many brain regions after severe global ischemia (Thilmann et al., 1986; Widmann et al., 1991). The recovery of protein synthesis after reperfusion appears to be different between vulnerable and less-vulnerable neurons. Although protein synthesis returns to normal levels in many regions at 12 hours after ischemia, it is persistently inhibited in the CA1 neurons before cell death, suggesting that this persistent blockage of protein translation could be an important factor contributing to selective neuronal death in this region (Thilmann et al., 1986). The failure of protein translation for a number of inducible genes has been observed in these vulnerable neurons after global ischemia, including the heat shock protein HSP70 (Vass et al., 1988; Simon et al., 1991), the immediate early gene NGFI—A (Kiessling et al., 1993), and the apoptosis-suppressor genes bcl-2 and bcl-x (Krajewski et al., 1995; Chen et al., 1997). However, it is unlikely that all protein synthesis is stopped in vulnerable neurons after ischemia. The protective effect against CA1 neuronal death offered by the administration of protein synthesis inhibitors after global ischemia (Papas et al., 1992) suggests that some proteins may be synthesized and play a role in effecting cell death. Among the toxic proteins may be the apoptosis-effector genes such as p53 and Bax, since they were found to be persistently induced in dying neurons after global ischemia or focal ischemia (Chopp et al., 1992; Krajewski et al., 1995; Wieloch et al., 1995; Chen et al., 1996). Taken together, these results suggest that the diminished synthesis capacity in dying ischemic neurons might result in a “selection” for adverse proteins to be synthesized. Such a selection for protein translation on induction of a lethal stimulus may reflect a cellular mechanism that controls gene expression and affects the fate of injured cells.
The induction of GADD45 protein in neurons after global ischemia may have a functional role. The induction of GADD45 on DNA damage was originally thought to serve as a regulator for cell cycle checkpoints, where GADD45 (as well as other growth arrest genes) transiently arrests cell growth to allow time for DNA repair to be completed (Jackman et al., 1994; Zhan et al., 1994). DNA-damaged cycling cells that fail to arrest at the cell cycle check point exhibit increased cell death and DNA fragmentation (Lau and Pardee, 1982). Recently, using an in vitro nuclear extract DNA-repair assay, Smith et al found that the GADD45 recombinant protein was able to stimulate DNA nucleotide excision repair (Smith et al., 1994), although this action of GADD45 on DNA repair was not observed using a whole-cell extract preparation (Kazantsev and Sancar, 1995). Smith et al suggested that GADD45 might interact with cellular DNA repair systems through binding to PCNA. PCNA is an important nuclear auxiliary protein that is required for the DNA nucleotide excision repair pathway (Nichols and Sancar, 1992; Shivji et al., 1992; Sancar, 1994). Subsequently, PCNA was found to be involved in the DNA base excision repair pathway as well (Matsumoto et al., 1994). Because the repair of oxidative DNA base damage relies on the base excision repair mechanism (Satoh and Lindahl, 1994), it is possible that GADD45 may also stimulate DNA base excision repair. More recently, the corresponding region of the PCNA molecule for GADD45 interaction has been identified (Hall et al., 1995). When this region is selectively mutated, cells exhibit increased sensitivity to ultraviolet radiation—induced DNA damage (Ayyagari et al., 1995), suggesting that GADD45 protein may indeed contribute to DNA repair in vivo. Although the present study does not prove a functional role for GADD45, the observations presented here and reported by others that the GADD45 gene is highly induced in the brain after injury, especially the observation that the encoded protein is persistently expressed in neurons that are injured but survived after ischemia or N-methyl-D-aspartate receptor neurotoxicity (Hughes et al., 1996; Jin et al., 1996), suggest that GADD45 could play a protective role, perhaps by accelerating DNA repair. Consequently, failure of expression of GADD45 protein (e.g., in dying neurons as shown by double-staining) may have a adverse effect on DNA repair and cell survival.
Numerous studies in non-nervous system tissues have defined a strong linkage between oxidative DNA damage, incomplete DNA repair, and cell death (Janssen et al., 1993). Likely, the DNA damage- and repair-related molecular events may also contribute to ischemic brain injury (Chopp et al., 1996). The results presented here show that one of the DNA repair genes is induced in neurons after global ischemia Beside the GADD45 gene, other DNA repair genes such as PCNA and DNA polymerase β are also inducible after ischemia (Tomasevic et al., 1995; Chopp et al., 1996; Stetler et al., 1996). However, whether the proteins encoded by these genes indeed participate in neuronal DNA repair and consequently have a role in cell survive remains to be determined.
Footnotes
Acknowledgments
The authors thank Michelle Huerbin and Kendra Basta for their excellent technical support, and Pat Saracco for secretarial support.