
Abstract
Hepatocyte growth factor (HGF) is one of the prospective agents for therapy against a variety of neurologic and neurodegenerative disorders, although the precise mechanisms for the effect of HGF remain to be elucidated. We showed that treatment with HGF protected hippocampal cornu ammonis (CA) subregion 1 neurons from apoptotic cell death after transient forebrain ischemia. Accumulating evidence indicates that ischemia-induced neuronal damage occurs via caspase-independent pathways. In the present study, we focused on the localization of apoptosis-inducing factor (AIF), which is an important protein in the signal-transduction system through caspase-independent pathways, to investigate the possible mechanism for the protective effect of HGF after transient forebrain ischemia. Hepatocyte growth factor attenuated the increase in the expression of AIF protein in the nucleus after transient forebrain ischemia. We further explored the upstream components of AIF translocation. Primary DNA damage induced by Ca2+ influx and subsequent NO formation are thought to be the initial events for AIF translocation, which results in the subsequent DNA damage by AIF. Hepatocyte growth factor prevented the primary oxidative DNA damage, as was estimated by using anti-8-OHdG (8-hydroxy-2'-deoxyguanosine) antibody. Oxidative DNA damage after ischemia is known to lead to the activation of poly(ADP-ribose) polymerase (PARP) and p53, resulting in AIF translocation. Marked increases in the PAR polymer formation and the expression of p53 protein after ischemia were effectively prevented by HGF treatment. In the present study, we first showed that HGF was capable of preventing neuronal cell death by inhibiting the primary oxidative DNA damage and then preventing the activation of the PARP/p53/AIF pathway.
Keywords
Introduction
Transient forebrain ischemia leads to the degeneration of vulnerable neurons in the brain, including pyramidal neurons in the hippocampal cornu ammonis (CA) subregion 1 (CA1) region. As the degeneration of neurons induced by cerebral ischemia results ultimately in dysfunction of the central nervous system (CNS), it is an important objective to explore strategies for protecting cells from cerebral ischemia-induced death. In this context, treatment with several neurotrophic factors have been attempted to prevent ischemic brain injury and to restore normal neuronal function.
Hepatocyte growth factor (HGF) is a multifunctional cytokine originally identified and purified as a potent mitogen for hepatocytes (Nakamura et al, 1984, 1987). Hepatocyte growth factor is known to evoke diverse cellular responses, including mitogenic, motogenic, morphogenic, angiogenic, and anti-apoptotic ones in various types of cells (Nakamura et al, 1984, 1989; Matsumoto and Nakamura, 1996). Hepatocyte growth factor and its receptor c-Met were recently found to be expressed in the CNS (Honda et al, 1995; Achim et al, 1997), and to promote the survival of hippocampal and cortical neurons during the aging of cells in culture (Honda et al, 1995; Hamanoue et al, 1996). In addition, exogenous HGF prevented neuronal cell death in the hippocampal CA1 region after transient forebrain ischemia in gerbils and attenuated the development of cerebral infarction after transient focal ischemia and widespread cerebral embolism in rats (Tsuzuki et al, 2001; Miyazawa et al, 1998; Hayashi et al, 2001; Date et al, 2004). These findings suggest that HGF has the ability to prevent cell injuries and to improve function in the CNS. Although such protective effects might be mediated by multipotent activities of HGF, including its antiapoptotic activity, their precise mechanisms remain unclear.
Although it is well known that caspase-dependent pathways play a role in apoptotic cell death after cerebral ischemia (Le et al, 2002; Niwa et al, 2001; Davoli et al, 2002; Wick et al, 2004), caspase inhibitors are likely to reduce ischemic injury after transient focal ischemia but not after a moderately long global ischemia (Li et al, 2000). The result indicates that caspase-independent pathways can contribute to cell death after transient forebrain ischemia. In this sense, accumulating evidence indicate that caspase-independent pathways are also involved in ischemia-induced neuronal damages (Zhang et al, 2005; Plesnila et al, 2004; Cao et al, 2003). An important protein in this pathway is thought to be the apoptosis-inducing factor (AIF), which is usually located in mitochondria in normal cells and acts as a mitochondrial oxidoreductase (Susin et al, 1999; Daugas et al, 2000). Once AIF is released from lesioned mitochondria, it produces reactive oxygen species (ROS) in the cytoplasm and also leads to large scale (~ 50 kbp) DNA fragmentation in the nucleus. This AIF-related apoptotic pathway is not affected by caspase inhibitors (Susin et al, 1999, 2000; Daugas et al, 2000; Cande et al, 2002). Cerebral ischemia appears to cause translocation of AIF from the mitochondria to the nucleus (Culmsee et al, 2005; Zhu et al, 2003; Plesnila et al, 2004; Zhao et al, 2004; Cao et al, 2003), suggesting that AIF plays a role in ischemia-induced neuronal cell death. Questions remain as to whether the inhibition of this caspase-independent pathway is involved in the protective effect of HGF in the ischemic brain, and if so, at what point in the process does HGF act. In the present study, we focused on the effect of HGF on the expression of AIF protein in the nucleus in the hippocampal CA1 region after transient forebrain ischemia and also explored the upstream components of AIF translocation. We first showed that HGF decreased nuclear translocation of AIF triggered by ischemia and reperfusion, which might be mediated by the prevention of primary oxidative DNA damage and the attenuation of subsequent activation of poly(ADP-ribose) polymerase (PARP) and p53.
Materials and methods
Recombinant Hepatocyte Growth Factor
Human recombinant HGF was purified from culture medium conditioned by Chinese hamster ovary cells transfected with an expression vector containing human HGF cDNA, as described earlier (Nakamura et al, 1989). The purity of HGF was > 98%, as determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
Animal Model
Male Wistar rats weighing 200 to 250 g (Charles River Japan Inc., Atsugi, Japan) were used in the present study. The animals were housed in a cage and maintained on a 12-h light/12-h dark cycle at a temperature of 23°C ± 1°C with a humidity of 55% ±5% throughout the experiment. The animals had free access to food and water according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the Guideline of Experimental Animal Care issued by the Prime Minister Office of Japan. All efforts were made to minimize animal suffering, to reduce the number of animals used, and to use alternatives to in vivo techniques, if available. The experimental protocol was approved by the Committee of Animal Care and Use of Tokyo University of Pharmacy and Life Science. Transient (15 mins) forebrain ischemia was produced by the four-vessel occlusion procedure for rats described previously (Takagi et al, 2003). In brief, rats were anesthetized intraperitoneally with 40 mg/kg sodium pentobarbital. The right and left second cervical vertebras were exposed, and both visible vertebral arteries were permanently electrocauterized. Two silk threads were placed around both common carotid arteries without interrupting the blood flow. Twenty-four hour after electrocauterization, anesthesia was induced with 3% enflurane and maintained with 1.5% enflurane in a mixture of oxygen/nitrous oxide (25%/75%). Bitemporal subdermal electroencephalogram (EEG) needle electrodes were placed in reference to a frontal subdermal electrode. After a baseline EEG level had been established, both common carotid arteries were exposed and occluded with aneurysm clips for 15 mins. Then, the clips were removed, and the rat was allowed to recover. Rectal temperature was continuously monitored during ischemia and was maintained at 37.0°C to 37.5°C with a heating pad. Only rats that showed a completely flat EEG and a loss of consciousness during the occlusion were chosen for use in the present study. Sham-operated animals received exactly the same surgical procedure, but without the arterial occlusion. Each set of animals received the same degree of surgical preparation and the same recovery paradigms to minimize variations that might result from surgical procedures.
In vivo Hepatocyte Growth Factor Treatment
Hepatocyte growth factor was diluted in physiologic saline and infused into the right hippocampal CA1 region by using an osmotic pump (Alzet model 1003D; DURECT Corp., Cupertino, CA, USA) attached to a 30-G needle implanted 3.5 mm posterior and 2.5 mm lateral to the bregma, and at a depth of 2.4 mm from the cortical surface. Before the start of infusion just after needle implantation, each osmotic pump was preincubated in physiologic saline at 37°C according to the instructions for use of the Alzet. The infusion of HGF was begun at 10 mins after the start of reperfusion at a flow rate of 1.0 μL/h and a concentration of 100 μg/mL (10 μg/3 days/animal). As a control, physiologic saline was used for the infusion.
Tissue Preparation
At various times after the start of reperfusion, animals were killed by decapitation, and their heads were quickly near-frozen in liquid nitrogen. The hippocampi were removed on ice, and hippocampal slices (730 mm) were prepared with a McIlewain tissue chopper (Brinkmann, Mickle Laboratory Engineering Co., Ltd, Gomshall, Surrey, UK). The hippocampal CA1 regions were dissected on ice in ice-cold 125 mmol/L Tris-HCl, pH 7.4, containing 320 mmol/L sucrose, 2 mmol/L sodium orthovanadate, 20 mmol/L sodium diphosphate decahydrate, 20 mmol/L DL-α-glycerophosphate, 0.1 mmol/L phenylmethylsulfonyl fluoride, and 5 mg/mL each of antipain, aprotinin, and leupeptin (homogenization buffer). The dissected CA1 region was homogenized in the ice-cold homogenization buffer. The samples were stored at −80°C until used and were thawed only once.
Western Immunoblotting
Hippocampal CA1 homogenates that had been solubilized by heating at 100°C for 5 mins in SDS sample buffer (10% glycerol, 5% β-mercaptoethanol, and 2% SDS in 62.5 mmol/L Tris-HCl, pH 6.8) were separated on 10% or 12% polyacrylamide gels and transferred to a polyvinylidene difluoride membrane. Protein blots were incubated with the appropriate antibodies, and the bound antibody was detected by the enhanced chemiluminescence method (Amersham Biosciences Inc., Piscataway, NJ, USA) as described by the manufacturer. Quantification was performed by using computerized densitometry and an image analyzer (ATTO Co., Tokyo, Japan). Care was taken to ensure that bands to be semiquantified were in the linear range of response. For removal of bound antibodies, immunoblots were heated for 30 mins at 65°Cin 62.5 mmol/L Tris-HCl buffer, pH 6.8, containing 2% SDS and 0.1mol/L β-mercaptoethanol. The efficacy of the stripping procedure was confirmed by reacting the stripped blot with secondary antibody alone to ensure that no bound antibodies had remained. Antibodies used were antiphospho-NR2B (Tyr 1472) (Chemicon, Temecula, CA, USA), anti-NR2B (clone 13; Transduction Laboratories, Lexington, KY, USA), anti-AIF (Chemicon, Temecula, CA, USA), anti-heat-shock protein (Hsp) 70 (Calbiochem, La Jolla, CA, USA), and anti-α-tubulin (Sigma-Aldrich, St Louis, MO) antibody.
Immunoprecipitation
For immunoprecipitation of AIF, hippocampal CA1 tissues were lysed in a buffer containing 10 mmol/L Tris-HCl, pH 7.5, 0.5% Triton X-100, 150 mmol/L NaCl, 2 mmol/L sodium orthovanadate, 0.1 mmol/L phenylmethylsulfonyl, 5 μg/mL each of antipain, aprotinin, and leupeptin. The lysates were preincubated for 1 h with L protein G-agarose beads and then centrifuged to remove any proteins that adhered nonspecifically to the protein G-agarose beads. The supernatant was then incubated at 4°C with anti-AIF antibody overnight. Next, protein G-agarose beads were added, and the incubation was continued at 4°C for 2 h. The immune complexes were isolated by centrifugation and washed, and the bound proteins were eluted by heating at 100°C in SDS sample buffer.
Histological Analysis
Animals were perfused transcardially with 4% paraformaldehyde (PFA) in 0.1 mol/L phosphate buffer (pH 7.4, phosphate buffer (PB)) under deep anesthesia. Their brains were quickly removed, cut into approximately 5-mm-thick coronal slabs, and postfixed overnight with 4% PFA in 0.1 mol/L PB. The slabs were embedded in paraffin and cut serially at 5 μm with a microtome. The coronal sections were then stained with cresyl violet acetate to assess neuronal damage. Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL)-positive cells were detected by using an in situ Apoptosis Detection Kit (MK500; Takara Bio Inc., Shiga, Japan). Surviving pyramidal cells and TUNEL-positive cells in the hippocampal CA1 region were counted under x 400 magnification (Olympus BX-52) in five to seven sections per animal. Results were expressed as the average number of cells per mm2 in the areas comprising the hippocampal CA1 pyramidal cell layer. For immunostaining, sections were incubated with 100 mmol/L Trisbuffered saline containing 0.1% Triton X-100 (TBST) for 30 mins, and then treated with 3% hydrogen peroxide for 5 mins to quench endogenous peroxidase. After blocking, the sections were incubated overnight at 4°C with mouse i anti-8-OHdG (8-hydroxy-2'-deoxyguanosine) (QED Bioscience, San Diego, CA, USA), mouse anti-poly(ADP-ribose) (PAR; Biomol, Plymouth Meeting, PA, USA), rabbit anti-AIF (Chemicon) or rabbit anti-p53 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) antibody. After having been washed, the sections were incubated with biotinylated anti-rabbit immunoglobulin G (IgG) antibody (DAKO, Carpinteria, CA, USA) for AIF and p53 or with biotinylated anti-mouse IgG antibody (DAKO) for 8-OHdG and PAR for 2 h and then with avidin: biotinylated enzyme complex solution (Vector) for 2 h. Color development was performed by incubating with 3,3'-diaminobenzidine and hydrogen peroxide (Vector). For 8-OHdG detection, sections were treated with RNase A (50 μg/mL in phosphate-buffered saline) at 37°C for 30 mins before blocking. Images were obtained by using an Olympus microscope (BX-52) or a Bio-Rad MRC 1024 confocal imaging system equipped with a krypton-argon laser and Nikon Diapot microscope, and processed by Adobe Photoshop (Adobe Systems, Mountain View, CA, USA). The microscopic observations were performed by a person unaware of the study group.
Statistics
The results were expressed as the means + s.e. Statistical comparison among multiple groups was evaluated by analysis of variance (ANOVA) followed by Scheffe's test or Fisher's protected least significant difference test. Differences with a probability of 5% or less were considered significant (P < 0.05).
Results
At first, we examined the effect of HGF on neuronal cell death in the hippocampal CA1 region of the four-vessel-occluded rats on day 3. Neuronal cell death in the hippocampal CA1 region after ischemia was significantly prevented by treatment with HGF at 10 μg/3 days/animal (Figures 1A-1C, Table 1). The dose used in the present study was based on the data obtained in our preliminary study, which showed that treatment at 10 μg/3 days/animal exerted the maximum protective effect. We next examined the effect of HGF on the number of TUNEL-positive cells in the hippocampal CA1 region after ischemia. The increase in the number of TUNEL-positive cells in the hippocampal CA1 region after ischemia was almost completely suppressed by HGF treatment (Figures 1D-1F, Table 1).
Effect of HGF on the number of viable neurons in the hippocampal region after transient forebrain ischemia followed by 3-day reperfusion (l/R3day)

CA1=cornu ammonis (CA) subregion 1; HGF = hepatocyte growth factor; I/R = ischemia/reperfusion; TUNEL = terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling.
The number of cresyl violet- and TUNEL-stained cells were counted. Values represented the means±s.e. n = 3-6.
P < 0.05 versus nonoperated naïve control;
P < 0.05 versus I/R3day, ANOVA with post hoc Scheffe.
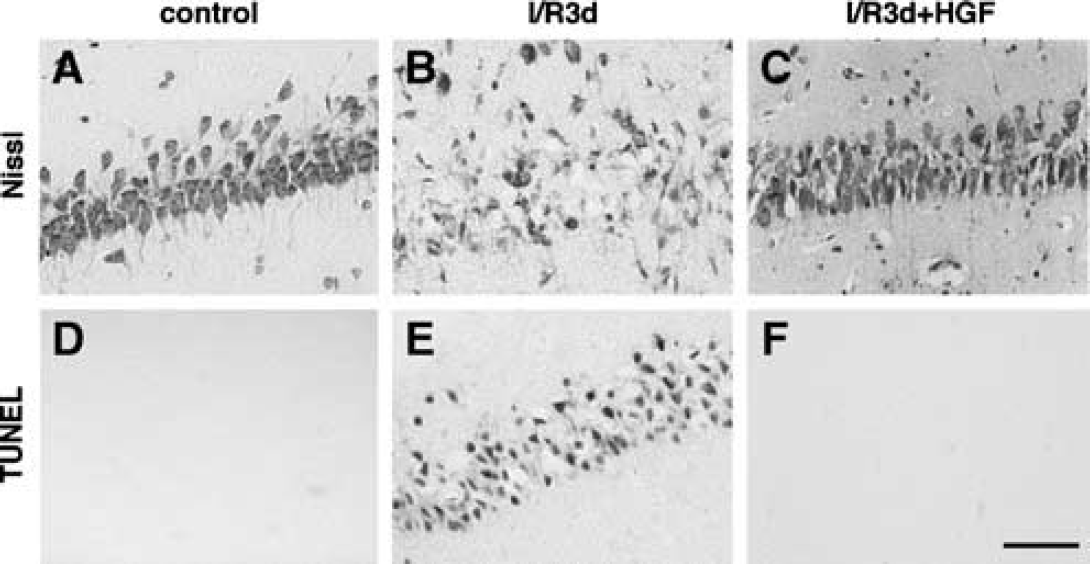
Effect of hepatocytes growth factor (HGF) on neuronal cell death in the hippocampal cornu ammonis (CA) subregion 1 (CA1) region after transient forebrain ischemia. (
To elucidate the mechanism for the antiapoptotic effect of HGF on the hippocampal CA1 neurons, we focused on the expression of AIF protein in the nucleus, which is one of the important proteins in the caspase-independent pathway. Immunoblotting showed that the total amount of AIF protein after ischemia was not altered regardless of treatment or not with HGF compared with that of nonoperated naive control rats (Figure 2I). Apoptosis-inducing factor in nonoperated naive rats was expressed in the neuronal cytoplasm (Figure 2A). The expression of AIF in the nucleus was evident at 24 h after the start of reperfusion (Figure 2C), and it became intensive at 36 h (Figure 2E). The number of AIF-positive nuclei increased after ischemia (Figures 2E and 2J), and this increase was attenuated by the HGF treatment (Figures 2G and 2J).
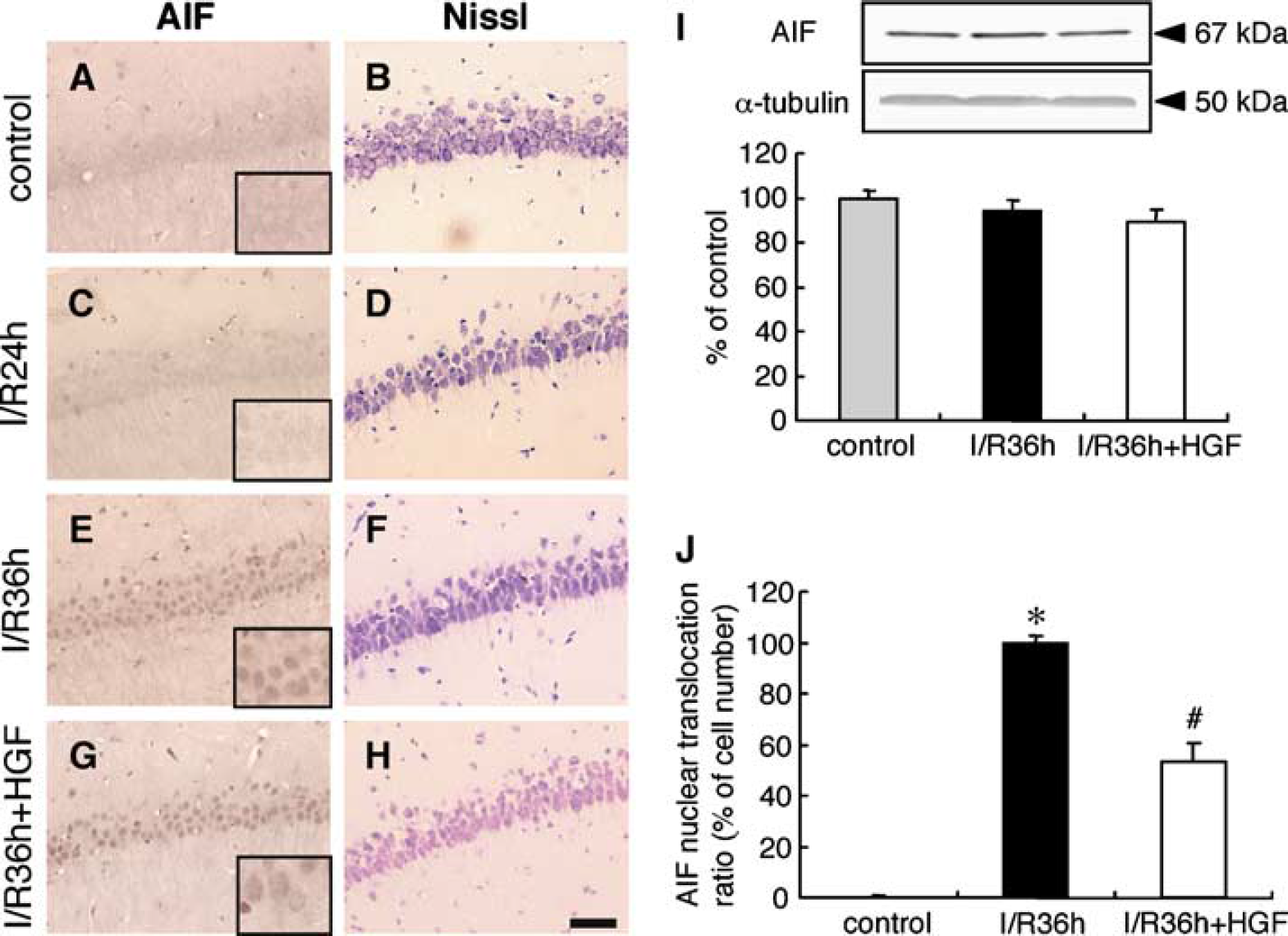
Effect of hepatocyte growth factor (HGF) on the expression of apoptosis-inducing factor (AIF) in the nucleus of the hippocampal cornu ammonis (CA) subregion 1 (CA1) region after transient forebrain ischemia. (
We next investigated the expression of Hsp70 protein after the start of reperfusion with or without HGF treatment, as Hsp70 is known to be an endogenous inhibitor of AIF. In nonoperated naïve rats, Hsp70 protein was barely expressed in the hippocampal CA1 region (Figure 3A). The amount of Hsp70 protein was significantly increased after transient ischemia, and the level of Hsp70 was not influenced by the HGF treatment (Figure 3A). Furthermore, we examined changes in the interaction of Hsp70 with AIF after ischemia with or without HGF treatment. Although the interaction of Hsp70 with AIF was elevated to 353.4% 7124.1% of the control value after ischemia, it was not influenced by HGF treatment (393.9% ± 81.6%) (Figure 3B)
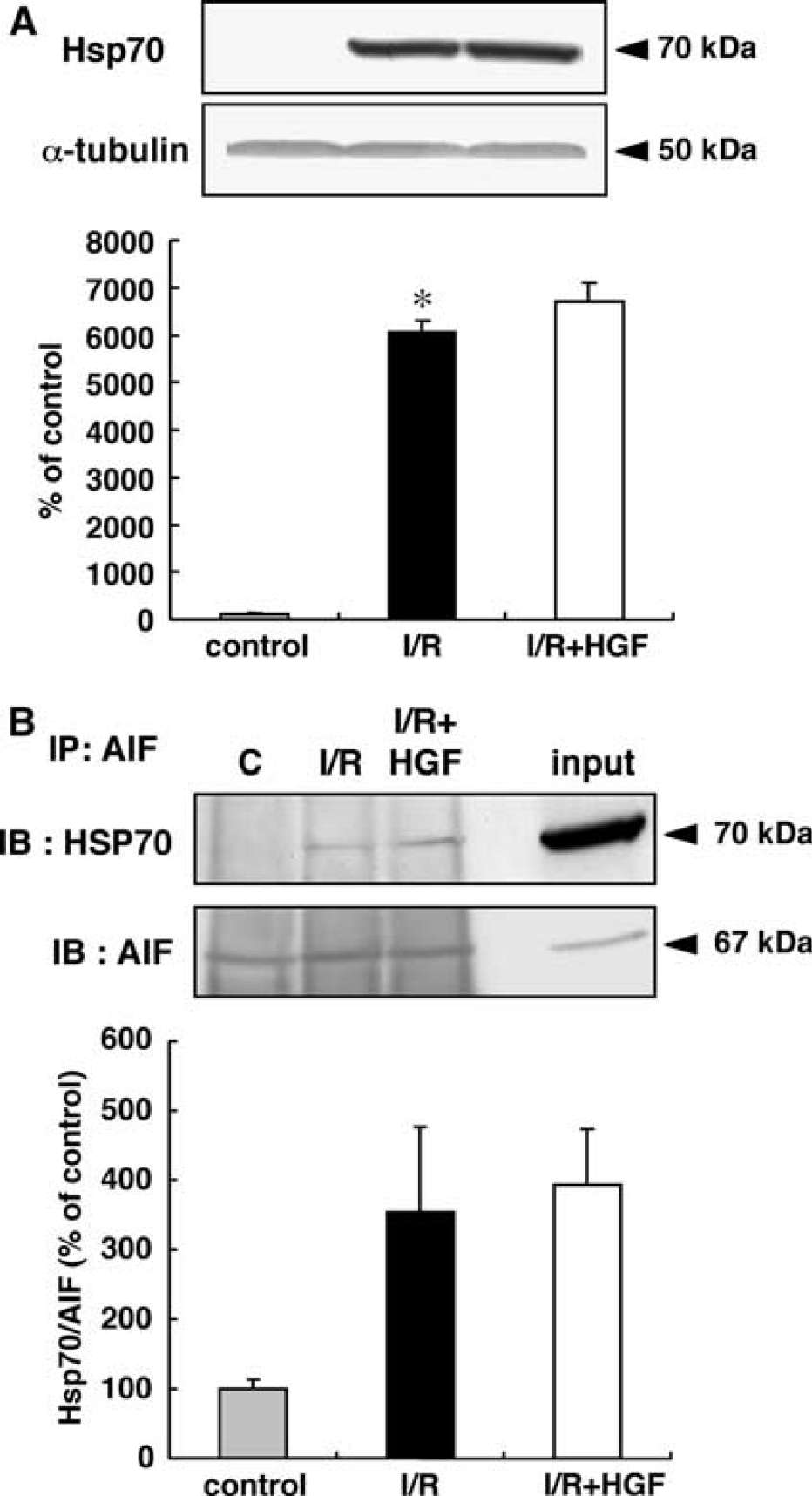
Effect of hepatocyte growth factor (HGF) on the expression of heat-shock protein 70 (Hsp70) and the interaction of Hsp70 with apoptosis-inducing factor (AIF) in the hippocampal cornu ammonis (CA) subregion 1 (CA1) region after transient forebrain ischemia. (
The activation of the N-methyl-D-aspartate (NMDA) receptors after ischemia leads to a marked increase in Ca2+ influx, which causes activation of nitric oxide synthase (NOS) and subsequent production of nitric oxide (NO). Using antityrosine phosphorylated NR2B antibody, we examined tyrosine phosphorylation of the NR2B subunit at the src site, Y1472, in the NMDA receptor after ischemia with or without HGF treatment as an indicator of its activity. The tyrosine phosphorylation of the subunit at 1 h of reperfusion was 10 times larger than that of control rats, and it returned to the control level by 24 h of reperfusion (Figure 4A). Treatment with HGF did not affect the ischemia-induced tyrosine phosphorylation of the NR2B subunit at any of the all time points investigated (Figure 4A).
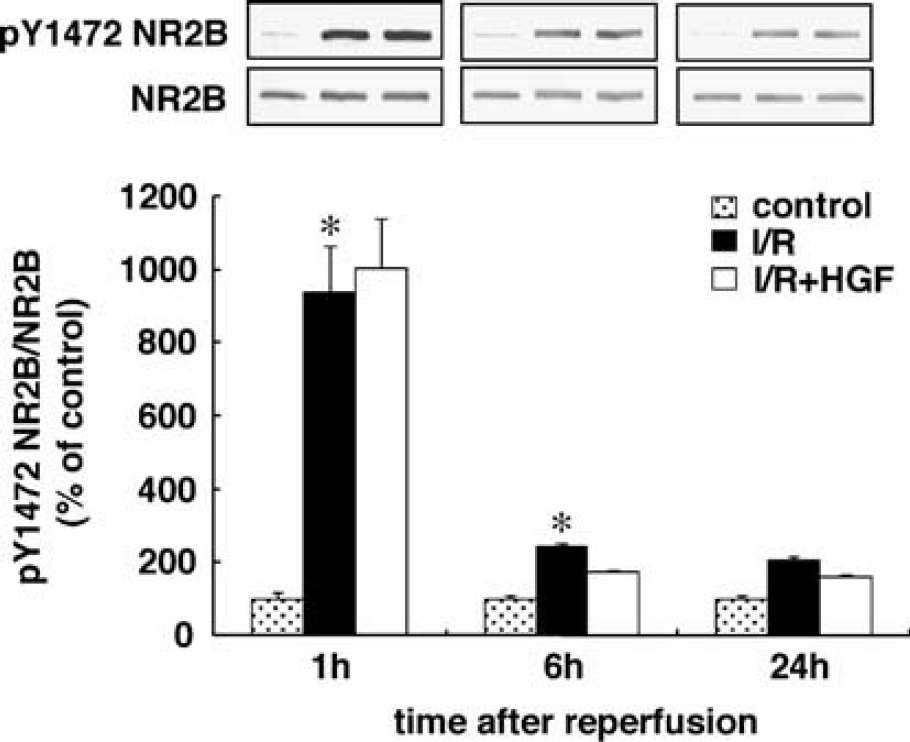
Effect of hepatocyte growth factor (HGF) on phosphorylation of the N-methyl-D-aspartate (NMDA) receptor in the hippocampal cornu ammonis (CA) subregion 1 (cA1) region after transient forebrain ischemia. Proteins (50 μg) from nonoperated naïve rats and four-vessel-occluded rats at 1, 6, and 24 h of reperfusion without (I/R) or with HGF (I/R + HGF) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and subjected to immunoblotting with anti-phospho-NR2B antibody [pY1472 NR2B]. The blots were then stripped and reprobed with anti-NR2B antibody [NR2B]. Bands corresponding to pY1472 NR2B or NR2B were scanned, and the scanned bands of pY1472 NR2B were normalized by NR2B on the same blot. Results are the mean percentages of the nonoperated naïve control± s.e. n = 3. *P < 0.05 versus nonoperated naïve control, analysis of variance (ANOVA) with post hoc Fischer's protected least significant difference.
Next, we examined the immunoreactivity of 8-OHdG, which is often used as a marker of oxidative DNA damage. We at first examined the time course of changes in its immunoreactivity after transient forebrain ischemia. The immunoreactivity of 8-OHdG was very faint in nonoperated naive rats (Figure 5A), whereas it increased at 30 mins of reperfusion (Figure 5B). The maximum increase in the immunoreactivity was detected at 6 h of reperfusion (Figure 5D), and the immunoreactivity gradually disappeared thereafter (Figures 5E and 5F). The prominent increase in 8-OHdG expression in the nucleus at 6 h of reperfusion was almost completely suppressed by the HGF treatment (Figure 5G).
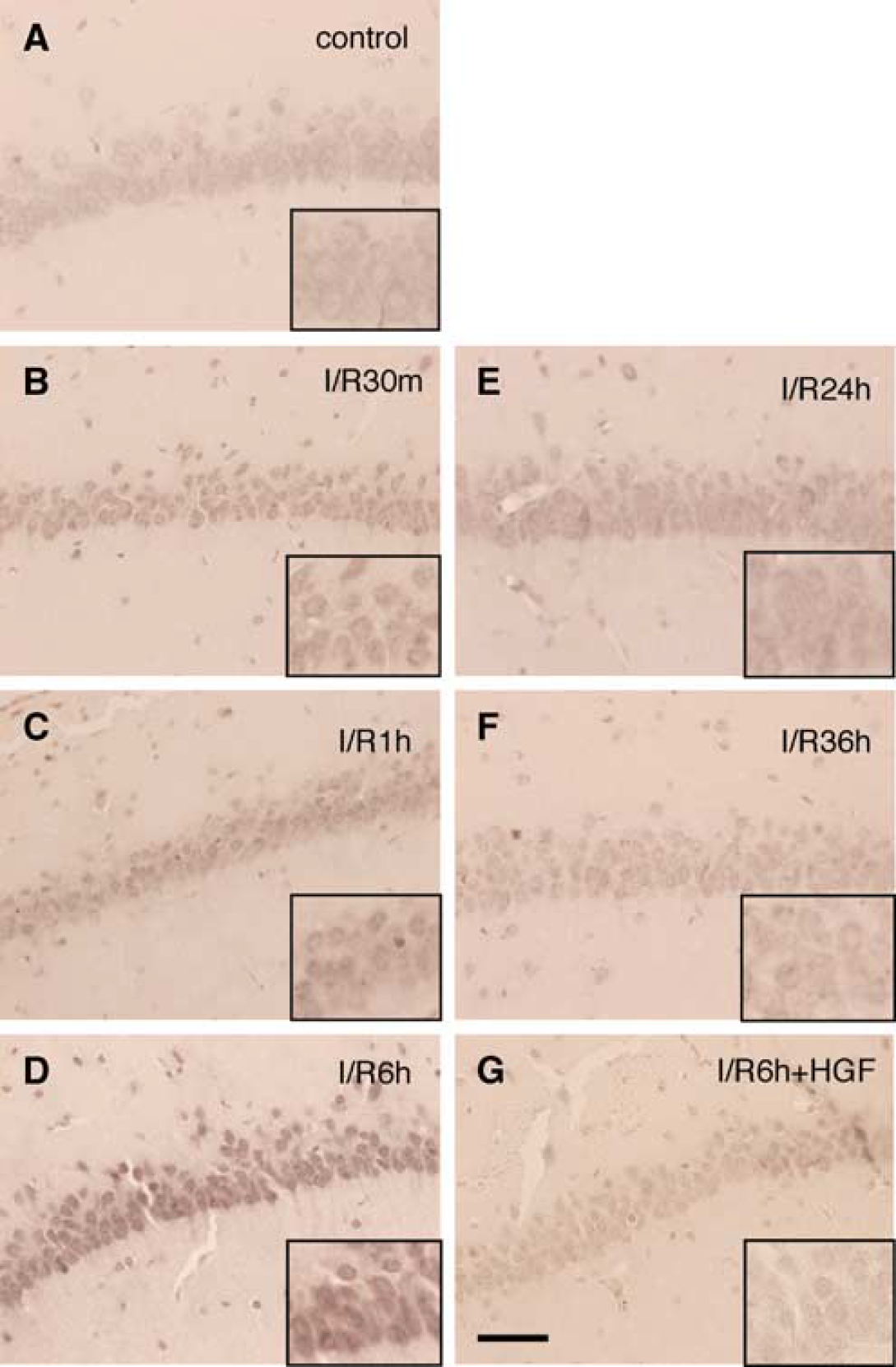
Effect of hepatocyte growth factor (HGF) on oxidative DNA damage in the hippocampal cornu ammonis (CA) subregion 1 (CA1) region after transient forebrain ischemia. (
Poly(ADP-ribose) polymerase is activated in response to DNA damage. We next examined poly(ADP-ribose) (PAR) polymer formation as a marker of PARP activity by using anti-PAR antibody.
PAR polymer formation in naiïve control was barely detected in the cytoplasm, whereas this formation was seen neither in the dendrites nor in the nuclei (Figure 6A). The immunoreactivity gradually rose, and the maximum increase in the nuclei was seen at 6 h of reperfusion (Figure 6D), which was comparable to the changes in the expression of 8-OHdG. Treatment with HGF suppressed ischemia-induced PAR polymer formation in the nuclei at 6 h of reperfusion (Figure 6F).
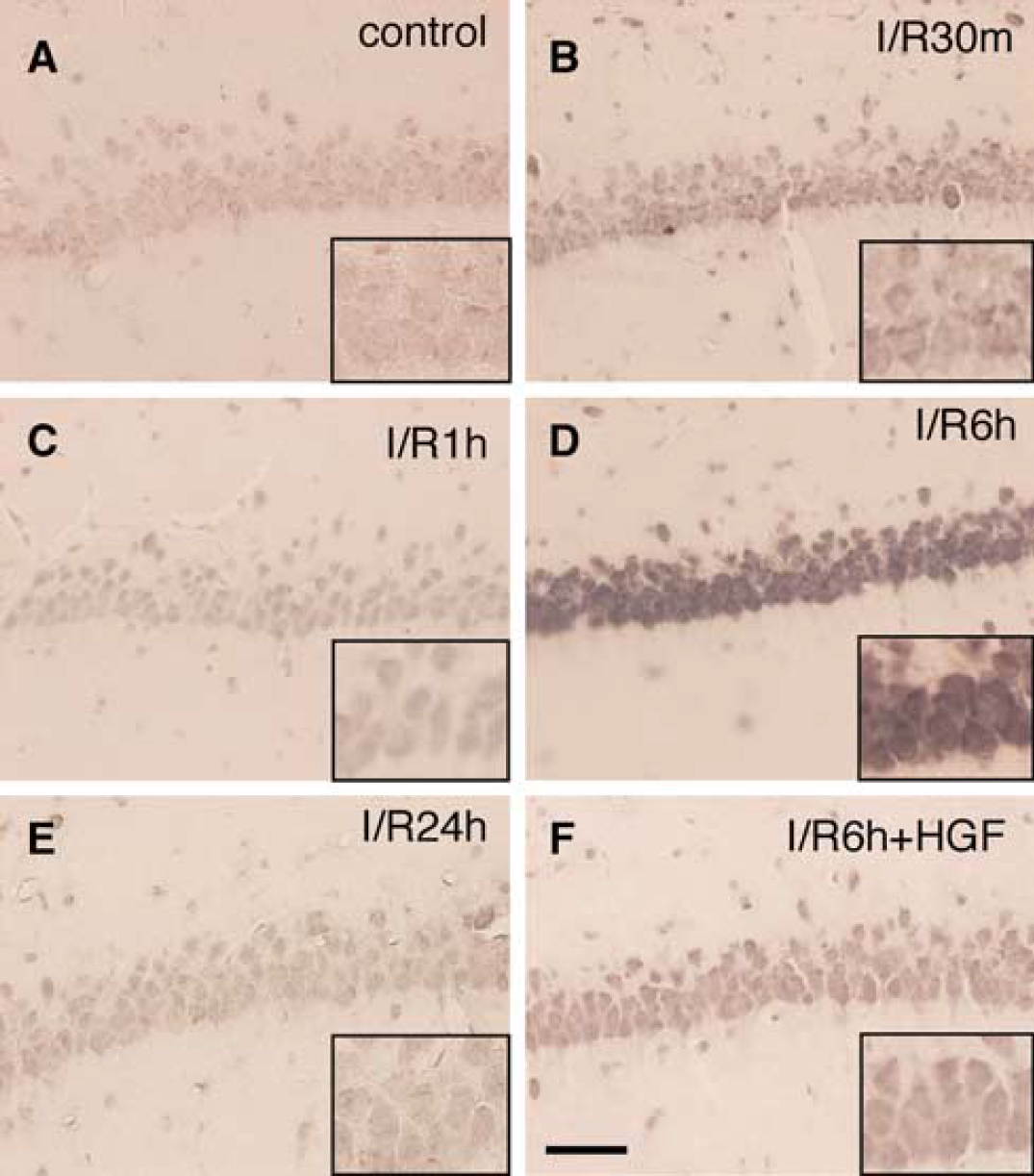
Effect of hepatocyte growth factor (HGF) on poly(ADP-ribose) polymer formation in the hippocampal cornu ammonis (CA) subregion l (CAl) region after transient forebrain ischemia. (
Oxidative DNA damage is known to induce p53 activation, leading to AIF translocation to the nucleus. So finally, we examined the expression of p53 protein after ischemia with or without HGF treatment. Faint cytoplasmic expression of p53 protein was found in the naiïve control (Figure 7A), whereas the immunoreactivity gradually increased up to 6 h of reperfusion (Figures 7B-7D). The increased immunoreactivity remained at 36 h of reperfusion (Figures 7E-7F). The notable increase in the expression of p53 at 6 h of reperfusion was prevented by HGF treatment, making the expression comparable to that for the nonoperated naiïve control (Figure 7G).
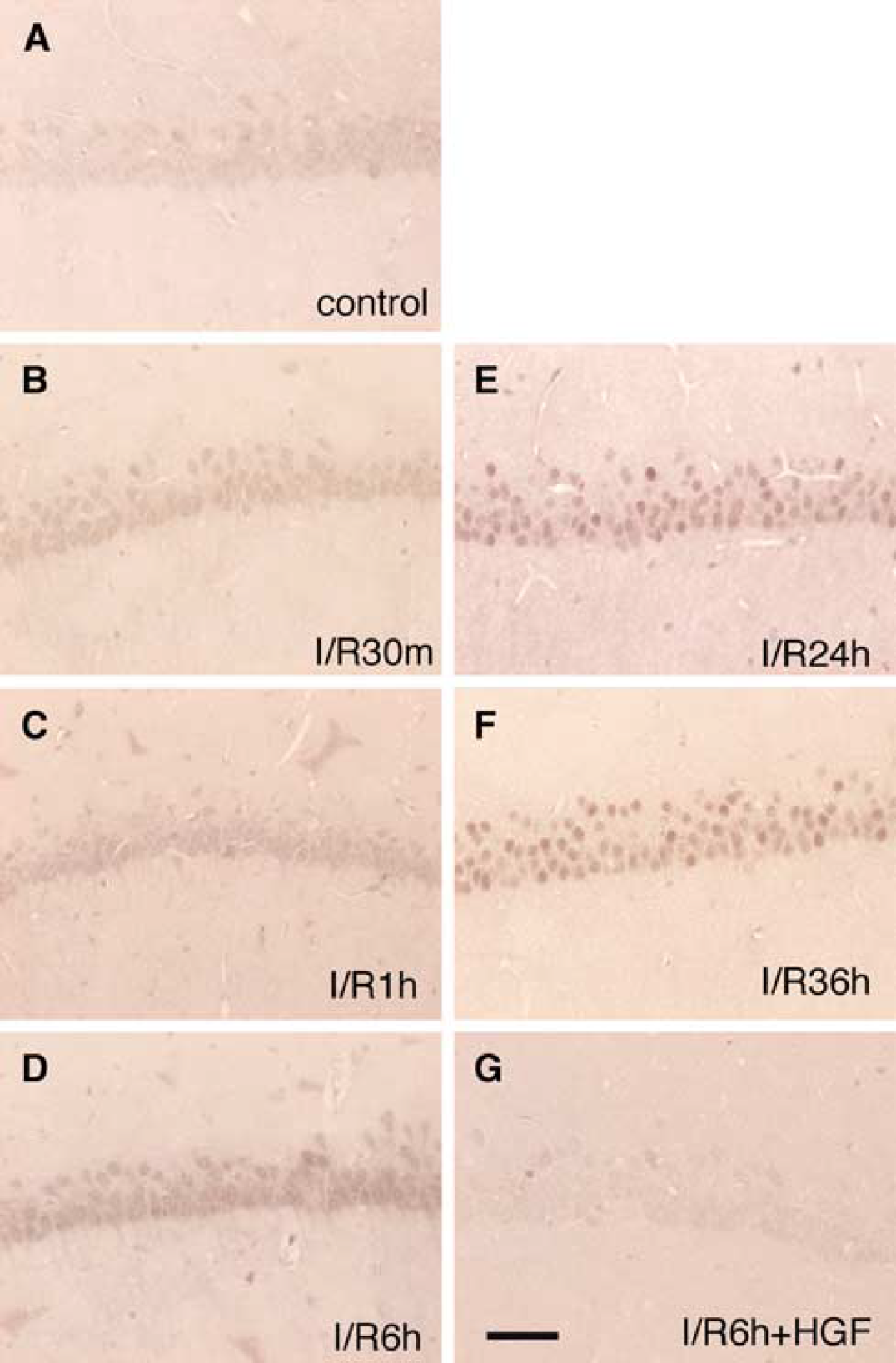
Effect of hepatocyte growth factor (HGF) on the expression of p53 in the hippocampal cornu ammonis (CA) subregion l (CAl) region after transient forebrain ischemia. (
Discussion
We showed that treatment with HGF protected hippocampal CA1 neurons from apoptotic cell death after transient forebrain ischemia. Although caspases have been identified as major molecules in the mechanism responsible for apoptotic cell death, accumulating evidence recently indicates that caspase-independent mechanisms may also play an important role in cell death (Rideout and Stefanis, 2001; Johnson et al, 1999; Lankiewicz et al, 2000; Zhan et al, 2001; Miller et al, 1997; D'Mello et al, 2000; Keramaris et al, 2000; Selznick et al, 2000). In the caspase-independent pathway, AIF is characterized as one of the crucial proteins (Cregan et al, 2004). Apoptosis-inducing factor is located in the mitochondrial intermembrane space in intact cells, but it is translocated to the nucleus after cerebral ischemia, and thereby causes DNA fragmentation in a caspase-independent manner (Culmsee et al, 2005; Zhu et al, 2003; Plesnila et al, 2004; Zhao et al, 2004; Cao et al, 2003). In the present study, we focused on the localization of AIF in cells to investigate the possible mechanism of the protective effect of HGF after transient forebrain ischemia. Our findings show that HGF attenuated the increase in the expression of AIF in the nucleus after ischemia without a change in the total amount of AIF, suggesting that the inhibition of AIF translocation to the nucleus contributes to the protective effect of HGF on apoptotic cell death.
To further investigate the mechanism for inhibiting AIF translocation after HGF treatment, we examined the expression of Hsp70 and its interaction with AIF. Only Hsp70 among Hsp family proteins, including Hsp10, 27, 60, 70, and 90, is regarded to be an endogenous inhibitor of AIF, as it directly binds to AIF and inhibits the import of AIF into the nucleus (Ravagnan et al, 2001; Gurbuxani et al, 2003). In this sense, it was recently reported that overexpression of Hsp70 proteins induced an increase in the interaction of Hsp70 with AIF, and reduced neonatal hypoxic/ischemic brain injury (Matsumori et al, 2005). We showed that although both the expression of Hsp70 and interaction with AIF were increased after transient cerebral ischemia, they were not influenced by HGF treatment. Therefore, it is conceivable that neither the amount of Hsp70 protein nor the interaction of Hsp70 with AIF contributes to the neuroprotective mechanism of HGF after transient forebrain ischemia.
Primary DNA damage induced by neuronal NOS (nNOS) activation and NO formation is thought to be an initial event leading to AIF release from mitochondria, which release results in subsequent secondary DNA damage (DNA fragmentation) by AIF (Yu et al, 2003). It is widely accepted that excessive Ca2+ influx through the activated NMDA receptor, which activation can be estimated by tyrosine phosphorylation of the NR2 subunit of the NMDA receptor (Wang and Salter, 1994; Kohr and Seeburg, 1996; Zheng et al, 1998; Chen and Leonard, 1996), leads to the activation of nNOS and subsequent production of NO (Castilho et al, 1998; Dawson et al, 1991). To determine whether protective effect of HGF required the phosphorylation-dependent activities of the NMDA receptor, we examined tyrosine phosphorylation of NR2B subunit of the NMDA receptor. Although tyrosine phosphorylation of NR2B subunit was significantly increased after the start of reperfusion compared with that of the naïve control, it was not influenced by HGF treatment. These results suggest that HGF treatment altered neither the intracellular Ca2+ concentration regulated by tyrosine phosphorylation of the NR2B subunit nor the production of NO after transient forebrain ischemia.
Calcium influx through the activated NMDA receptor elicits production of not only NO but also mitochondrial ROS, leading to formation of peroxynitrite (ONOO−-) and subsequent hydroxyl radical, which eventually results in oxidative DNA damage (Yu et al, 2003). To assess oxidative DNA damage after ischemia with or without HGF treatment, we measured 8-OHdG as an indicator of oxidative DNA damage, as its expression is elevated after oxidative DNA damage (Pastoriza Gallego and Sarasin, 2003; Toyokuni et al, 1997). In fact, the expression of 8-OHdG was shown earlier to be increased after transient forebrain ischemia (Won et al, 2001; Hwang et al, 2004; Baek et al, 2000). In agreement with these findings, we showed that the expression of 8-OHdG was elevated at the early period after the start of reperfusion. It is noteworthy that this elevated 8-OHdG expression was almost completely suppressed by HGF treatment. Our results suggest that HGF inhibited AIF translocation to the nucleus by preventing the primary oxidative DNA damage after ischemia.
Oxidative DNA damage is also known to lead to the activation of PARP and p53 after ischemia (Komjati et al, 2004; Koh et al, 2004; Nagayama et al, 2000; Banasiak and Haddad, 1998; McGahan et al, 1998; Huang et al, 1995; Renolleau et al, 1997; Tomasevic et al, 1999). Poly(ADP-ribose) is a DNA repair enzyme that reveals its activity by utilizing nicotinamide adenine dinucleotide+ as a substrate. Therefore, excessive DNA damage induces a marked activation of PARP to repair DNA, and thereby depletes energy, which results in the release of cytochrome c, endonuclease G, and AIF (Yu et al, 2003; Meli et al, 2003). In addition, activation of p53 induced by DNA damage elicits ROS production and subsequent mitochondrial membrane disruption, which are associated with cytochrome c- independent apoptosis (Li et al, 1999). It has also been shown that AIF translocation is involved in p53-mediated neuronal injury (Cregan et al, 2004). Taking these findings into consideration, PARP and p53 activation after ischemia might be one of the steps in the AIF-dependent apoptotic pathway. Therefore, we further investigated the activity of PARP, which was assessed by the immunohistO2−-chemistry using an anti-PAR antibody to detect PAR polymer formation, and the expression of p53 protein after ischemia with or without HGF treatment. Marked increases in the PAR polymer formation and the expression of p53 protein after ischemia were effectively prevented by HGF treatment, suggesting that HGF reduced AIF translocation after ischemia by inhibiting PARP and p53 activation.
Questions remain as to how HGF can suppress primary oxidative DNA damage. It has been shown that HGF protected cardiomyocytes from H2O2-stimulated apoptosis by increasing Bcl-XL expression (Nakamura et al, 2000) and by activating the MAP kinase kinase-mitogen-activated protein kinase pathway (Kitta et al, 2001). Furthermore, extracellular signal regulated kinase activation downregulated p53 in cancer cells, thereby reducing ROS production and subsequent depolarization of the mitochondrial membrane (Ostrakhovitch and Cherian, 2005). Recently, activation of the phosphatidylinositol 3'-kinase-Akt pathway induced by HGF protected hepatocytes from hypoxia-reoxygenation-induced oxidative stress and apoptosis by inhibiting the activation of rac1 small GTPase (Ozaki et al, 2003). Hepatocyte growth factor itself is unlikely to exert a direct effect on the redox state (Ozaki et al, 2003). Whereas HGF prevented ceramide-induced apoptosis by increasing catalase expression, the signaling cascade via c-Met to induce the expression of catalase remains unclear (Kannan et al, 2004). Therefore, further studies will be required to determine signal-transduction pathways via c-Met, which may inhibit the primary oxidative DNA damage that occurs in the ischemic brain in vivo.
We recently showed that HGF protected cultured hippocampal neurons against NMDA-induced excitotoxicity via the partial prevention of caspase-3 activity and the inhibition of AIF translocation to the nucleus (Ishihara et al, 2005). Therefore, although the inhibition of AIF-dependent pathway contributes to the protective effects of HGF, we cannot fully rule out the possibility that HGF can prevent cell death through the inhibition of caspase-dependent pathway in the ischemic brain. Alternatively, our results suggest that the potent protective effects of HGF on apoptotic cell death after transient forebrain ischemia might be mediated by the inhibition of AIF translocation in addition to a prevention of caspase-dependent pathway.
Although we suggest that suppression of the primary oxidative damage at the early stage after transient forebrain ischemia is, at least, involved in the protective effects of HGF, whether HGF inhibits the translocation of AIF by attenuating oxidative DNA damage remains to determine. It was recently showed that Bcl-2 transfection in the peri-infarct region blocked AIF translocation to the nucleus and prolonged cortical neuron survival (Zhao et al, 2004). Therefore, it is possible that HGF inhibits translocation of AIF after transient forebrain ischemia mediated by an expression of Bcl-2 family proteins, such as Bcl-2.
Although HGF has the ability to prevent ischemic brain injuries and is a prospective agent for therapy against a variety of neurologic and neurodegenerative disorders, the intracellular signaling associated with its protective effects is not fully understood. It is thus an important objective to elucidate the molecular basis of the protective effects of HGF under pathologic conditions. In the present study, we showed that HGF was capable of preventing in vivo ischemia-induced neuronal cell death by inhibiting the primary oxidative DNA damage and then preventing activation of the PARP/p53/AIF pathway.