
Abstract
Accumulating evidence on the molecular and cellular basis of ischemiaireperfusion-induced neurodegeneration suggests that oxidative stress is involved. Heme oxygenase (HO) and cyclooxygenase (COX) play physiologically important roles in the CNS. Conversely, HO and COX also can increase oxidative stress. Recent studies suggest that c-Jun phosphorylation is an important step in some forms of stress-induced neuropal apoptosis. In this study, the authors tried to clarify the association of HO and COX with c-Jun phosphorylation. Inducible forms of HO and COX (HO-1 and COX-2, respectively) were transiently induced in CA1 pyramidal neurons after ischemia. c-Jun also was induced in pyramidal neurons throughout the hippocampal formation, but its phosphorylation was limited to CAL In contrast, these molecules were constitutively expressed at low levels. Most (84%) of the CA1 pyramidal neurons examined expressed HO-1, COX-2, or both, and such expression showed good co-localization with c-Jun phosphorylation. These results suggest the following: (1) c-Jun phosphorylation was associated with ischemia/reperfusion-induced neuronal apoptosis; (2) HO-1 and COX-2 were induced in CA1 pyramidal neurons, which undergo cell death; and (3) most CA1 pyramidal neurons expressed HO-1, COX-2, or both, which strongly suggests that these are candidates for neuron killers.
Heme oxygenase (HO: heme, hydrogen-donor:oxygen oxidoreductase (α -methene-oxidizing, hydroxylating); E.C. 1.14.99.3) promotes oxidation of the heme molecule in concert with NADPH-cytochrome P-450 reductase followed by the specific cleavage of heme into biliverdin, carbon monoxide, and iron (Maines, 1988). HO has at least two isomers: an inducible and a constitutive type (HO-1 and HO-2, respectively). One metabolite of heme, biliverdin, is further converted to bilirubin, which is a powerful antioxidant (Stocker et al., 1987), and HO-1 induction protects the kidney against oxidative damage after ischemia and reperfusion (Maines, 1993). In contrast to these helpful functions of HO, HO also produces free iron, which may act as a strong source of oxidative stress in diseases of the CNS in humans (Gelman, 1995) and is induced in CA1 pyramidal neurons, which undergo apoptosis after transient forebrain ischemia (Matsuoka et al., 1998b). Cyclooxygenase (COX: 8, 11, 14-eicosatrienoate, hydrogen-donor:oxygen oxidoreductase; E.C. 1.14.99.1), also referred to as prostaglandin (PG) endoperoxide H2 synthase, catalyzes the formation of PGH2 from arachidonic acid (Vane et al., 1998). COX has at least two isomers: a constitutive and an inducible type (COX-1 and COX-2, respectively). Cyclooxygenase produces PGH2, which is a seed in the PG cascade, and the resulting PG have various physiologic functions (Kaufmann et al., 1997). However, the effects of PG in ischemia/reperfusion-induced neurodegeneration still are unclear. In addition, COX also may increase oxidative stress (Vane et al., 1998). Reactive oxygen species, an oxidative radical, is markedly increased by COX (Morita et al., 1995) and may be involved in ischemia-induced neurodegeneration (Pellegrini-Giampietro et al., 1990; Simonian and Coyle, 1996). Therefore, the effects of the induction of HO-1 and COX-2 are still of concern.
Jun is a component of a transcription factor, activator protein-1 (AP-1), which binds and activates transcription at an AP-1 element. The transcription activity of c-Jun is regulated by phosphorylation at Ser63 and Ser73 (Binetruy et al., 1991; Smeal et al., 1991). Extracellular signals, including growth factor, transforming oncoproteins, and ultraviolet irradiation, stimulate phosphorylation of c-Jun at Ser63/73 and activate c-Jun-dependent transcription. Mutation of Ser63173 renders c-Jun nonresponsive to mitogenic and stress-induced signaling pathways (Ui et al., 1998). The mitogen-activated protein kinase homologue, Jun N-terminal kinase (JNK), also referred to as stress-activated protein kinase, binds to the N-terminal region of c-Jun and phosphorylates c-Jun at Ser63/73. In addition, the activity of JNK is stimulated by the same signals that activate c-Jun (Dérijard et al., 1994; Kyriakis et al., 1994). Recent findings suggest that JNK is a key kinase in some forms of stress-induced neuronal apoptosis (Bossy-Wetzel et al., 1997; Eilers et al., 1998; Herdegen et al., 1998; Watson et al., 1998). It is still a concern, whether necrosis or apoptosis underlie the ischemia-induced delayed neuronal death. However, cells undergoing apoptosis are associated with cleavage of genomic DNA, and these changes have been detected in the pyramidal neurons of CA1, weakly 3 days after treatment and strongly 7 days after transient forebrain ischemia (Nitatori et al., 1995). Therefore, CA1 hippocampal neurons may undergo apoptosis after ischemia. However, it is still unclear whether c-Jun and its phosphorylation participate in ischemia/reperfusion-induced neurodegeneration, and how HO-1 and COX-2 are associated with c-Jun phosphorylation.
Therefore, we tried to clarify the association of HO-1 and COX-2 with c-Jun phosphorylation. For this purpose, we analyzed the localization of HO-1 and COX-2 and its association with phosphorylated c-Jun after transient forebrain ischemia.
MATERIALS AND METHODS
Animals and materials
Male Wistar rats, weighing 270 to 330 g, were purchased from Charles River (Atsugi, Japan). The animals were acclimated to and maintained at 23°C under a 12-hour light-dark cycle. All animals were housed in standard laboratory cages and had free access to food and water throughout the study period. The experimental protocol was approved by the Committee for Animal Research at Kyoto Pharmaceutical University.
Rabbit anti-rat heat shock protein-32 (HO-1) polyclonal antibody was from Stress Gen (Victoria, Canada); mouse anti-rat COX-2 monoclonal antibody was from Transduction Laboratories (Lexington, KY, U.S.A.); rabbit anti-c-Jun polyclonal antibody and rabbit anti-Ser73-phosphorylated c-Jun polyclonal antibody were from New England BioLabs (Beverly, MA, U.S.A.); and mouse anti-rat microtubule associated protein (MAP)-2 monoclonal antibody and fluorescein isothiocyanate- and rhodamine-labeled secondary antibodies were from Chemicon International (Temecula, CA, U.S.A.). Hoechst 33258 dye was from Molecular Probes (Eugene, CA, U.S.A.). The Vectastain ABC Elite kit was from Vector Laboratories (Burlingame, CA, U.S.A.).
The anti-HO-1 antibody was raised using purified rat liver HO-1 and showed no cross-reaction with HO-2. The anti-COX-2 antibody was raised using a recombinant protein corresponding to amino acids 368-604 of rat COX-2. The anti-c-Jun antibody was raised using a synthetic peptide corresponding to amino acids 1-79 of human c-Jun. The antiphosphorylated c-Jun antibody was raised using a synthetic phospho-Ser73 peptide corresponding to amino acids 68–77 (LLKLAS*ELERC) of human c-Jun (asterisk indicates phosphorylation). This antiphosphorylated c-Jun antibody detects phosphorylated c-Jun at Ser73 yet will not react with nonphosphorylated c-Jun.
Animal model
After the rats were fasted overnight with free access to water, they were subjected to transient forebrain ischemia. Rats were anesthetized briefly with 3.5% halothane, intubated, and connected to a Starling-type respirator. Anesthesia was maintained with 0.5% halothane and 30% oxygen in nitrous oxide. The femoral artery and vein were exposed and silicone catheters were inserted to monitor arterial blood pressure, arterial gas, and blood glucose levels. After isolation of the bilateral common carotid arteries, the rats were allowed to remain in a steady state for 20 minutes while maintaining the rectal temperature at 37 ± 0.2°C, artcrial Pao2 at 100 to 150 mm Hg, and Paco2 at 20 to 40 mm Hg. Transient forebrain ischemia was induced by clamping the bilateral carotid arteries and lowering the blood pressure to 50 mm Hg for 8 minutes and withdrawing blood from the venous catheter. After this 8 minutes of ischemia, the blood pressure was restored to normal levels by rapid reinfusion of the shed blood, and the bilateral carotid clamps were removed.
Tissue preparation and immunohistochemistry
On days 1, 3, and 7 after ischemia, the animals were killed, and sections were prepared as previously described (Matsuoka et al., 1998a). Some of sections were stained with hematoxylin and eosin to examine the histologic changes. The free-floating sections were incubated with antibodies against MAP-2 (1:3,000), HO-1 (1:10,000), COX-2 (250 ng IgG/mL), c-Jun (1:200), or phosphorylated c-Jun (1:200) for 4 days at 4°C. Antibodies were detected with the ABC Elite kit, and labeling was revealed by incubation with 50 mmol/L Tris-HCl buffer (pH 7.6) containing 0.02% 3,3′ -diaminobenzidine and 0.0045% hydrogen peroxide (DAB solution) with nickel enhancement using 0.6% nickel ammonium sulfate to obtain dark blue-stained positive products. For double immunostaining of phosphorylated c-Jun with HO-1 or COX-2, sections were incubated with anti-phosphorylated c-Jun antibody and visualized as described earlier (immunoreactivity was indicated by dark blue products). The sections were treated with 0.3% hydrogen peroxide to inactivate peroxidase activity and then incubated with anti-HO-1 or anti-COX-2 antibody. The second-cycle antibody was detected by incubation with DAB solution without nickel enhancement to obtain brown-stained positive products. The immunostained sections were scanned using a high-resolution camera (ProgRes 3008, Carl Zeiss, Jena, Germany), and the photographs were printed on a color photo printer (Pictrography 3000, Fuji Film, Tokyo, Japan). For double immunostaining of HO-1 and COX-2, sections were co-incubated with two primary antibodies and then detected by fluorescein isothiocyanate- and rhodamine-labeled secondary antibodies. For quantitative analysis of a cell population that expresses HO-1, COX-2, or both, the double-immunostained sections were counter-stained with Hoechst 33258 (1 μg/mL for 10 minutes), which has high affinity for DNA. Immuno-stained samples were scanned under a laser scanning confocal microscope (excitation 488 nm, emission 510 to 525 nm for detection of fluorescein isothiocyanate; and excitation 543 nm, emission over 590 nm for detection of rhodamine) (LSM 410, Carl Zeiss). To minimize the attenuation of fluorescence, each field was exposed to light for the same amount of time, and the laser intensity was set at 3% of maximum. The laser intensity, brightness, and contrast were set at the same values.
Image analysis and statistics
For the quantitative analysis, sections at 2.56 mm caudal from the bregma were used. c-Jun-immunoreactive cells and phosphorylated c-Jun-immunoreactive cells were counted by an image analyzer (WinRoof, Mitani Corp., Fukui, Japan) and presented as the number of immunoreactive cells per square millimeter. We classified the cell with brightness over 50 in eight-bit color scale as being immunopositive. Both HO-1- and COX-2-immunoreactive cells were counted by an image analyzer (WinRoof) and presented as the percentage of immunoreactive cells in CA1 pyramidal neurons; Hoechst 33258 staining was used. Data are presented as the mean ± SD of five rats. The significance of differences was determined by an analysis of variance (ANOV A) with the Bonferroni/Dunn post hoc test (Stat View, Abacus Concepts, Berkeley, CA, U.S.A.).
RESULTS
Changes of CA1 pyramidal neurons in the hippocampus after ischemia
Histologic changes were examined by using hematoxylin and eosin staining. With hematoxylin and eosin staining, the nuclear structures are stained dark purple or blue, and practically all cytoplasmic structures and inter-subcellular substances are stained varying shades of pink (Bloom and Fawcett, 1975). Although the loss of pyramidal neurons was not detected on day I after the 8-minute period of forebrain ischemia, degenerative neurons that showed the changes in cell shape and eosinophilic somata with picnotic nuclei were observed in the CA1 subfield (Fig. 1B). Three days after ischemia, almost all CA1 pyramidal neurons were degenerated, and a few neurons were lost (Fig. 1C). After 7 days, almost all CA1 pyramidal neurons were lost (Fig. 1C), although sham-operation had no effect (Fig. 1A). Microtubule-associated protein-2 is a major component of cytoskeletal proteins in neurons (Vallee et al., 1984), and MAP-2 immunohistochemical study has been used as an index of neurodegeneration in animal models, including ischemia (Matesic and Lin, 1994). Seven days after ischemia, MAP-2 immunoreactivity was lost in the CA1 subfield (Fig. 1F), although sham operation had no effect (Fig. 1E).
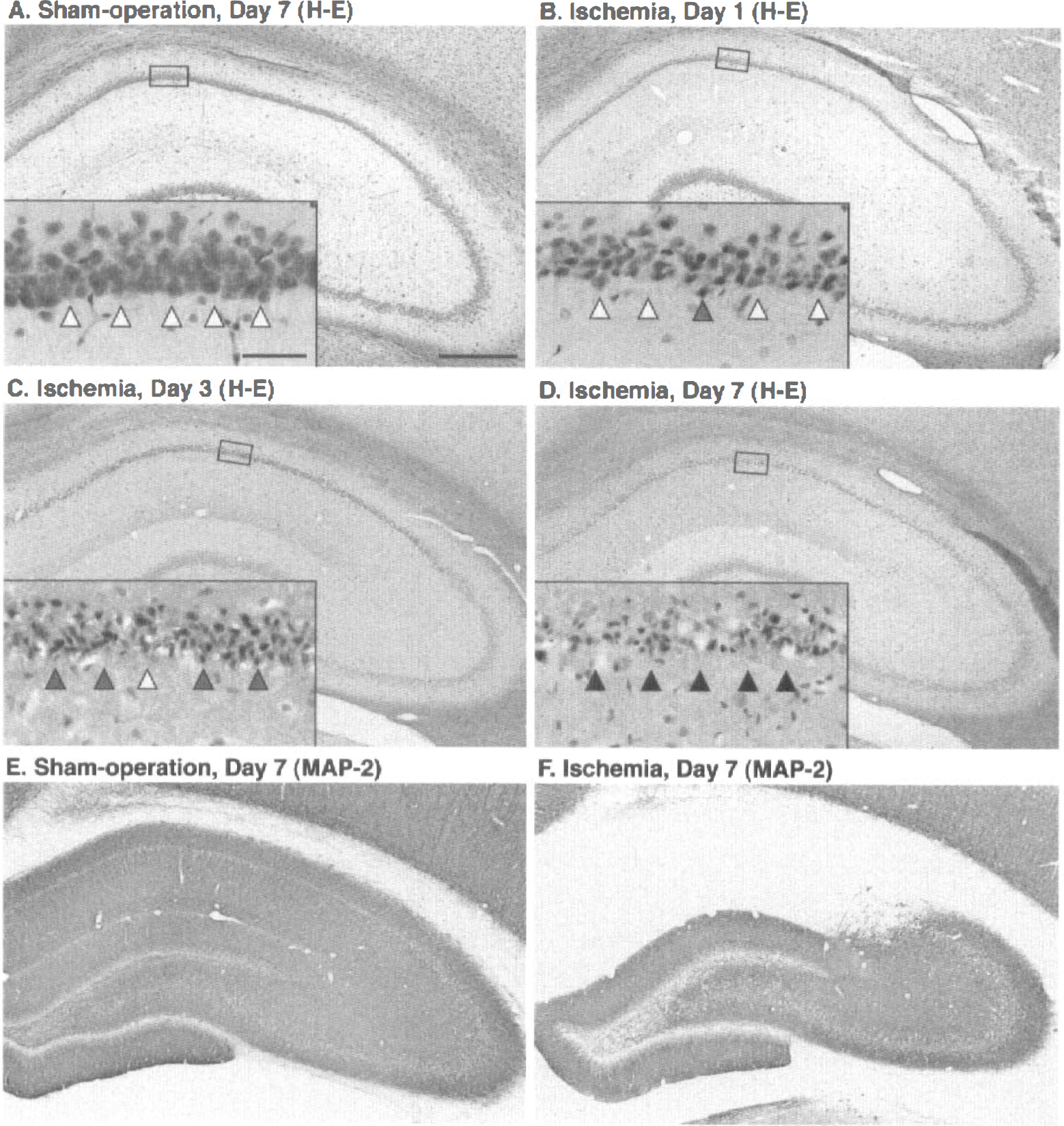
Histologic changes after transient forebrain ischemia in rat hippocampus. Sections were stained with hematoxylin and eosin. Boxed area is shown with higher magnification in the inset. Open and hatched arrowheads indicate the normal and degenerated pyramidal neurons, respectively. Closed arrowheads indicate the neuronal cell loss. Bar = 500 μm for
Induction of HO-1 and COX-2 in the hippocampus after ischemia
In the sham-operated rat hippocampus, slight HO-1 and COX-2 immunoreactivity was observed, that is, HO-1 was expressed in some of the neurons in the hilus of the dentate gyrus (DG) (Fig. 2A), and COX-2 was expressed in some of the neurons in CA3, the hilus of DG, and DG (Fig. 2B). The HO-1 immunoreactivity was markedly induced in CA1 pyramidal neurons 1 day after ischemia (Fig. 2C) and in activated microglia 3 to 7 days after ischemia (data not shown), as previously reported (Matsuoka et al., 1998b). Also, COX-2 immunoreactivity was markedly induced in CA1 pyramidal neurons 1 day after ischemia (Fig. 2D); COX-2 immunoreactivity also was induced in CA3, the hilus of DG, and DG, but the intensity of the immunoreactivity in these subfields was much weaker than that in CA1 (Fig. 2D). The COX-2 immunoreactivity in CA1 then gradually disappeared, and only faint immunoreactivity was detected at 7 days after ischemia (Figs. 2G and 2H). In contrast to HO-1 induction, COX-2 was induced only in neurons and not in glial cells up to 7 days after ischemia. Quantitatively, 36% and 69% of CA1 pyramidal neurons were expressed HO-1 and COX-2, respectively, at 1 day after transient forebrain ischemia (Table 1).
Expression of HO-1 and COX-2 in CA1 pyramidal neurons after ischemia

The distribution of cells expressing HO-1 or COX-2 in CA1 pyramidal neurons was quantitatively analyzed. Data are presented as the mean ± standard deviation of five rats. COX-2, cyclooxygenase-2; HO-1 heme oxygenase-1.
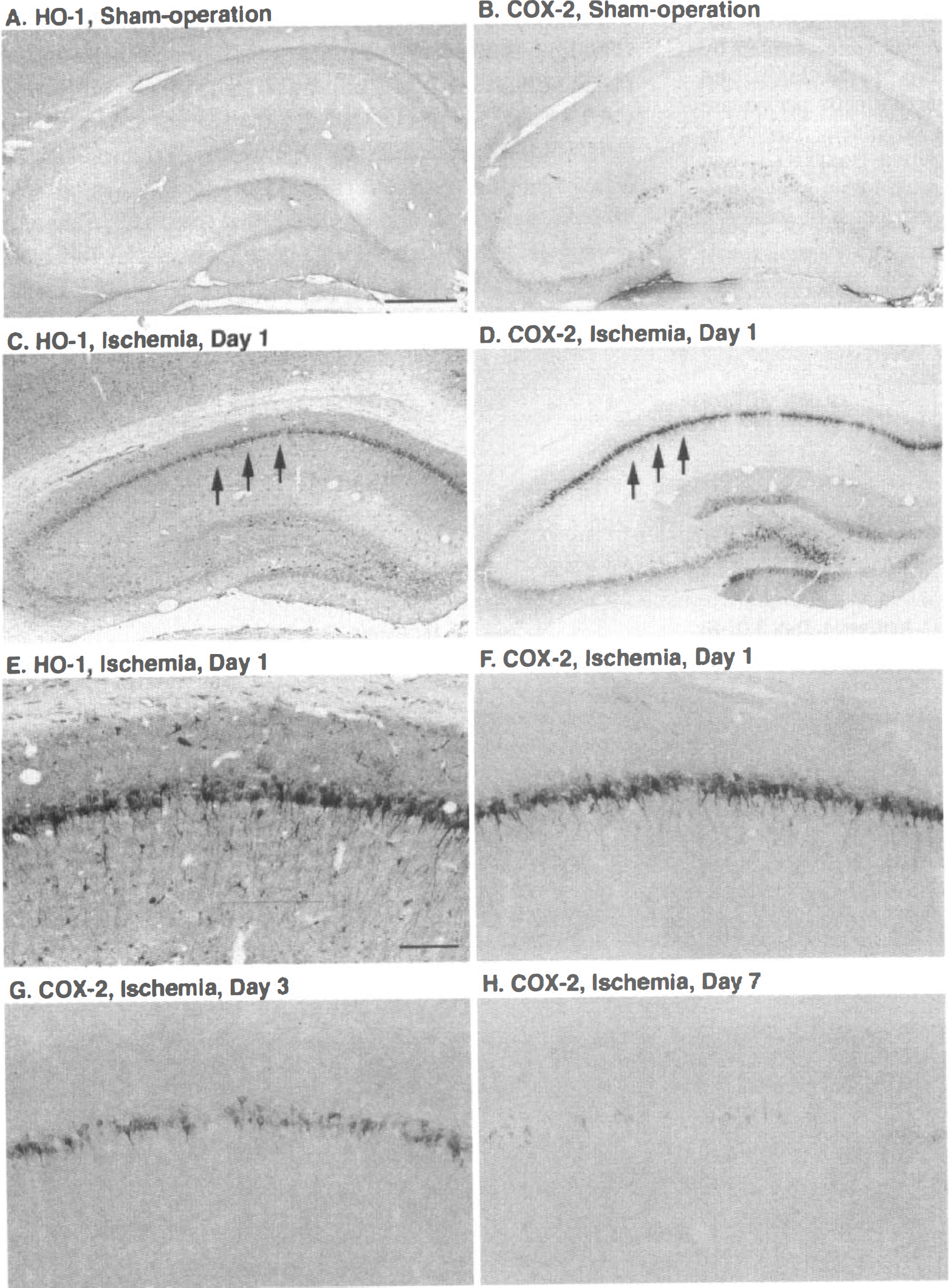
Induction of heme oxygenase-1 (HO-1) and cyclooxygenase-2 (COX-2) in the hippocampus after ischemia. Both HO-1 (
Induction of c-Jun and its phosphorylation in CA1 pyramidal neurons after ischemia
In the sham-operated rat hippocampal formation, c-Jun was constitutively expressed in the granular neurons of DG but was not detected in other subfields (Fig. 3A). One day after ischemia, c-Jun was induced in CA1 pyramidal neurons (Fig. 3C). Although c-Jun phosphorylation was not detected in the sham-operated rat hippocampal formation (Fig. 3B), c-Jun phosphorylation was induced in CA1 after ischemia (Fig. 3D). In contrast to the marked induction of c-Jun and its phosphorylation in the CA1 subfield, these were slight in CA3 and did not induce neuronal apoptosis (Figs. 3E versus 3G, and 3F versus 3H, respectively).
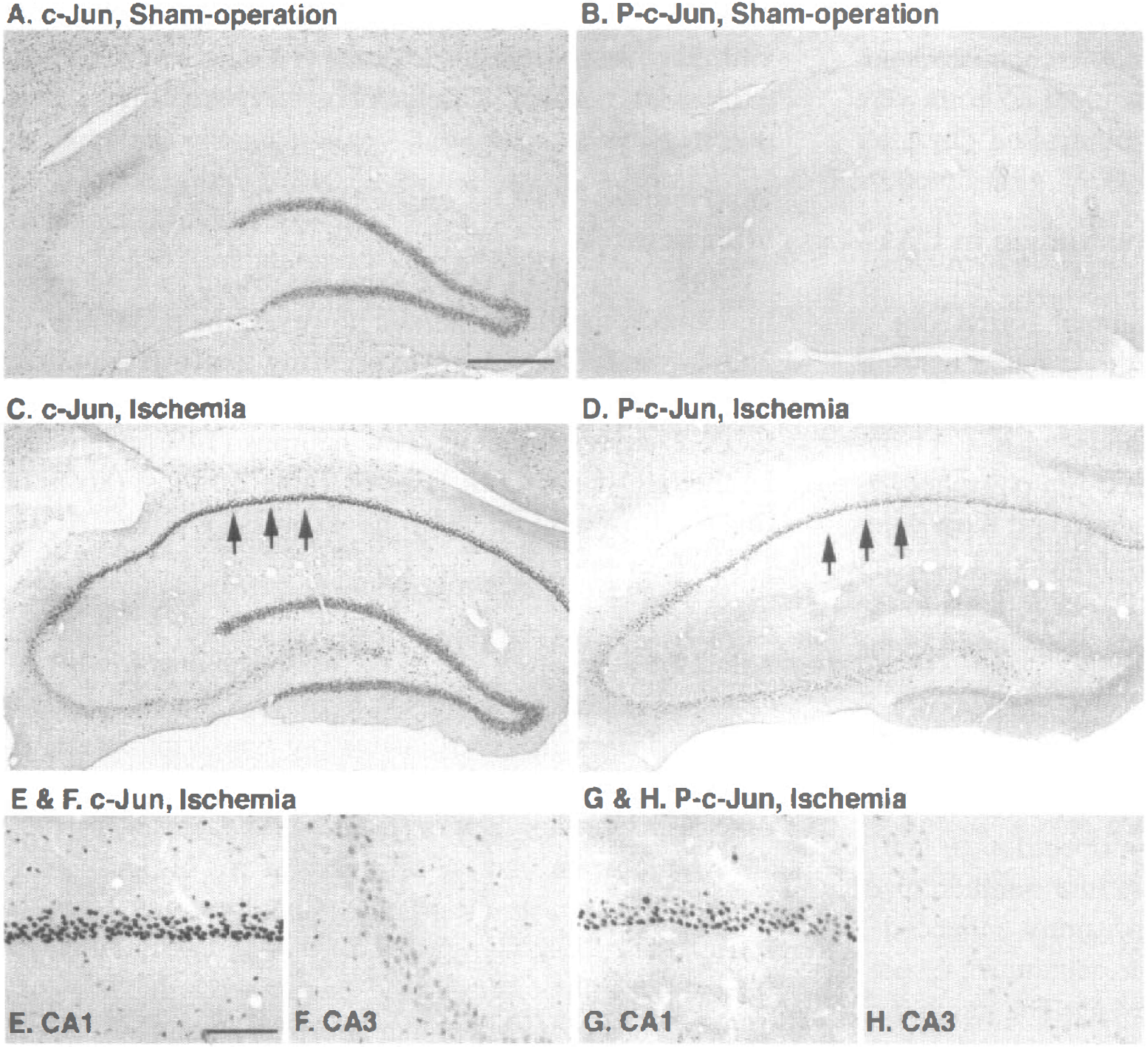
Induction and phosphorylation of c-Jun in the hippocampus after ischemia. c-Jun (
The number of c-Jun-immunoreactive cells in CA1 was limited 1 to 3 days after a sham operation (6 and 4 cells/mm2, respectively). In contrast, the number of c-Jun-immunoreactive cells was significantly enhanced 1 to 3 days after ischemia (395 and 401 cells/mm2, respectively, P < 0.001 by ANOV A) (Fig. 4A). The number of phosphorylated c-Jun-immunoreactive cells also was limited after a sham operation (7 and 8 cells/mm2, respectively). However, the number of phosphorylated c-Jun-immunoreactive cells was significantly enhanced 1 day after ischemia (323 cells/mm 2, P < 0.001 by ANOV A) (Fig. 4B). This number gradually decreased but still was significantly enhanced 3 days after ischemia (101 cells/mm 2, P < 0.001 by ANOV A) (Fig. 4B). Eighty-two percent and 25% of c-Jun-expressing cells were phosphorylated 1 and 3 days after ischemia, respectively.
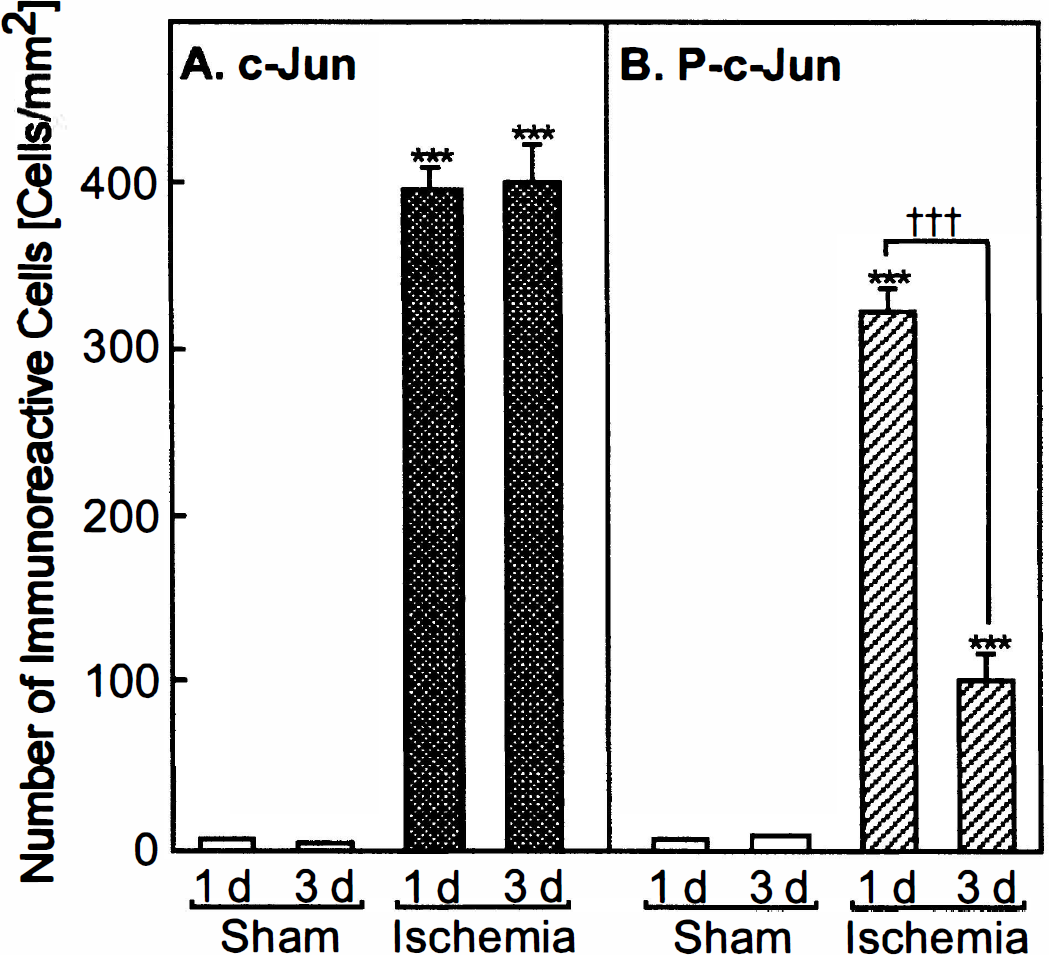
Number of c-Jun-immunoreactive cells
Co-localization of phosphorylated c-Jun with HO-1 and COX-2 after ischemia
Since the relation between phosphorylated c-Jun and HO-1 or COX-2 is unclear, we examined the co-localization of phosphorylated c-Jun with HO-1 and COX-2. Both HO-1 and COX-2 (indicated by brown in Fig. 5) were strongly co-localized with phosphorylated c-Jun (indicated by dark blue) (Figs. 5A and 5C, and 5B and 5D, respectively). Neither HO-1- nor COX-2-expressing neurons without phosphorylated c-Jun were detected.
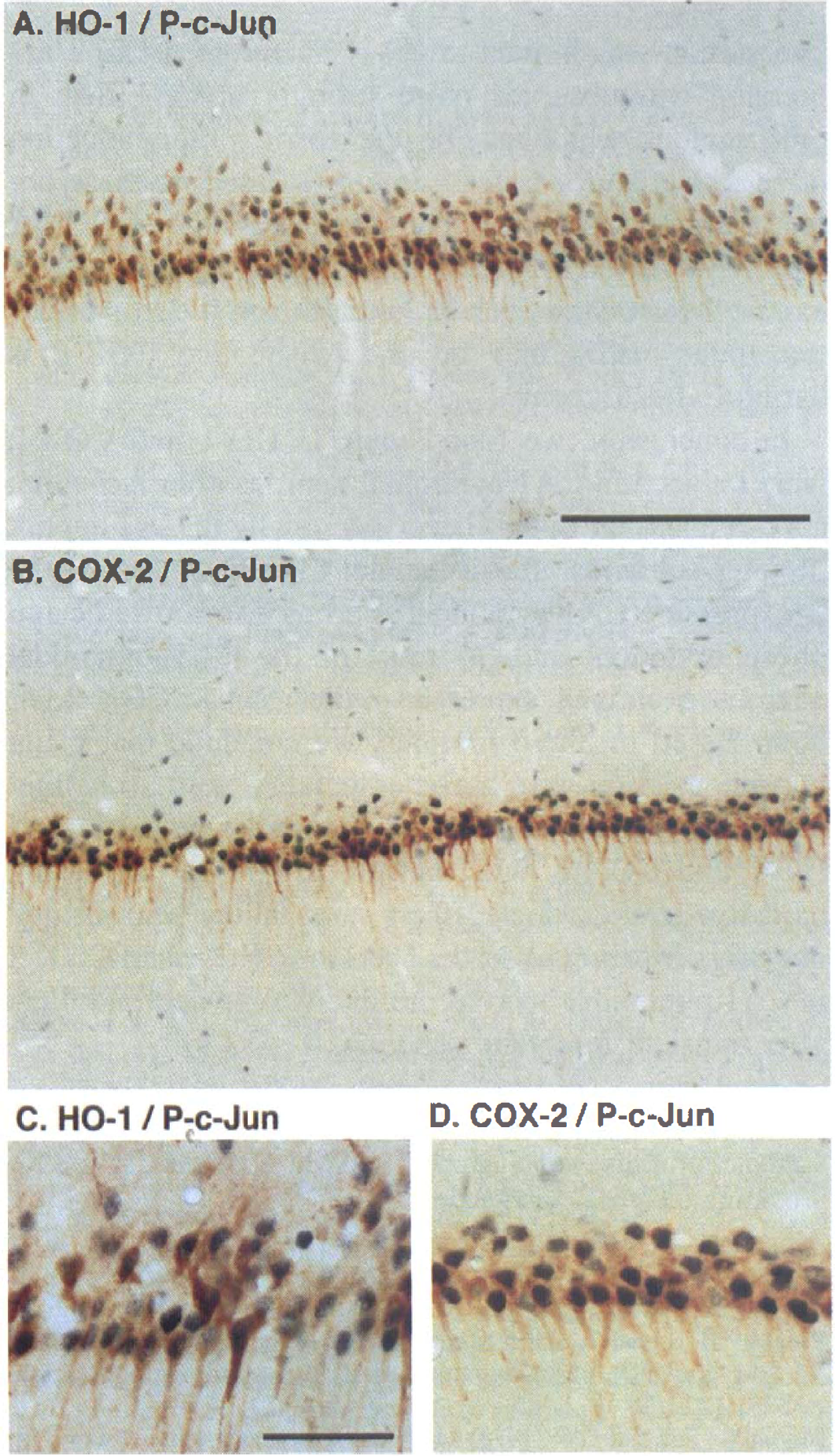
Co-localization of phosphorylated c-Jun with HO-1 and COX-2 after ischemia. Double immunostaining of phosphorylated c-Jun (P-c-Jun) with HO-1 (
Distribution of HO-1 and COX-2 in CA1 after ischemia
Both HO-1 and COX-2 were expressed in CA1 pyramidal neurons after ischemia; however, the population of HO-1– and COX-2–co-expressing cells is unclear. Therefore, we examined the population of HO-1– and COX-2-expressing cells. Few neurons (14%) expressed neither HO-1 nor COX (Fig. 6; Table 2). Most (86%) of the CA1 pyramidal neurons expressed either HO-1, COX-2, or both.
Colocalization of HO-1 and COX-2 in CA1 pyramidal neurons after ischemia

The distribution of cells expressing HO-1 and/or COX-2 in CA1 pyramidal neurons was quantitatively analyzed. Data are presented as the mean ± standard deviation of five rats. COX-2, cyclooxygenase-2; HO-1 heme oxygenase-1.
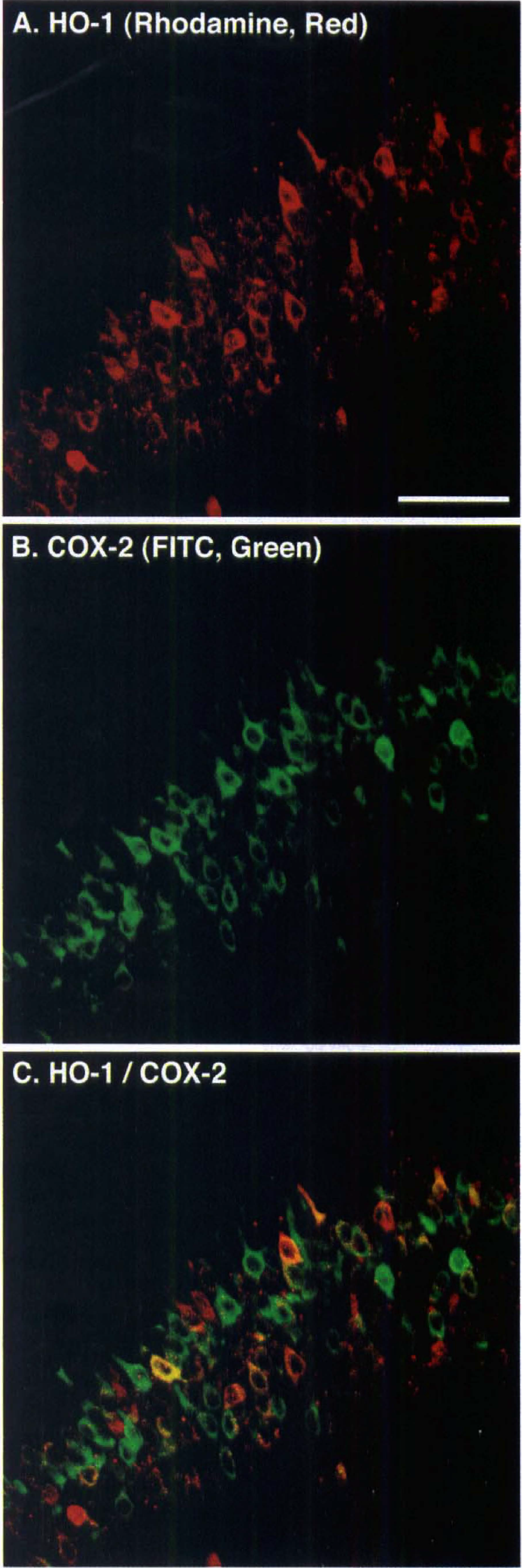
Distribution of HO-1 and COX-2 in CA1 pyramidal neurons after ischemia. Double immunostaining of HO-1 and COX-2 1 day after ischemia. Notice that HO-1 was detected by rhodamine (red) (
DISCUSSION
As a component of the AP-1 complex, c-Jun is known to dimerize c-Jun, c-Fos, activating transcription factor-2, cyclic AMP responsive element binding protein, and other transcription factors (Karin, 1995; Herdegen et al., 1998). Phosphorylation of c-Jun at Ser63 and Ser73 markedly enhances the transcriptional activity of AP-1 (Hibi et al., 1993; Dérijard et al., 1994), and its sensitivity varies with its dimerized partners (Kallunki et al., 1996). It has been shown that phosphorylation of c-Jun at Ser63 or 73 by JNK activation is involved in some forms of stress-induced neuronal apoptosis in cultured cells in vitro (Bossy-Wetzel et al., 1997; Eilers et al., 1998; Herdegen et al., 1998; Watson et al., 1998) and that JNK inhibitor attenuated 1-methyl-4-phenyl tetrahydropyridine-mediated neuronal loss in vivo (Saporito et al., 1999). In addition, mice that are deficient in the JNK3 gene show attenuated excitatory amino acid-induced neurodegeneration (Yang et al., 1997). We also found that injection of kainate induced c-Jun phosphorylation, specifically in neurons in CA3, which undergo apoptosis (Matsuoka et al., 1999b). Our current results indicate that ischemia induced transient phosphorylation of c-Jun, specifically in cells that undergo apoptosis, and that c-Jun was phosphorylated at an early stage in ischemia-induced neurodegeneration. These observations suggest that transient forebrain ischemia induces neuronal death through an apoptotic process in the CA1 subfield and that c-Jun phosphorylation is closely associated with this phenomenon. On the other hand, JNK participated with induction of HO-1 and COX-2 (Oguro et al., 1998; Subbaramaiah et al., 1998, respectively), therefore, phosphorylated c-Jun also may participated with induction of these proteins.
Whereas most protein synthesis in CA1 is inhibited after transient forebrain ischemia (Kiessling et al., 1986), certain proteins, such as heat shock protein, are induced (Nowak, 1993). Even in the untreated and vehicle-injected rat hippocampus, HO-1 and COX-2 mRNA and its protein are expressed at a low level (Vincent et al., 1994; Yamagata et al., 1993, respectively). In contrast, HO-1 and COX-2 were markedly and transiently induced in CA1 pyramidal neurons after transient forebrain ischemia.
It is still a concern whether apoptosis or necrosis underlies transient forebrain ischemia-induced delayed neuronal death. However, cells that undergo apoptosis are associated with cleavage of genomic DNA, and this can be detected before neuronal cell loss after ischemia (Nitatori et al., 1995). Because HO-1 produces powerful antioxidants such as biliverdin and bilirubin, HO-1-expressing cells may have some self-defense mechanism (Stocker et al., 1987). This result seems ambiguous, since HO-1-expressing CA1 pyramidal neurons undergo apoptosis. Iron also is produced by HO-1, which could lead to increased oxidative stress if not handled properly. Glutathione, an endogenous antioxidant, is part of a system of cellular defenses, and the level of glutathione is a factor in neuron survival after physical trauma (Lucas et al., 1998). The concentration of glutathione in neurons is lower than that in glial cells, which suggests that the self-defense system of neurons is weaker (Raps et al., 1989). Heme oxygenase-1 was induced in activated microglia on days 3 to 7 after ischemia, and cell death of microglia was not detected (Matsuoka et al., 1998b, 1999a). The intraperitoneal injection of hemin, an HO inducer, induced HO-1 expression in the hippocampus but did not reduce ischemia-induced neurodegeneration (Takizawa et al., 1998). In addition, HO-1-expressing cells also showed c-Jun phosphorylation, which is an important step in neuronal apoptosis. Based on these ob-servations and reports, we consider that HO-1 increases oxidative stress, which affects surrounding neurons and may trigger neuronal death. Therefore, HO-1 is a double-edged sword, and the effects of HO-1 expression may be related to self-defense. We showed here that the co-localization of HO-1, COX-2, and phosphorylated c-Jun, as well as the question regarding the function of HO-1 and COX-2 in ischemia-induced neurodegeneration, remain to be elucidated in future study.
Cyclooxygenase produces PGH2, a seed in the PG cascade. In ischemia/reperfusion-induced neurodegeneration, the local injection of PGE2 increased neuronal damage (Thornhill and Smith, 1998), whereas a PGI2 analogue had a neuroprotective effect (Matsuda et al., 1997). Thus, the effect of PG in ischemia-induced neurodegeneration remains unclear. On the other hand, COX markedly increased reactive oxygen species production (Morita et al., 1995), which is involved in ischemia-induced neurodegeneration (Pellegrini-Giampietro et al., 1990). In this study, COX-2 was induced in neurons at an early stage of neuronal death. In addition, a COX-2–selective inhibitor has been shown to prevent the delayed neuronal death in the CA1 subfield (Nakayama et a1., 1998) and to reduce the infarct volume in middle cerebral artery occlusion model (Nogawa et al., 1997). Therefore, COX-2 expression in CA1 pyramidal neurons was harmful through an increase in oxidative stress. Cyclooxygenase-2 was predominantly induced in CA1 and also was weakly induced in CA3, the hilus of DG, and DG, in contrast to the limited expression of HO-1 in CA1 Conversely, COX-2 has been reported to also have helpful effects in that COX-2 expression inhibits apoptosis (von Knethen and Brune, 1997). In addition, a report indicates that COX-2 expression induced increased adhesion to extracellular matrix protein such as laminin (Tsujii and DuBois, 1995). Laminin is constitutively expressed in hippocampal pyramidal neuronal layers (Chen and Strickland, 1997). Furthermore, laminin interacts with plasminogen and its activator (Salonen et al., 1984). An excitatory amino acid, kainate, induces plasminogen (Matsuoka et al., 1998c), and laminin is degraded before neuronal cell loss (Chen and Strickland, 1997). In addition, laminin was specifically lost in CA1 pyramidal neurons after transient forebrain ischemia (Matsuoka Y. et al., unpublished observation). Recent reports suggest that neurogenesis persists in the DG of rodents throughout their adult life (Gage et al., 1998), and this was enhanced by excitatory amino acid-induced neurodegeneration (Gray and Sundstrom, 1998). Expression of COX-2 requires the activation of c-Jun phosphorylation (Guan et al., 1998). After transient forebrain ischemia, c-Jun phosphorylation was limited to CA1. Therefore, COX-2 expression was observed more strongly in CA1 than in other regions, which may be one reason for the induction of selective neuronal death in CAL Based on these observations, we consider that the expression of COX-2 may have different effects that depend on the state of extracellular matrix such as laminin, and the loss of extracellular matrix may be one reason that COX-2 is harmful after ischemia.
In conclusion, we found that (1) HO-1 and COX-2 were induced in CA1 pyramidal neurons after ischemia; (2) c-Jun was phosphorylated specifically in CA1 but not in other subfields after ischemia; (3) HO-1- and COX-2-expressing CA1 pyramidal neurons also showed c-Jun phosphorylation; and (4) most of the CA1 pyramidal neurons examined expressed either HO-1, COX-2, or both. Based on these findings, we conclude that c-Jun phosphorylation is closely associated with ischemia/reperfusion–induced neuronal apoptosis in vivo, and HO-1 and COX-2 have harmful effects by increasing oxidative stress, which affects surrounding neurons and may trigger neuronal death. Therefore, HO-1 and COX-2 in CA1 pyramidal neurons are likely to be neuron killers after transient forebrain ischemia.
Footnotes
Acknowledgments:
The authors thank Professor Takashi Taniguchi for his encouragement and Mr. Kazuyuki Takata for providing technical assistance.