
Abstract
Hemoglobin (Hb) toxicity precipitates secondary brain damage following intracerebral hemorrhage (ICH). CD163 is an anti-inflammatory Hb scavenger receptor and CD163-positive macrophages/microglia locally accumulate post-bleed, yet no studies have investigated the role of CD163 after ICH. ICH was induced in wildtype and CD163−/− mice and various anatomical and functional outcomes were assessed. At 3 d, CD163−/− mice have 43.4 ± 5.0% (p = 0.0002) and 34.8 ± 3.4% (p = 0.0003) less hematoma volume and tissue injury, respectively. Whereas, at 10 d, CD163−/− mice have 49.2 ± 15.0% larger lesions (p = 0.0385). An inflection point was identified, where CD163−/− mice perform better on neurobehavioral testing and have less mortality before 4 d, but increased mortality and worse function after 4 d (p = 0.0389). At 3 d, CD163−/− mice have less Hb, iron, and blood–brain barrier dysfunction, increased astrogliosis and neovascularization, and no change in heme oxygenase 1 (HO1) expression. At 10 d, CD163−/− mice have increased iron and VEGF immunoreactivity, but no significant change in HO1 or astrogliosis. These novel findings reveal that CD163 deficiency has distinct temporal influences following ICH, with early beneficial properties but delayed injurious effects. While it is unclear why CD163 deficiency is initially beneficial, the late injurious effects are consistent with the key anti-inflammatory role of CD163 in the recovery phase of tissue damage.
Introduction
Intracerebral hemorrhage (ICH) is an especially severe stroke subtype that results in significant morbidity and mortality, yet no treatments are currently available.1,2 Hematoma clearance following ICH is required for recovery of local homeostasis and neurologic function. 3 This process involves hemolytic events that result in large quantities of extracellular hemoglobin (Hb). 4 Subsequently, as the heme-iron of Hb is converted from ferrous to ferric form, Hb is oxidized and becomes unstable, quickly releasing its heme moieties.5,6 Extracellular Hb/heme/iron initiates a neurotoxic cascade of free radical-induced damage, oxidative stress, and inflammation.4,5,7 While the inflammatory response and associated cellular activation is initially helpful in the clean-up process after ICH, resolution of neuroinflammation is necessary or additional secondary brain damage will occur.3,8
CD163 is a scavenger receptor expressed on cells of the monocyte lineage that is classically known for its function of facilitating the safe clearance of extracorpuscular Hb, thereby preventing the toxic sequelae of extracellular Hb/heme/iron. 9 CD163 is also a marker of alternatively activated anti-inflammatory macrophages/microglia that are abundant during the resolution phase of the inflammatory process, and has been postulated to participate in angiogenic repair mechanisms.10–12 It is of considerable interest that no studies have evaluated the role of CD163 after ICH, given its aforementioned functions and reports using human autopsy specimens and a porcine model demonstrating that CD163-positive macrophages/microglia accumulate in the brain following ICH.13–15
Using CD163-deficient mice, we reveal that CD163 has distinct temporal influences on ICH outcomes, with early deleterious properties and delayed beneficial effects. We also investigated potential local mechanisms for these biphasic outcomes including Hb clearance and the heme degradation pathway, blood–brain barrier (BBB) integrity, gliosis, and angiogenesis.
Materials and methods
Mice
All animal procedures were approved by the University of Florida Institutional Animal Care and Use Committee, conducted in accordance with the National Institutes of Health PHS policy on Humane Care and Use of Laboratory Animals, and reported following ARRIVE guidelines. Mice were bred and maintained in our temperature-controlled (23 ± 2℃) animal facilities on a reverse light cycle (12-h light/dark), so neurobehavioral testing could be conducted during the awaken phase. Three cohorts of C57BL/6 N wildtype (WT) and CD163 knockout (CD163−/−) male mice were used. The 3-d study used n = 20 WT (4.1 ±0.3 mo) and n = 19 CD163−/− (4.7 ± 0.4 mo) mice. The 10-d study used n = 30 WT (3.1 ± 0.2 mo) and n = 23 CD163−/− (3.6 ± 0.1 mo) mice. For determination of initial hemorrhage volume, n = 5 WT and n = 4 CD163−/− mice age-matched to the 3- and 10-d cohorts were used. Sample sizes were calculated to achieve a power of 80% using data from a prior analogous study (standard deviation of 0.7652, effect size Cohen’s d of 1.2034). Sample sizes were estimated to be n = 10 and n = 20 for the 3- and 10-d endpoints, respectively, and were subsequently adjusted to account for an expected mortality rate of 30–40%. The CD163−/− mice were originally generated by Dr. Søren K Moestrup. 9 They develop normally, have no gross anatomical or behavioral abnormalities, and are visually indistinguishable from WT littermates. Computer-generated random numbers were used with a unique code linking to the individual animal. No mice were excluded from this study. All surgical procedures and anatomical and functional outcomes were performed and assessed in a blinded manner.
ICH model
ICH was induced using our described model. 16 Briefly, mice were anesthetized with isoflurane and immobilized in a stereotactic frame. An injection of 0.04 U collagenase type VII-S (Sigma) dissolved in 0.4 µl of sterile water was performed at a 40° angle from the vertical plane into the left hemisphere at 0.2 µl/min using an automated injector. The injection site was 3.6 mm ventral from the skull surface at 0.0 mm anterior and 3.8 mm left, relative to bregma. The needle was left in place for 5 min and then slowly removed over a 15-min period to prevent backflow. Rectal temperature was maintained at 37.0 ± 0.5℃ during surgery. Mice were allowed ad libitum food and water before and after surgery and allowed to fully recover in temperature- and humidity-controlled chambers postoperatively. All possible efforts were made to reduce animal suffering. Bupivacaine was used for analgesia, hydration and wet chow were provided immediately postoperatively and daily thereafter, and animals were euthanized at humane endpoints. For euthanasia, mice were anesthetized and transcardially perfused with ice-cold phosphate-buffered saline (PBS) followed by 4% paraformaldehyde.
Initial hemorrhage volume
Potential differences in collagenase-induced bleed volumes in WT and CD163−/− mice were evaluated by measuring brain Hb content in both groups at 5-h post-ICH as we have described with slight modifications.17,18 Briefly, mice were anesthetized and transcardially perfused with ice-cold PBS. After quick removal of the brain, the olfactory bulbs, cerebellum, and contralateral hemisphere were discarded and the ipsilateral hemisphere was snap frozen. Samples were subsequently thawed, homogenized, and centrifuged at 14,000 rpm and 4℃ for 30 min, and the protein content of the supernatant was estimated using the bicinchoninic assay (ThermoFisher Scientific). Hb content was determined by enzyme-linked immunosorbent assay (Biomatik) according to manufacturer instructions after diluting samples 1:20,000 in the kit-provided sample diluent. Results are expressed as the concentration of Hb normalized to total protein content.
Functional outcomes
Functional outcomes were assessed by open field locomotor activity, rotarod, and neurological deficit scores (NDS) daily post-ICH. 16 Behavioral tests were performed in the same order and at the same time of day, with 1 h of rest between tests.
Histology and quantification
Histology and quantification were conducted as we have described. 16 Briefly, 10 sets of 16 sections equally distributed throughout the hematoma and anteroposterior brain regions were processed. Cresyl violet staining was used to assess lesion and hematoma volume, tissue injury, percent ipsilateral hemispheric enlargement, and hematoidin-pigment (bilirubin). Perls’ iron staining was used to evaluate iron. Primary antibodies used for immunohistochemistry include heme oxygenase 1 (HO1; 1:3000, Enzo Life Sciences), Hb (1:500, MP Biomedicals), immunoglobulin G (IgG) (1:300, Vector Laboratories), glial fibrillary acidic protein (GFAP), (1:1000, Dako), platelet endothelial cell adhesion molecule 1 (PECAM) (1:400, Santa Cruz), and vascular endothelial growth factor (VEGF) (1:500, Santa Cruz). For a given stain and endpoint, slides for all animals were simultaneously stained. All slides were scanned using an Aperio ScanScope CS and analyzed with ImageScope software (Leica Biosystems).
For quantification of total brain pathology (lesion and hematoma volume, tissue injury, ipsilateral hemispheric enlargement, Hb, iron, bilirubin, IgG, and HO1), all sections were quantified. For GFAP, five sections representing maximal lesion area were analyzed. For 3-d PECAM and VEGF, three sections representing maximal lesion area were used. For 10-d VEGF, the section representing maximal lesion area was evaluated. Lesion Volume: injured brain areas were outlined. Using these areas, known distances between sections, and section thickness, a total brain lesion volume was calculated. Percent ipsilateral hemispheric enlargement: ipsilateral and contralateral hemispheres were outlined, volumes were calculated similar to lesion volume, and the following equation was used, 100 × ((ipsilateral-contralateral)/contralateral). A positive pixel count algorithm was used for quantification of appropriately outlined brain regions for hematoma volume, iron, bilirubin, and immunohistochemical stains. These algorithms were tuned for each stain such that the appropriate signal level was detected. 16 Hematoma volume: after running the algorithm, the number of blood-positive pixels was converted into an area using pixel size and a volume was calculated in an identical manner as for lesion volume. Tissue injury: hematoma volume was subtracted from total lesion volume. Hb: data are presented as ipsilateral hemisphere signal normalized for contralateral. GFAP: cortical astrogliosis was analyzed by placing 1000 × 1000 pixel boxes in the motor cortex. Perihematomal astrogliosis was analyzed by circling the ipsilateral and contralateral striatum, and the lesion area was excluded such that the ipsilateral analysis represents perihematomal quantification rather than a total striatal analysis. Hemispheric astrogliosis was analyzed by circling of the ipsilateral and contralateral hemispheres, again excluding the lesion area. The perihematomal area was quantified to evaluate the local response to the ICH, and the cortex was quantified due to its connection with the striatum/perihematoma and its involvement in motor function. The global response to the ICH was assessed by hemispheric astrogliosis. PECAM: cortical and hematomal PECAM was analyzed by outlining the motor cortices and hematoma region, keeping a constant distance away from the damaged area, respectively. Ipsilateral data are reported. VEGF: was analyzed identical to PECAM, except data are presented as relative ipsilateral to contralateral signal.
Statistics
Statistics were performed using SAS-JMP in consultation with a biostatistician. Mortality was evaluated using a χ2 test and a log-rank test for Kaplan–Meier estimates. Neurobehavioral endpoint and anatomical data were analyzed by unpaired two-tailed Student’s t-tests. Neurobehavioral regressions were analyzed by repeated measures linear mixed modeling to account for identified baseline differences between groups and allow estimations of mortality dropouts. All data sets were checked for normality and differences in variance, and data were transformed or a nonparametric test was used as applicable. Data are expressed as mean ± SEM with p < 0.05 considered statistically significant.
Results
ICH-induced brain damage
CD163 has distinct temporal influences on ICH-induced brain damage (Figure 1(a) and (g)). At 3 d, CD163−/− mice have 33.2 ± 4.5% smaller lesion volumes (13.9 ±1.0 mm3 vs. 9.3 ± 0.6 mm3, p < 0.0001, Figure 1(c)), 43.4 ± 5.0% reduced hematoma volumes (3.2 ± 1.2 mm3 vs. 1.8 ± 0.5 mm3, p = 0.0002, Figure 1(b) and (d)), and 34.8 ± 3.4% less tissue injury (10.6 ± 3.9 mm3 vs. 6.9 ± 1.2 mm3, p = 0.0003, Figure 1(e)). No significant difference in percent ipsilateral hemispheric enlargement was seen (WT: 10.0 ± 1.4%, CD163−/−: 7.6 ± 1.4%, p = 0.2327, Figure 1(f)). At 10 d, CD163−/− mice show 49.2 ± 15.0% larger lesion volumes (1.8 ±0.3 mm3 vs. 2.7 ± 0.3 mm3, p = 0.0385, Figure 1(i)). Hematoidin-pigment (bilirubin) appears around 10 d post-ICH,
5
and no difference was seen between groups (WT: 15.7 ± 7.0 × 104 A.U., CD163−/−: 16.8 ±5.7 × 104 A.U., p = 0.9233, Figure 1(h) and (j)).
CD163 deficiency temporally influences ICH-induced brain damage. Representative Cresyl violet stained brain sections are shown for WT and CD163−/− mice at (a) 3 d and (g) 10 d after collagenase-induced ICH. Within genotype and study endpoint, images are from the same animal, where left-to-right corresponds to anterior-to-posterior sections. Representative high magnification images are shown for (b) blood content at 3 d and (h) hematoidin content at 10 d for WT and CD163−/− mice. At 3 d, CD163−/− mice have significantly less (c) overall ICH-induced brain damage, (d) residual blood volume, and (e) tissue injury. (f) No significant difference is seen in ipsilateral hemisphere enlargement at 3 d. (i) At 10 d, CD163−/− mice have significantly more ICH-induced brain damage. (j) No difference is seen in the amount of hematoidin-pigment (bilirubin) content. At 3 d, comparisons include n = 20 WT and n = 19 CD163−/− mice. At 10 d, comparisons include n = 11 WT and n = 12 CD163−/− mice. *p < 0.05, ***p < 0.001, ****p < 0.0001.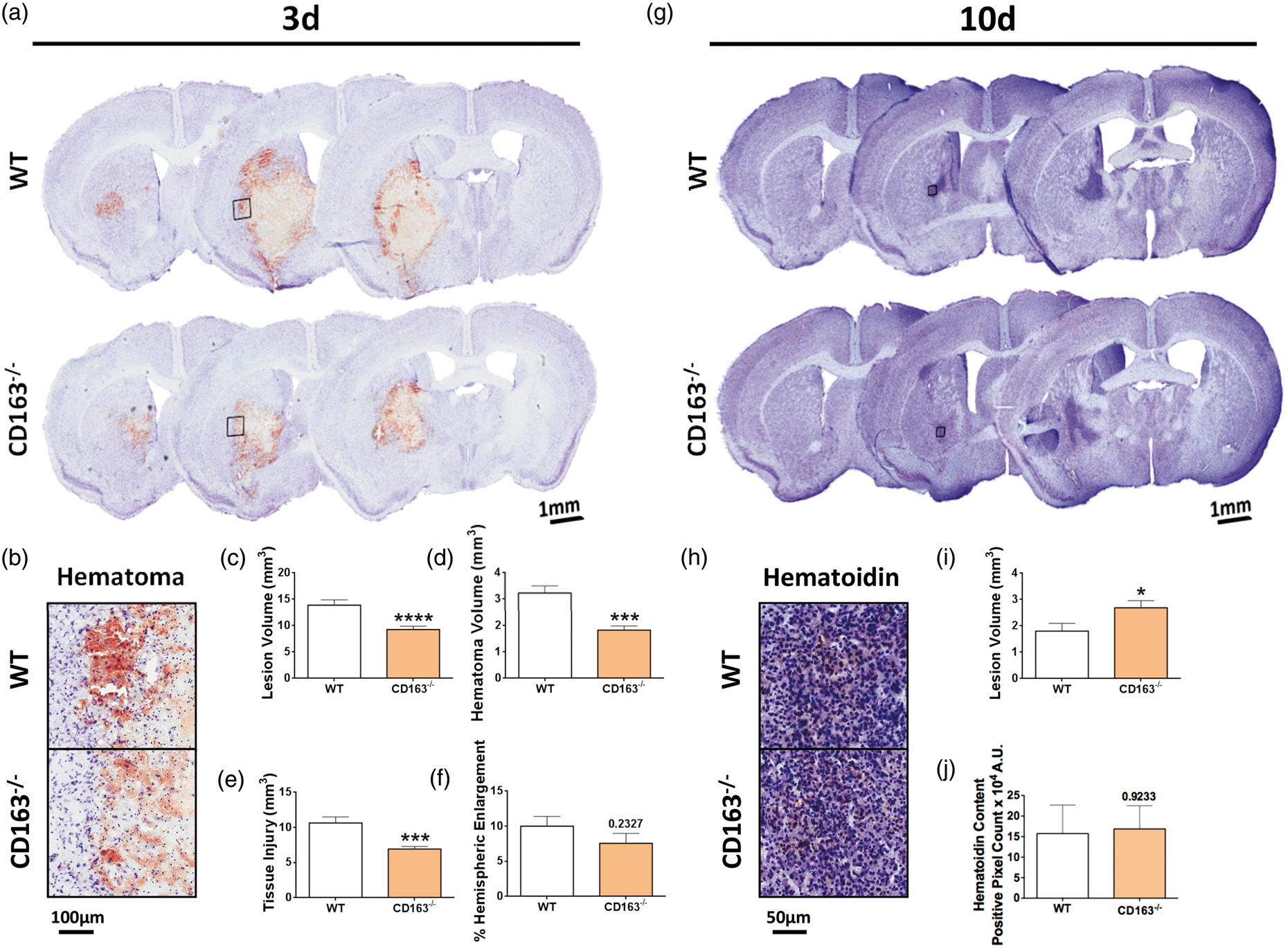
Mortality
Over the course of 10 d, no significant difference was observed between the Kaplan–Meier estimates of survival (p = 0.2533, Figure 2(a)), and no difference in 10-d endpoint mortality was observed between WT (43.3%, 13/30) and CD163−/− (39.1%, 9/23) mice (p = 0.7566). However, further inspection of mortality over the course of these 10 d revealed a temporal disproportion, where mortality on or before 4 d and after 4 d significantly differed between groups (p = 0.0389, Figure 2(b)). WT mice accounted for the majority of deaths on or before 4 d, where 76.9% of the total deaths were WT mice versus only 23.1% that were attributed to CD163−/− mice. Whereas CD163−/− mice accounted for the majority of deaths after 4 d, with 33.3% of the total deaths after this time attributed to WT mice and 66.7% to CD163−/− mice (Figure 2(b)).
CD163 deficiency temporally influences mortality and functional outcomes after ICH. WT and CD163−/− mice underwent collagenase-induced ICH and were survived to 10 d. (a) Log-rank analysis demonstrates no difference in Kaplan–Meier survival estimates between groups. (b) However, a temporal disproportion in mortality was identified, where mortality on or before 4 d and after 4 d significantly differed between groups. WT mice account for the majority of deaths on or before 4 d and CD163−/− mice account for the majority of deaths after 4 d. (c) Regression analysis reveals that WT mice recover neurologic function significantly faster than CD163−/− mice. (d) Endpoint analyses show that CD163−/− mice have reduced neurological deficits at 3 d, but greater deficits at 10 d. (e–j) On measures of locomotor activity, regression analyses (e,g,i) show no differences in the rate of recovery between groups, and endpoint analyses (f,h,j) show no differences at 3 d. However, at 10 d, CD163−/− mice have reduced ambulatory distance and stereotypic time, and increased resting time, while WT mice demonstrate values approximating that of their baseline function. (k,l) On accelerating Rotarod performance, (K) regression analysis shows no difference in the rate of recovery between groups, but at the 3-d endpoint, CD163−/− mice exhibit improved latency to fall. No differences are seen between groups at the 10-d endpoint. The identified baseline differences in locomotor activity and rotarod performance between groups were statistically accounted for by linear mixed modeling, which also allows estimations of mortality dropouts. P values for regression analyses (c,e,g,i,k) represent slope comparisons. All comparisons include n = 23 WT and n = 17 CD163−/− mice. *p < 0.05, **p < 0.01.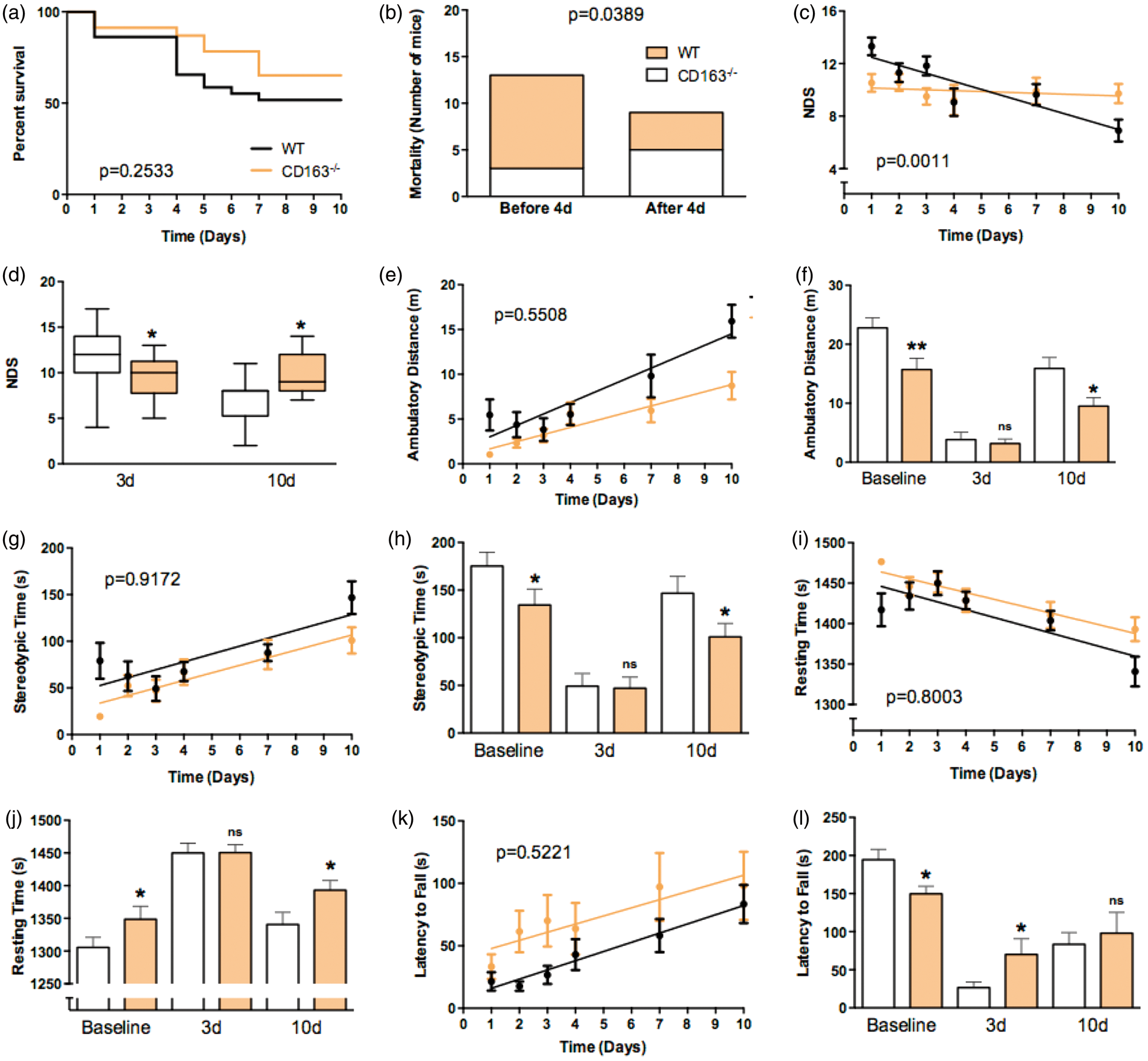
Functional outcomes
In accordance with ICH-induced brain damage and mortality, CD163 deficiency has temporal influences on functional outcomes (Figure 2). At the 3-d endpoint, CD163−/− mice display less neurological deficits (11.8 ± 0.7 vs. 9.5 ± 0.6, p = 0.0488, Figure 2(d)) and improved latency to fall on an accelerating Rotarod (83.5 ± 15.3 s vs. 98.1 ± 27.2 s, p = 0.0421, Figure 2(l)). At the 10-d endpoint, CD163−/− mice show greater neurological deficits (6.9 ± 0.8 vs. 9.7 ± 0.7, p = 0.0387, Figure 2(d)), reduced ambulatory distance (15.9 ± 1.8 m vs. 9.5 ± 1.4 m, p = 0.0129, Figure 2(f)) and stereotypic time (146.8 ± 17.5 s vs. 101.0 ± 14.1 s, p = 0.0497, Figure 2(h)), and increased resting time (1341 ± 18.4 s vs. 1393 ± 14.8 s, p = 0.0378, Figure 2(j)). Neurological deficit regression analyses over 10 d reveal that the WT mice recover faster while CD163−/− mice remain stable, leading to a mortality- and anatomical-corroborating inflection point at approximately 4-d post-ICH (WT slope: −0.6 ± 0.14, CD163−/− slope: −0.1 ± 0.1, p =0.0011, Figure 2(c)). Similar regression analyses over 10 d for open field locomotor activity and performance on an accelerating Rotarod were not significantly different between WT and CD163−/− mice (Figure 2(e), (g), (i), and (k)). It should be noted that the absence of CD163 led to significantly reduced baseline function on rotarod and all measures of open field activity. Baseline differences were statistically accounted for when evaluating post-ICH function such that the comparisons provided above are directly evaluating differences between WT and CD163−/− mice after ICH, independent of their baseline differences.
Initial hemorrhage volume
Since different initial bleed volumes in WT and CD163−/− mice after collagenase-induced ICH could temporally influence anatomical and functional outcomes, we assessed brain Hb content at 5-h post-collagenase injection. Brain Hb content was not significantly different between WT and CD163−/− mice (WT: 3.0 ± 1.0 ng Hb/µg protein, CD163−/−: 1.4 ± 0.3 ng Hb/µg protein, p = 0.1780).
Hb
To begin understanding the role of CD163 in Hb clearance after ICH, immunohistochemical staining for Hb was performed at 3 d (Figure 3(c)). CD163−/− mice display 30.0 ± 7.8% less Hb (4.6 ± 0.4 A.U. vs. 3.2 ±0.4 A.U., p = 0.0167, Figure 3(d)). When individually normalized for lesion volume, this difference is no longer observed (p = 0.8710).
CD163 deficiency reduces BBB dysfunction and Hb content. Representative images for WT and CD163−/− mice showing (a) IgG and (c) Hb immunohistochemistry 3-d post-ICH. Square selections on low-magnification images denote the location of magnified regions. (b) CD163−/− mice have significantly less BBB dysfunction. (d) CD163−/− mice display significantly less Hb. Comparisons include n = 7 WT and n = 14 CD163−/− mice for IgG and n = 5 WT and n = 7 CD163−/− for Hb. *p < 0.05, **p < 0.01.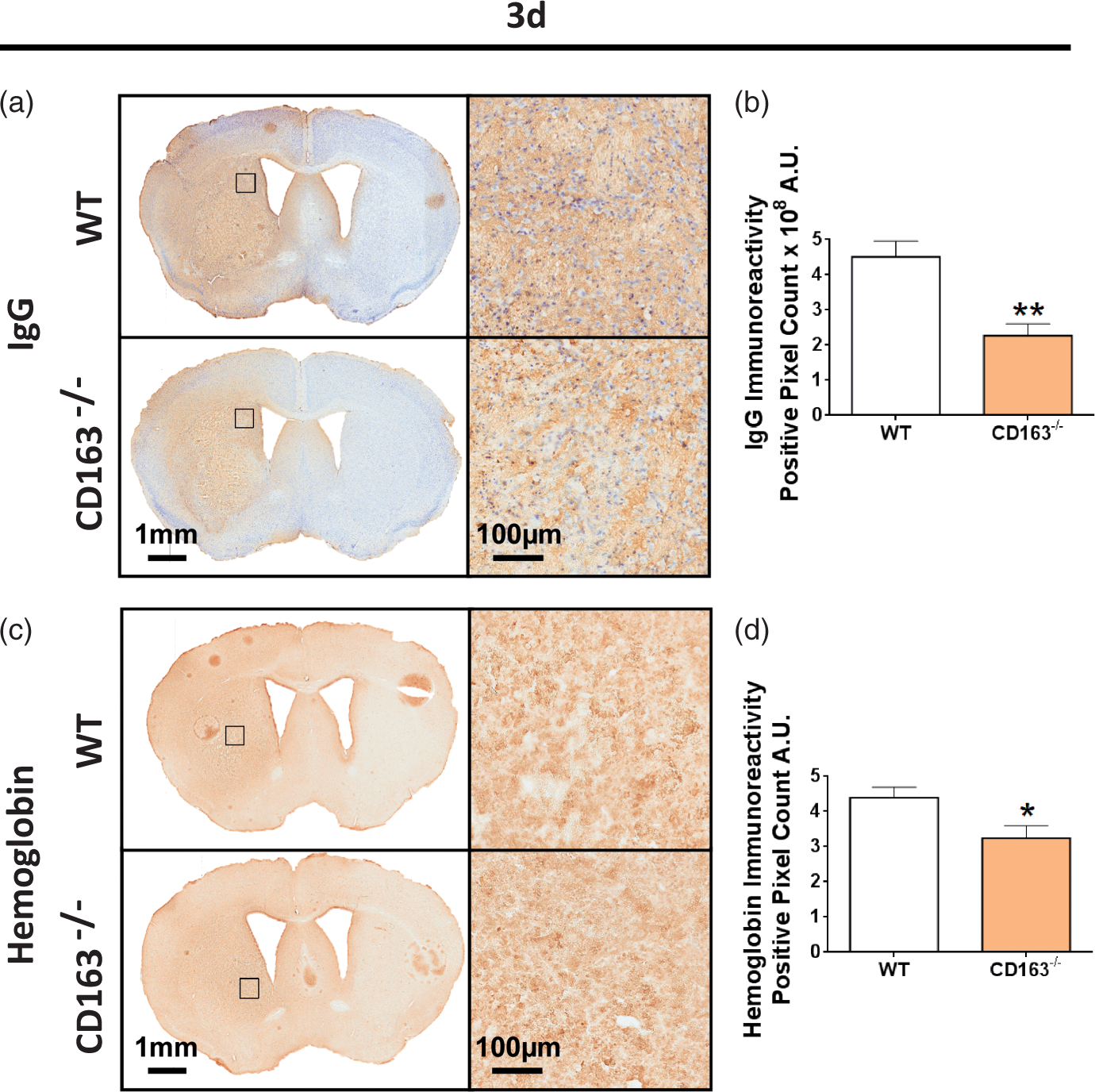
HO1 and iron
Furthermore, HO1 expression and iron content were evaluated by histology. HO1 expression was higher and primarily constrained to perihematomal areas at 3 d (Figure 4(a)) as compared to 10 d (Figure 4(e)) where expression was lower and more diffuse throughout the lesion. No difference in HO1 expression was seen at 3 d (WT: 2.8 ± 0.7 × 106 A.U., CD163−/−: 2.7 ± 0.9 × 106 A.U., p = 0.8384, Figure 4(b)) or 10 d (WT: 6.6 ± 2.3 × 105 A.U., CD163−/−: 8.2 ± 1.6 ×105 A.U., p = 0.3408, Figure 4(f)). When individually normalized for lesion volume, still no difference is seen at 3 d (p = 0.4727) or 10 d (p = 0.9530).
Effect of CD163 deficiency on HO1 and Perls’ iron. Representative images showing HO1 expression for WT and CD163−/− at (a) 3 d and (e) 10 d and iron deposition at (c) 3 d and (g) 10 d. Square selections on the low magnification images denote the location of magnified regions. HO1 images at 3 d and 10 d were counterstained with Cresyl violet and hematoxylin, respectively. Perls’ iron images were counterstained with nuclear fast red. (b,f) For both endpoints, no difference in HO1 expression is seen. (d) At 3 d, CD163−/− mice have significantly less iron. (h) At 10 d, CD163−/− mice have significantly more iron. At 3 d, comparisons include n = 14 WT and n = 16 CD163−/− mice for Perls’ iron and n = 10 WT and n = 10 CD163−/− mice for HO1. At 10 d, comparisons include n = 9 WT and n = 12 CD163−/− mice for Perls’ iron and n = 7 WT and n = 9 CD163−/− mice for HO1. *p < 0.05, ***p < 0.001.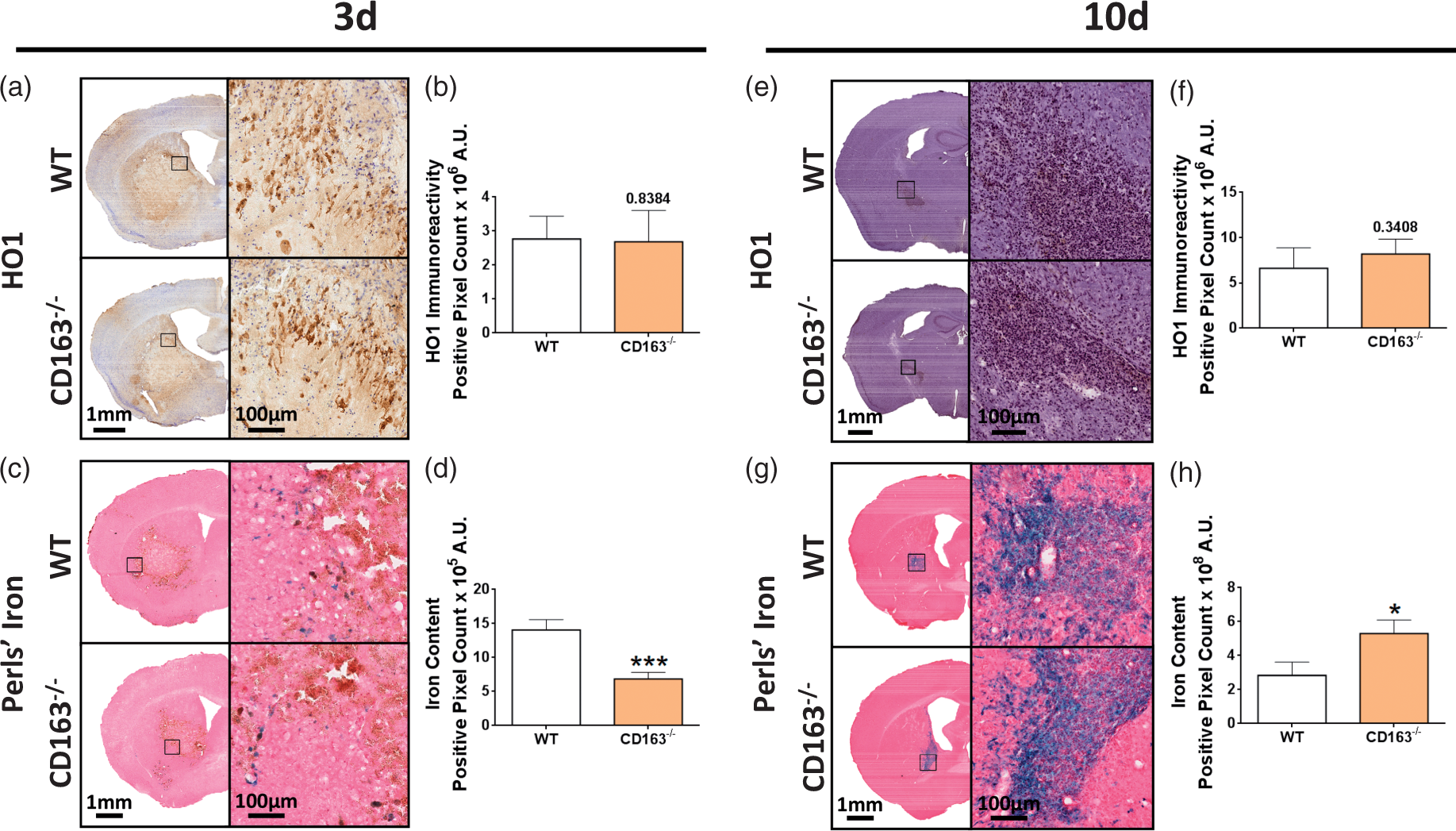
Iron was only observed in injured brain regions at both 3 d (Figure 4(c)) and 10 d (Figure 4(g)), and at 10 d, Perls’ staining was seen in glia mostly concentrated around vessels. CD163−/− mice exhibit 51.4 ±7.0% less iron at 3 d (14.1 ± 1.5 × 105 A.U. vs. 6.8 ± 1.0 × 105 A.U., p = 0.0004, Figure 4(d)) and 86.5 ± 28.5% more iron at 10 d (2.8 ± 0.8 × 108 A.U. vs. 5.3 ± 0.8 × 108 A.U., p = 0.0221, Figure 4(h)). When individually normalized for lesion volume, this difference is no longer observed at 3 d (p = 0.3632) and 10 d (p = 0.2306), suggesting that the differences between groups are likely due to the underlying lesion size differences.
BBB integrity
Immunohistochemical staining for IgG was performed to assess BBB dysfunction at 3 d (Figure 3(a)). CD163−/− mice have 49.7 ± 7.4% improved BBB integrity (4.5 ± 0.5 × 108 A.U. vs. 2.3 ± 0.3 × 108 A.U., p =0.0036, Figure 3(b)). When individually normalized for lesion volume, significance is lost (p = 0.0526).
Astrogliosis
Immunohistochemical staining for GFAP was performed to evaluate astrogliosis for WT and CD163−/− mice at 3 d (Figure 5(a) and (b)) and 10 d (Figure 5(f) and (g)). At 3 d, CD163−/− mice demonstrate 125.3 ± 55.8% (0.7 ± 0.3 × 10−2 A.U. vs. 1.5 ± 0.4 ×10−2 A.U., p = 0.0419, Figure 5(c)), 52.1 ± 25.2% (2.9 ± 0.2 × 10−2 vs. 4.3 ± 0.7 × 10−2 A.U., p = 0.0348, Figure 5(d)), and 47.5 ± 25.8% (2.2 ± 0.2 × 10−2 A.U. vs. 3.3 ± 0.6 × 10−2 A.U., p = 0.0348, Figure 5(e)) increased ipsilateral cortical, perihematomal, and hemispheric astrogliosis, respectively. No significant difference in contralateral cortical (WT: 0.3 ± 0.2 ×10−2 A.U., CD163−/−: 0.4 ± 0.1 × 10−2 A.U., p = 0.2556, Figure 5(c)), perihematomal (WT: 0.4 ± 0.1 × 10−2 A.U., CD163−/−: 0.8 ± 0.3 × 10−2 A.U., p = 0.1439, Figure 5(d)), and hemispheric (WT: 0.9 ± 0.1 × 10−2 A.U., CD163−/−: 1.3 ± 0.3 × 10−2 A.U., p = 0.1667, Figure 5(e)) astrogliosis is seen.
CD163 deficiency temporally influences astrogliosis. Representative images showing ipsilateral and contralateral cortical, perihematomal, and hemispheric GFAP immunoreactivity at 3 d for (a) WT and (b) CD163−/− mice and at 10 d for (f) WT and (g) CD163−/− mice. Square selections on the low magnification center whole-brain images denote the location of magnified regions. (c–e) At 3 d, CD163−/− mice demonstrate significantly increased ipsilateral (c) cortical, (d) perihematomal, and (e) hemispheric astrogliosis. (h–j) At 10 d, CD163−/− mice tend to show reduced astrogliosis in the ipsilateral (h) cortex and (j) hemisphere, but no difference is seen in the (i) perihematomal area. At 3 d, comparisons include n = 5 WT and n = 8 CD163−/− mice. At 10 d, comparisons include n = 4 WT and n = 6 CD163−/− mice. *p < 0.05.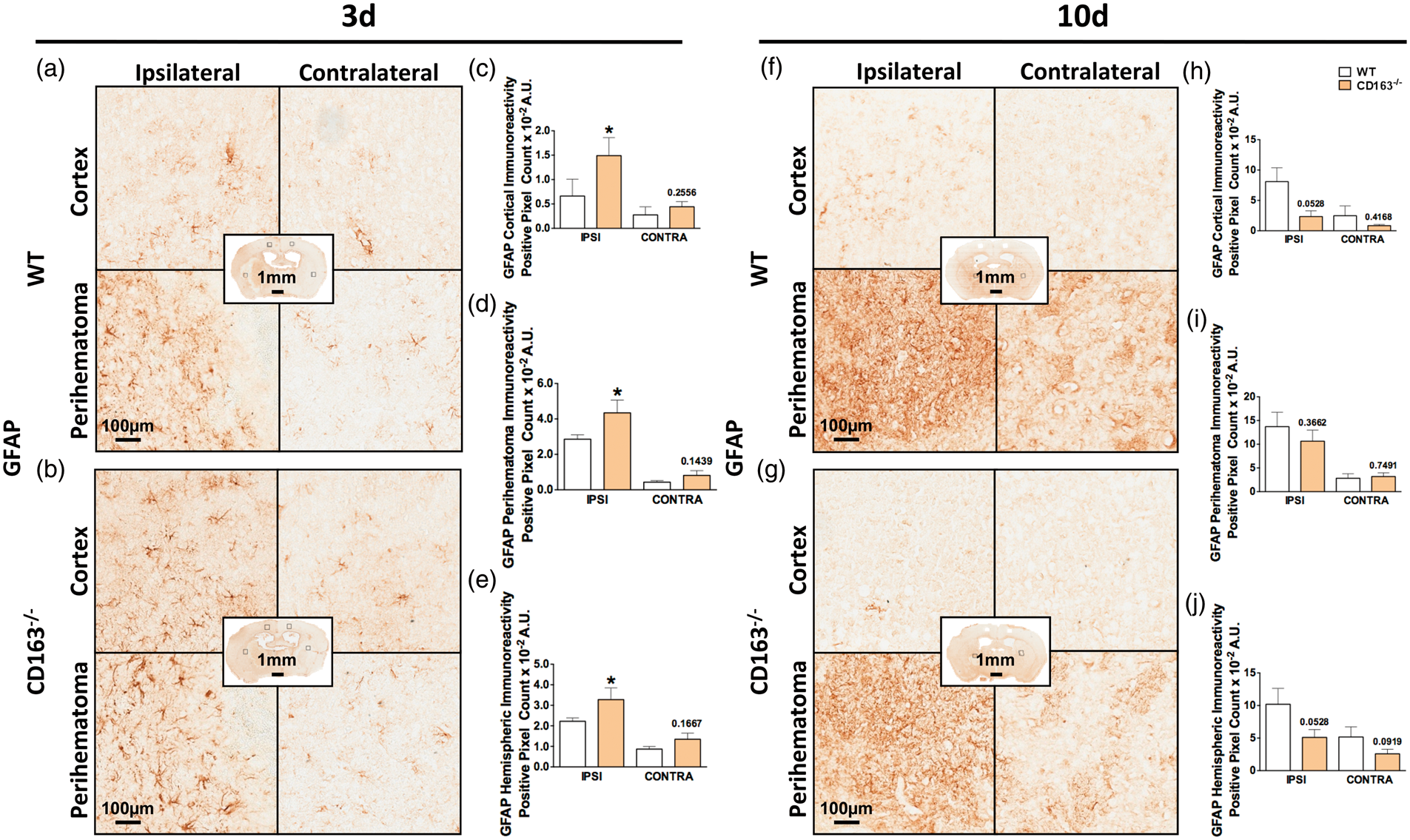
At 10 d, CD163−/− mice tend to display 560.2 ±91.1% decreased ipsilateral cortical astrogliosis (8.1 ±2.3 × 10−2 A.U. vs. 2.3 ± 0.9 × 10−2 A.U., p = 0.0528, Figure 5(h)). No difference is seen in contralateral cortical astrogliosis (WT: 2.5 ± 1.6 × 10−2 A.U., CD163−/−: 0.8 ± 0.2 × 10−2 A.U., p = 0.4168, Figure 5(h)), ipsilateral perihematomal astrogliosis (WT: 13.7 ± 3.0 × 10−2 A.U., CD163−/−: 10.7 ± 2.3 × 10−2 A.U., p = 0.3662, Figure 5(i)), or contralateral perihematomal astrogliosis (WT: 2.8 ± 1.0 × 10−2 A.U., CD163−/−: 3.2 ± 0.8 × 10−2 A.U., p = 0.7491, Figure 5(i)). CD163−/− mice also tend to show 50.0 ± 11.8% reduced ipsilateral hemispheric astrogliosis (10.2 ± 2.4 × 10−2 A.U. vs. 5.1 ± 1.2 × 10−2 A.U., p = 0.0528), but no difference in contralateral hemispheric astrogliosis is seen (4.3 ± 1.4 × 10−2 A.U. vs. 2.6 ± 0.7 × 10−2 A.U., p = 0.0919).
Angiogenesis/neovascularization
To investigate angiogenesis/neovascularization, immunohistochemical staining for VEGF and PECAM was performed (Figure 6(a), (d), and (g)). At 3 d, quantification of hematomal regions reveals no difference in PECAM (WT: 5.7 ± 0.7 × 105 A.U., CD163−/−: 5.2 ±0.3 × 105 A.U., p = 0.5160, Figure 6(c)). After individual normalization for lesion volume, still no difference is seen (WT: 5.0 ± 0.5 × 104 A.U., CD163−/−: 6.4 ±0.5 × 104 A.U., p = 0.1439). CD163−/− mice exhibit increased neovascularization in the ipsilateral motor cortex (2.8 ± 0.6 × 10−3 A.U. vs. 4.9 ± 0.6 × 10−3 A.U., p = 0.0439, Figure 6(b)).
Effect of CD163 deficiency on angiogenesis/neovascularization. Representative images showing ipsilateral cortical and hematomal immunoreactivity of (a) PECAM at 3 d, (d) VEGF at 3 d, and (g) VEGF at 10 d for WT and CD163−/− mice. Square selections on the low magnification center hemi-brain images denote the location of magnified regions. (b) At 3 d, CD163−/− mice have significantly increased neovascularization in the motor cortex. (c,e,f) No difference in hematomal neovascularization or cortical and hematomal VEGF expression is seen. (h–i) At 10 d, CD163−/− mice have increased hematomal VEGF expression, but no difference in cortical expression. At 3 d, comparisons include n = 5 WT and n = 7 CD163−/− mice for PECAM and n = 7 WT and n = 7 CD163−/− mice for VEGF. At 10 d, comparisons include n = 6 WT and n = 10 CD163−/− mice. *p < 0.05.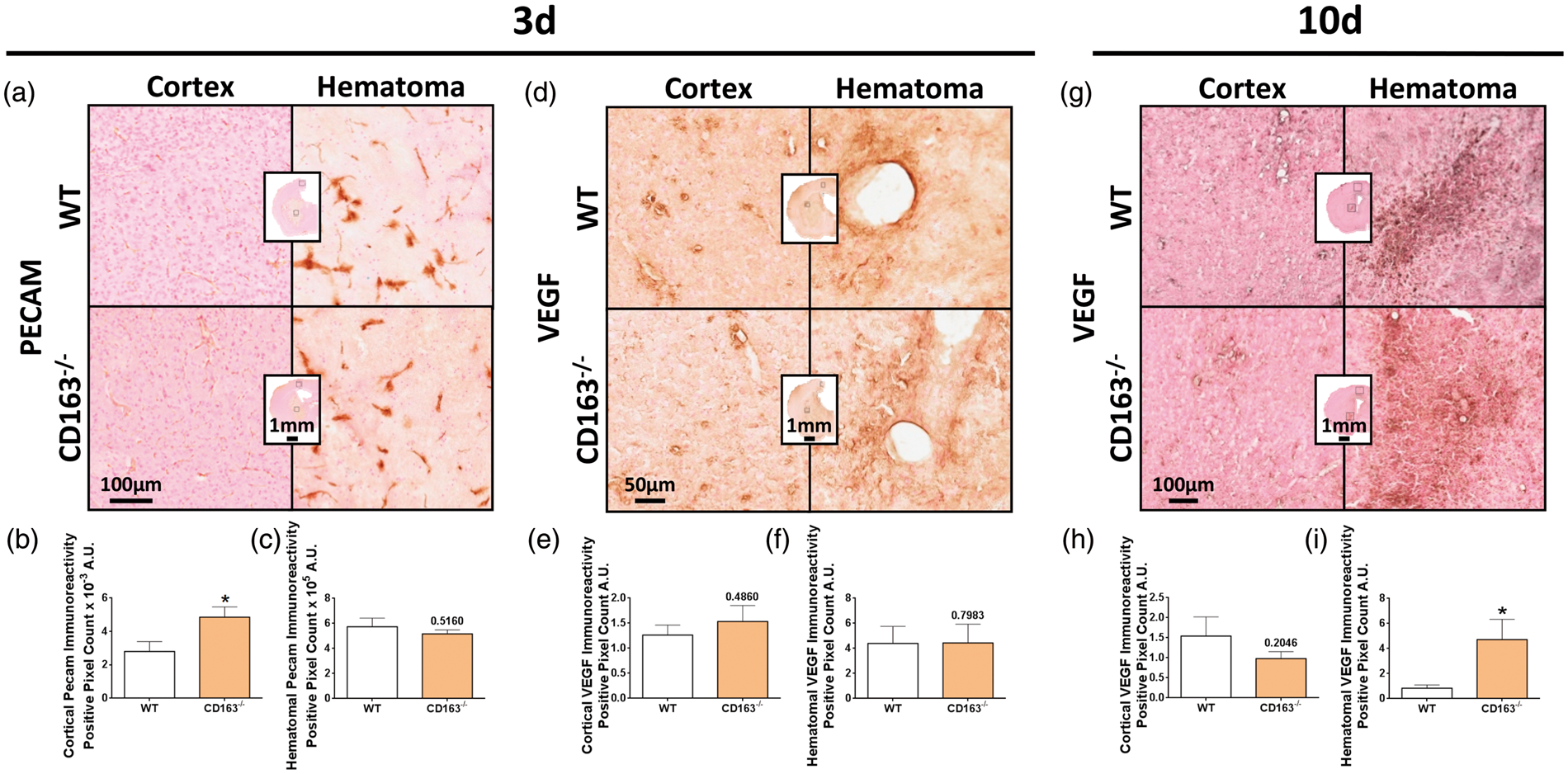
At 3 d, no difference in VEGF expression is seen in the ipsilateral motor cortex (p = 0.7983, Figure 6(e)) or hematomal regions (p = 0.4860, Figure 6(f)). After individual normalization for lesion volume, still no difference in hematomal VEGF expression is seen (p = 0.4853). At 10 d, no difference in cortical VEGF expression is observed (p = 0.2046, Figure 6(h)), but CD163−/− mice demonstrate increased hematomal VEGF expression (0.8 ± 0.3 A.U. vs. 4.7 ± 1.6 A.U., p = 0.0243, Figure 6(i)). After individual normalization for lesion volume, significance is lost (WT: 0.4 ±0.1 A.U., CD163−/−: 2.0 ± 0.7 A.U., p = 0.1407).
Discussion
This study is the first to evaluate the contribution of CD163 to ICH outcomes. Acutely, CD163−/− mice display significantly smaller lesions with reduced hematoma volumes and tissue injury, whereas larger lesions are seen at 10 d. Temporally, these differences in anatomical damage correlate with functional outcomes, where CD163−/− mice have fewer neurological deficits and improved performance on an accelerating rotarod acutely, but in the long term have greater deficits and worse ambulatory ability. Regression analyses of functional outcomes and mortality identified the inflection point to be 4-d post-ICH. At or before this time, CD163−/− mice do well and have significantly less mortality, but after this point, they have worse neurologic function and increased mortality. At 3 d, CD163−/− mice have significantly less Hb, iron, and BBB breakdown, increased cortical, striatal, and hemispheric astrogliosis and cortical neovascularization, and no detectable change in HO1 expression. At 10 d, CD163−/− mice have increased iron and hematomal VEGF immunoreactivity, no change in HO1 expression, and tend to have decreased astrogliosis.
Hematoma absorption involves erythrophagocytosis and detoxification/clearance of hemolysis products. Hemolysis begins after 24-h post-ICH resulting in the accumulation of cytotoxic extracorpuscular Hb, 4 and Hb is an important instigator of delayed poor outcomes after ICH. 5 Haptoglobin (Hp) is an endogenous plasma protein that tightly binds and detoxifies Hb. 19 The resting central nervous system (CNS) Hb-binding capacity is estimated to be 50,000-fold lower than that of the large capacity systemic system. 20 Although Hp enters the brain as part of the hemorrhage, the collective levels are inadequate to handle the massive hemolytic release of Hb. Additionally, compared to humans, mice have around a fivefold lower plasma Hb-binding capacity.21,22
CD163 is the scavenger receptor for Hp-Hb complexes and also clears uncomplexed Hb under severe hemolytic conditions associated with Hp depletion, thereby serving as its own fail-safe Hb scavenger receptor.9,10 A soluble form of CD163 exists and is generated by ADAM17-mediated shedding of the ectodomain from the membrane-bound form of the receptor, although soluble CD163 is likely not significantly contributing to the outcomes of the present study since the mice used here do not have the consensus sequence for ADAM17-mediated ectodomain cleavage. 23 In mice, the estimated Kd values for complexed and uncomplexed Hb to membrane-bound CD163 are 18 and 61 nM, respectively. 9 It should be noted that an apparent greater difference in kinetic parameters exists in humans, with complexed and uncomplexed Hb having Kd values of 19 and 198 nM, respectively. 9
Basal CNS CD163 expression is extremely low and restricted to perivascular macrophages.15,24–27 Therefore, the capacity for Hb uptake immediately following the bleed must be limited. However, during acute hemorrhagic or non-hemorrhagic CNS pathology, CD163-positive macrophages/microglia accumulate within and surrounding the lesions.15,24–27 Specifically, following ICH, no significant changes in CD163 are seen throughout the first several hours. Statistically significant but small increases in CD163 expression levels and the number of CD163-positive macrophages/microglia are observed beginning at 24-h post-bleed, and both the expression level and number of cells continue to steadily rise thereafter. These changes are observed within the brain parenchyma and perivascular areas in and around the clot and also in regions more remote from the lesion.13–15 With this distant accumulation/expression and lack of predominance in hemorrhagic brain lesions, a few have speculated that there is a primary anti-inflammatory role for CD163, particularly in the resolution of inflammation.15,25,27 Although a clear interaction with Hb clearance exists, as receptor-mediated endocytosis of Hb coordinately increases CD163 expression and several other molecules involved in heme degradation, as well as IL-6 and IL-10 secretion.10,24,27 Therefore, CD163 has two anti-inflammatory consequences, (a) removal of pro-inflammatory Hb and (b) polarization of microglia/macrophages to an anti-inflammatory phenotype with an altered cytokine profile.
The current study is consistent with previous speculation that CD163 has primarily an anti-inflammatory role after acute brain injury. If Hb clearance was the main operating mechanism, CD163 deficiency would result in increased Hb and worse outcomes at 3-d post-ICH, where hemolytic events are pronounced and CD163-positive microglia/macrophages are elevated. However, CD163−/− mice have less Hb and smaller lesion volumes associated with reduced tissue injury, implying that CD163−/− mice acutely have an augmented Hb clearance ability. It is possible that these results are due to underlying compensatory mechanisms, which may have a greater influence early when CD163-positive macrophages/microglia are not in peak quantity. Although the smaller hematoma volumes combined with lower Hb levels could hint at enhanced erythrophagocytosis in the absence of CD163, an intriguing paradoxical notion, since M2-alternatively activated macrophages have high phagocytic capacity and participate in wound healing by producing IL-10, chemotactic, angiogenic, and extracellular matrix components. 28 It is possible that lifting the anti-inflammatory feedback loop, that CD163 initiates, polarizes their phenotype to a further enhanced phagocytic potential and/or erythrophagocytosis represents a distinct under-characterized phagocytic category. IL-4, a canonical M2 stimuli, induces tissue macrophage accumulation and erythrophagocytosis, but paradoxically decreases CD163 expression,10,29 indirectly suggesting that CD163-negative M2 macrophages could have enhanced erythrophagocytic potential. The temporal interacting complexity between CD163, macrophage polarization, activation stimuli, and erythrophagocytosis is not clear and warrants further investigation. Chronically, CD163−/− mice display increased brain damage, an intuitive finding given the well-characterized role for CD163-positive macrophages/microglia in the resolution of tissue inflammation, which is damaging if chronic and untamed. Importantly, these distinct temporal anatomical outcomes correlate with the 4-d inflection point identified on functional repeated measures parameters, including both mortality and neurobehavioral testing. Last, we have confirmed that the susceptibility to collagenase-induced bleeding is not different between WT and CD163−/− mice, with both groups displaying the same initial bleed volume. Therefore, the results presented here are not as a result of differences in lesion evolution; rather, the absence of CD163 more likely results in biphasic ICH outcomes.
The other outcome measures used in this study aimed to further characterize Hb clearance and heme degradation, inflammation, and the additional roles for CD163 suggested in other systemic conditions. No difference in HO1 expression (an inducible heme-metabolizing enzyme) was seen between groups; however, iron levels followed the temporal nature of this study with less and more iron seen at 3 d and 10 d, respectively. There are several possible explanations for these findings. CD163 deficiency would not allow the coordinately increased expression of Hb degradation molecules (e.g. HO1) upon Hp-Hb or Hb internalization, although heme itself (among many other factors) is an HO1-inducer. Thus, there are two main differential forces on HO1 induction between the groups, where WT mice would have more HO1 induction due to the presence of CD163, and CD163−/− mice would have more induction due to the hypothesized increased erythrophagocytosis-derived intracellular heme. It is also possible that cell-type specific changes in HO1 are occurring (since the methods used here would not distinguish in this case) and/or that the results are independent of CD163 and result from the underlying lesion size differences at a given study endpoint, as suggested by the lack of differences seen in iron content after normalizing results for lesion volume. Although iron redistribution/clearance/storage mechanisms are also likely different between groups. Interestingly, using an inducer and inhibitor of HO1, an analogous distinct temporal role for HO1 after ICH was recently reported. 30 HO1 was injurious early after ICH, increasing brain damage, iron deposition, and neurologic deficits, but was beneficial in later stages, increasing hematoma clearance, angiogenesis, and recovery of neurologic function. 30 While these results do not directly relate to the findings of no difference in HO1 expression in the present study (due to the different experimental paradigms), it is intriguing that a similar temporal influence on ICH outcomes was separately found for HO1 and CD163 given the coordinated regulation of these two important molecules in Hb clearance and the heme degradation pathway.
Significant differences in astrogliosis, BBB integrity, and angiogenesis were also observed in CD163 deficient mice after ICH. At 3 d, the reduced Hb, iron, and tissue injury correlate well with the improved BBB integrity in CD163−/− mice. In other settings, CD163 has been associated with angiogenesis, although these have been mostly associations linked through the known angiogenic properties of alternatively activated macrophages, rather than causative. Interestingly, here, CD163−/− mice show increased cortical neovascularization acutely with trends toward reduced cortical VEGF at 10 d, and increased hematomal VEGF expression at 10 d, which suggests a more direct angiogenic role for CD163. Last, glial scar formation is an important consideration following ICH, and CD163−/− mice again tend to display this temporal switch in outcomes, in this case, astrocyte activation and morphological changes.
In conclusion, we demonstrate a unique temporal role for CD163 after ICH. Although some questions remain unanswered, we believe the findings of this study open the door for future studies that should focus on further exploring the refined mechanisms and therapeutic potential of targeting the CD163 receptor.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: NIH grants F31NS086441 (JLL) and R01NS046400 (SD), the McKnight Brain Research Foundation, Brain and Spinal Cord Injury Research Trust Fund (SD), and the American Heart Association 17GRNT33450010 (SD).
Acknowledgments
The authors wish to thank the University of Florida Center for Translational Research in Neurodegenerative Disease and the McKnight Brain Institute, and members of the Doré lab for their technical expertise, with a special thanks to Harrison Phillips.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
JLL and SD conceived and designed the study. JLL, ASL, CLA, and BS performed experiments. JLL, ASL, and TV analyzed data. SKM provided the CD163−/− mice. JLL, ASL, PS, SKM, and SD interpreted results and wrote the manuscript. All authors have approved the manuscript for publication.