
Abstract
Apolipoprotein E (apoE) is a multifunctional protein with an expanding role in the neurobiology of disease. Although originally described in the context of cholesterol metabolism, interest in the neurobiology of apoE has intensified following the association between apoE genotype and risk of developing Alzheimer's disease. Recent clinical observations also suggest that apoE genotype may influence recovery after a variety of neurological insults. Thus, in addition to the study of disease-specific mechanisms by which apoE may modulate susceptibility of developing Alzheimer's disease, there has been an increasing focus on its role in modulating the CNS response to acute injury. Although the neurobiology of apoE in the injured brain remains incompletely defined, there is evidence to suggest neurotrophic, immunomodulatory, and antioxidant effects.
Apolipoprotein E (apoE) is a 299–amino acid protein with multiple biological properties. There are three common human isoforms of apoE, designated E2, E3, and E4, which are encoded by three different alleles with frequencies of 7%, 78%, and 15%, respectively (Roses et al., 1996; Roses et al., 1995). The three different apoE isoforms differ by a single amino acid substitution: the E2 isoform has cysteine residues at positions 112 and 158, whereas the E3 isoform has an arginine at position 158, and the E4 has an arginine at both positions. Although initially described as an important mediator of cholesterol metabolism, apoE also has been demonstrated to have immunomodulatory properties in vitro, and may regulate smooth muscle and endothelial cell growth and differentiation (Vogel et al., 1994; Majack et al., 1988; Pepe and Curtiss, 1986; Macy et al., 1983; Avila et al., 1982; Curtiss and Edgington, 1981; Edgington and Curtiss, 1981; Hui et al., 1980; Curtis and Edgington, 1978). Also, apoE synthesis is locally upregulated after peripheral nerve injury, and is upregulated by astrocytes and oligodendrocytes after CNS injury, which has prompted speculation that this protein might be involved in neural injury or repair (Aamar et al., 1992; Stoll et al., 1989; Ignatius et al., 1986; Stoll and Mueller, 1986; Snipes et al., 1986; Muller et al., 1985).
APOLIPOPROTEIN E AND ALZHEIMER'S DISEASE
The brain provides the largest extrahepatic source of apoE synthesis, and interest in the role of apoE in the CNS was renewed after the observation that the E4 isoform is associated with increased risk of developing sporadic and late-onset familial Alzheimer's disease (Strittmatter et al., 1993a; Saunders et al., 1993). The mechanism by which apoE influences susceptibility of developing Alzheimer's disease remains to be elucidated. An active area of investigation focuses on the hypothesis that one of the normal functions of apoE within neurons is to contribute to microtubular integrity and stabilization of the neuronal cytoskeleton. In Alzheimer's disease, it is suggested that the widespread neuronal disruption is a consequence of disruption of this normal apoE function (Roses et al., 1995). In support of this hypothesis, several reports have documented isoform-specific interactions between apoE and the microtubule binding protein tau (Fleming et al., 1996; Strittmatter et al., 1994) Isoform-specific interactions also have been observed between apoE and β-amyloid, raising speculation that apoE might influence amyloid deposition, metabolism, or fibrillogenesis (Gallo et al., 1994; Sanan et al., 1994; Strittmatter et al., 1993). The hypothesis that apoE is involved in the early stages of amyloid deposition is supported by our observations that most amyloid deposits after head injury are Aβ42(43)-positive and immunostain with apoE (Horsburgh et al., 1997b). The presence of the APOE4 allele also has been associated with deposition of β amyloid after head injury (Nicoll, 1995).
APOLIPOPROTEIN E AND RECOVERY AFTER CNS INJURY
In addition to its association with Alzheimer's disease, several clinical observations suggest an isoform-specific role for apoE in determining outcome after acute CNS injury. In 1995, Nicoll and associates reported a neuropathologic study on a series of patients who died within 30 days after severe head injury. The authors found that the frequency of the APOE4 allele was significantly higher in individuals with amyloid deposition than in those without. However, the mean age of the group with amyloid deposition (high APOE4 allele frequency) was 52 years, whereas that of the other group (control APOE4 frequency) was 28 years. The initial interpretation of this study was that the APOE4 allele was associated with deposition of amyloid after head injury. However, an alternate explanation for these findings is that plaques form at a young age in people possessing the APOE4 allele, and that people with APOE4 are more likely to die after CNS trauma. This study provides indirect evidence that apoE genotype affects recovery after closed head injury (Nicoll et al., 1995). Direct evidence that apoE genotype influences outcome after head injury has been provided from a prospective study of 90 individuals studied 6 months after sustaining a head injury. Individuals possessing an APOE4 allele were found to be twice as likely to have a poor outcome after head injury compared with individuals without an APOE4 allele, as assessed using the Glasgow outcome scale (Teasdale et al., 1997). Two reports also have documented an association between the E4 isoform and poor prognosis after closed head injury (Sorbi et al., 1996; Seliger et al., 1997). In a preliminary study of boxers, possession of APOE4 allele was associated with chronic traumatic brain injury in high-exposure boxers (Jordan et al., 1997), further supporting role for apoE genotype on determining outcome after brain injury.
There are also preliminary clinical data that apoE genotype affects prognosis after intracerebral hemorrhage. In a recent study, Alberts and others performed genotyping on a consecutively ascertained, prospective series of patients with nonaneurysmal intracerebral hemorrhage. Although there was no clear association between apoE genotype and incidence of intracranial hemorrhage, patients with the APOE4 allele had a much higher mortality than those with the more common apoE genotypes E2/E3 or E3/E3 (68% versus 19%). Of patients who survived their hemorrhage, those without the APOE4 allele had improved functional recovery (Alberts et al., 1996). Thus, although there is conflicting evidence concerning the effect of apoE on risk of intracerebral hemorrhage, recovery appears to be strongly related to apoE genotype (Greenberg et al., 1996; Nicoll et al., 1996).
Additional evidence that apoE genotype is relevant to acute brain injury is taken from patients undergoing cardiopulmonary bypass for cardiac surgery procedures. Neuropsychiatric changes occur in as many as 50% to 60% of these patients (Borowicz et al., 1996; Bruggemans et al., 1995). In a recent study, a significant association was found between apoE4 dose and postoperative cognitive deficits, as measured by a battery of neuropsychiatric tests (Tardiff et al., 1997; Newman et al., 1995). This observation also supports the contention that apoE either modifies risk of CNS injury or reparative responses after acute CNS insult.
Several studies have produced conflicting results regarding the association between apoE genotype and incidence of thromboembolic stroke (Basun et al., 1996; Kuusisto et al., 1995; Couderc et al., 1993). In a large, retrospective study examining elderly men, Basun and colleagues found no association between stroke incidence and apoE genotype (Basun et al., 1996; Pedro-Botet et al., 1992). However, a reduced frequency of E3/E4 was observed in patients who had survived a prior stroke, which is consistent with the hypothesis that although apoE genotype may not alter risk for stroke, it may alter recovery once it occurs. The hypothesis that apoE influences recovery from acute CNS injury in an isoform-specific fashion also is consistent with the recent observation that patients with the APOE4 allele are more likely to be cognitively impaired after stroke (Slooter et al., 1997; Snowden et al., 1997).
APOLIPOPROTEIN E LOCALIZATION WITHIN THE CNS
In rodents, apoE immunoreactivity is largely confined to astrocytes, although small extracellular deposits have been described in the perivascular and periarachnoid spaces (Boyles et al., 1985). Neuronal apoE immunoreactivity has not been reported in normal rodent brains. However, alterations in the cellular localization of apoE occurs after acute brain injury in rodents (Hall et al., 1995; Kida et al., 1995; Horsburgh and Nicoll, 1996a,b; Horsburgh et al. 1997c). Most notably, apoE immunoreactivity is detected in neurons after different forms of ischemic injury, although the appearance of intraneuronal apoE seems to be dependent on the type and severity of the ischemic insult. In a rat model of cardiac arrest, neuronal apoE is observed at 6 hours after global ischemia (Kida et al., 1995), whereas in a transient ischemia model, neuronal apoE is not detected until at least 24 hours after the initial insult (Horsburgh and Nicoll, 1996b). In general, apoE immunoreactivity after transient global ischemia is localized to selectively vulnerable brain regions such as the caudate and CA1 region of hippocampus (Horsburgh et al. 1996b). After injury, apoE immunoreactivity is initially increased in astrocytes and neuropil (preceding neuronal degeneration) and later increases in neurons and processes that are degenerating (Fig. 1). These temporal alterations in the localization and levels of apoE are consistent with an upregulation of apoE by astrocytes, release into the extracellular space, and subsequent uptake by neurons. It is also consistent with a slower rate of upregulation of neuronal expression from a low baseline. Intraneuronal accumulation of apoE also has been reported in a rat model of subdural hematoma within 30 minutes of the initial injury (Horsburgh et al., 1997c). The significance of apoE accumulation in injured neurons is unknown, although it has been speculated that it may be an initial protective response to injury by providing cholesterol and lipids for neuronal repair.
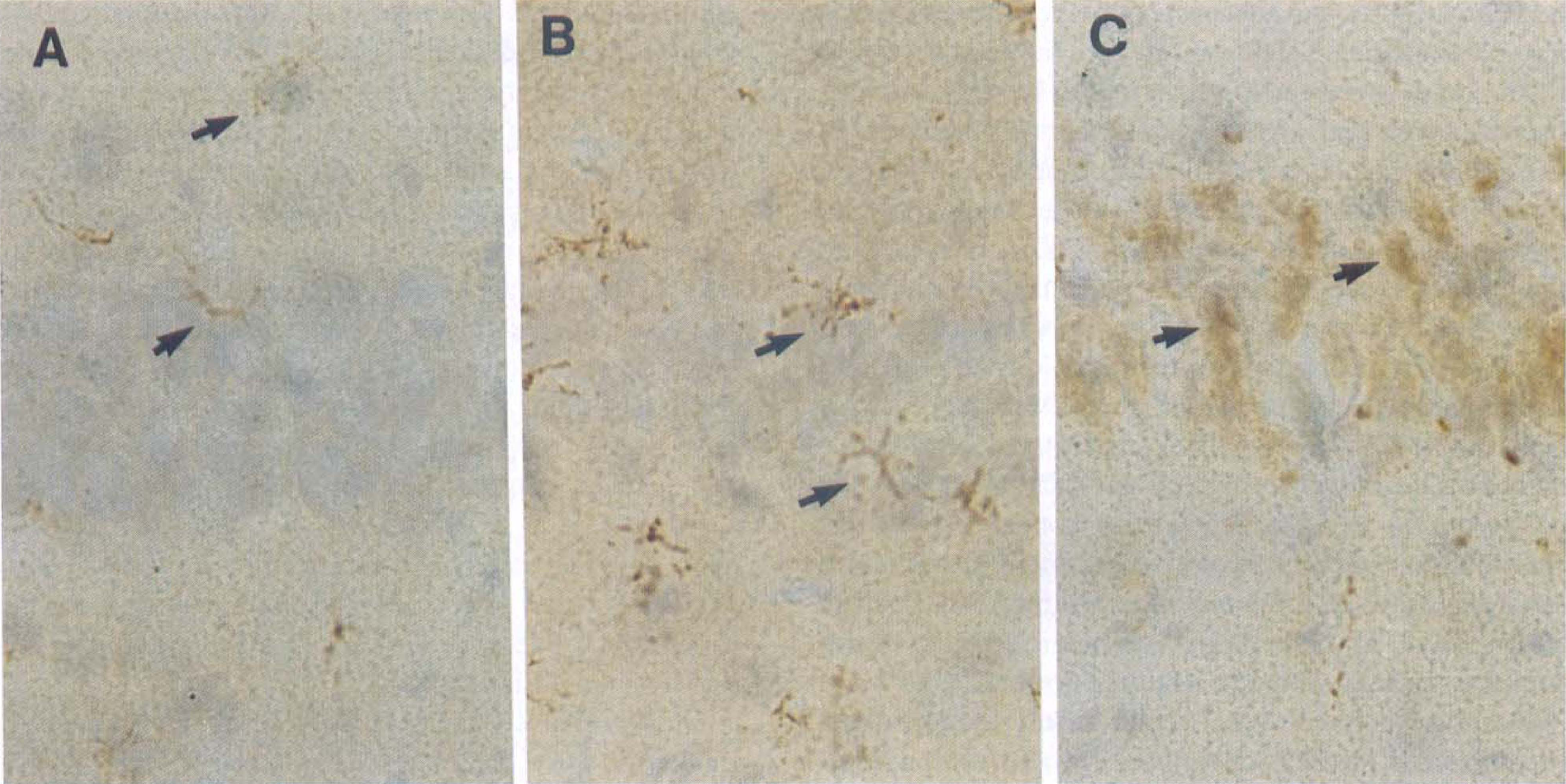
Apolipoprotein E (apoE) immunostaining of rat hippocampus at the CA1 pyramidal cell layer of sham-operated animals (
Although rodent models of acute brain injury have been instrumental in providing fundamental information regarding the response of apoE to CNS injury, they do not permit the effect of genotype to be studied (only one apoE isoform has been identified in rat and mouse and is most analogous to the human E4 protein) (Rajavashisth et al., 1985). This obviously is crucial in trying to understand the role of apoE in human disease.
In humans, apoE is produced primarily by astrocytes. In contrast to healthy rodent brain, where neuronal immunoreactivity is not normally observed, several studies demonstrate that neuronal apoE is present both in patients with chronic neurologic disease and normal aged controls (Han et al., 1994a,b; Benzing and Mufson, 1995). We also have observed apoE immunoreactivity in young neurologically normal individuals (Horsburgh 1997, unpublished observations). It remains to be defined whether apoE genotype is associated with extent of neuronal apoE.
Paralleling the rodent models, human intraneuronal apoE is increased after brain injury. In a preliminary study, we demonstrated marked increases in neuronal apoE immunoreactivity in areas showing acute neuronal injury after global ischemia, hypoglycemia, and status epilepticus (Nicoll et al., 1997). Recently, in a cohort of patients who sustained a period of global ischemia as a result of cardiac arrest, apoE immunoreactivity was shown to be significantly increased in hippocampal pyramidal neurons compared with neurologically normal controls (Fig. 2) (Horsburgh et al., 1997a). Neuronal apoE is most likely internalized from the extracellular space, since apoE has been demonstrated to be internalized by neurites in vitro, and astrocytes, but not neurons, have been demonstrated to produce their own apoE mRNA (Poirer et al., 1993; Poirer et al., 1991). Studies are underway in transgenic animals that express neuronal apoE to determine the genotype effect, if any, on the pathologic mechanism and apoE response after ischemic injury (Xu et al., 1996).
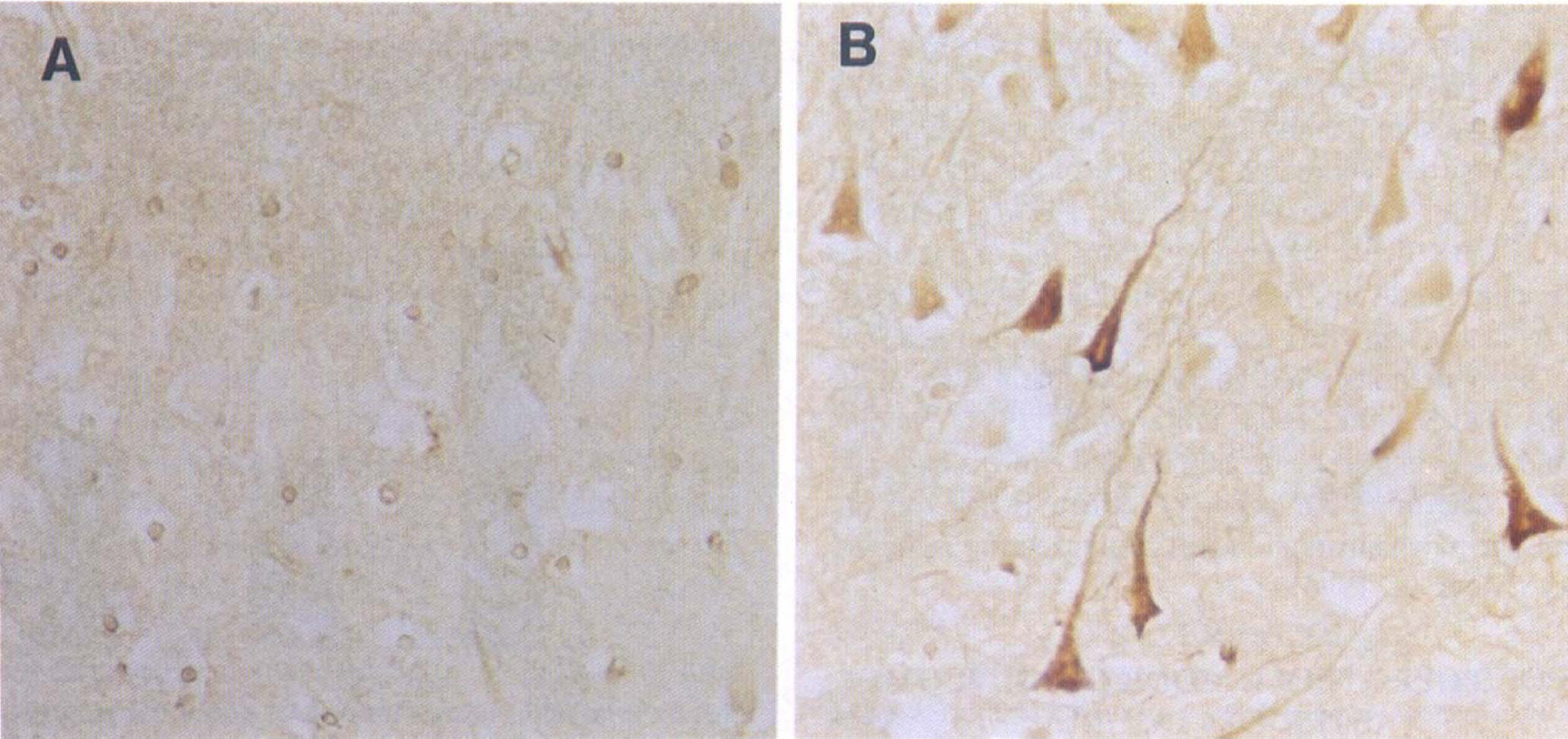
Apolipoprotein E immunostaining in the CA1 hippocampal sector of a 62-year-old neuropathologically normal individual (
APOLIPOPROTEIN E MODIFIES OUTCOME AFTER CNS INJURY
Despite the provocative clinical observations suggesting a role for apoE in the recovery from acute CNS insult, there have been relatively few animal models systematically examining this issue. After entorhinal cortex lesions, the ability of synapses to regenerate is retarded in apoE-deficient mice (Masliah et al., 1996). Intracerebral injection of apoE has been reported to partially restore synaptic repair in apoE mice (Masliah et al., 1997). These findings suggests that apoE may play an important role in the maintenance and regeneration of synapses. We also have examined the role of apoE in modifying outcome in a murine stroke model. When subjected to focal ischemia and reperfusion, apoE-deficient mice have significantly larger infarct volumes than age- and sex-matched controls (Fig. 3) (Peidrahita et al., 1992; Laskowitz et al., 1997b). We also have data showing apoE-deficient mice have significantly increased neuronal damage after an episode of global ischemia compared to wild-type littermate controls (Horsburgh et al., 1998, unpublished observations).
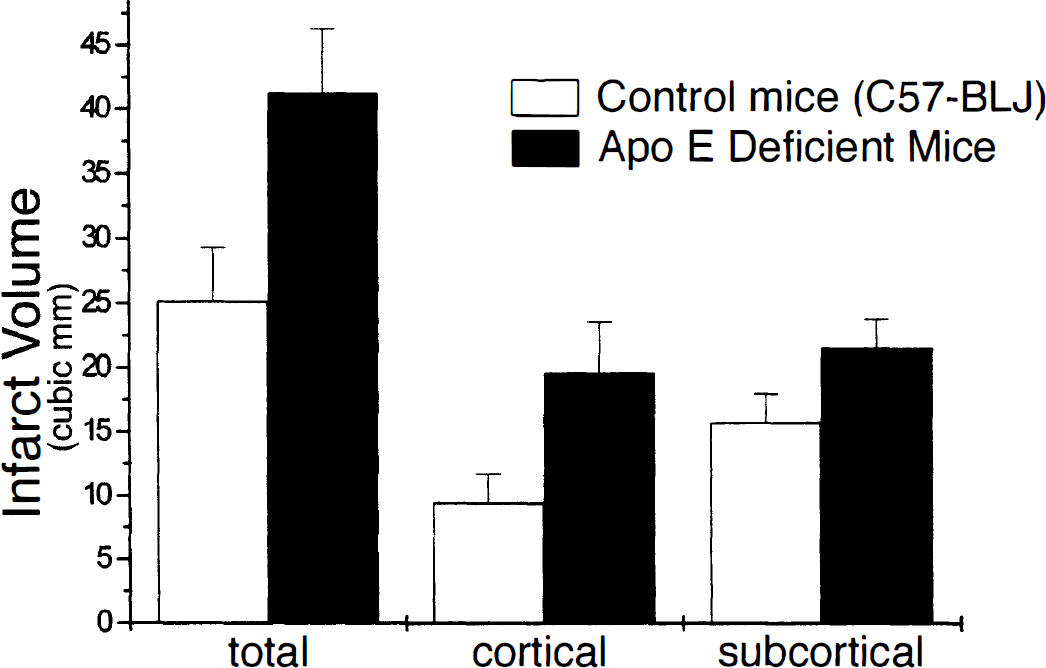
Total infarct volumes were 64% greater in apoE-deficient mice after transient middle cerebral artery occlusion compared with matched controls (P = 0.02). The apoE-deficient mice also had increased cortical and subcortical infarct volumes (P = 0.03, and 0.07, respectively). Data presented as mean ± SEM.
Of significance has been the recent development of transgenic mice that express the human E3 and E4 isoforms (but not their native murine apoE) (Xu et al., 1996). These mice express comparable amounts of apoE appropriately in glia and other tissue, and serum cholesterol is corrected, suggesting the presence of functional apoE protein. Moreover, these mice, which are bred using human genomic DNA, provide a model of “humanized” apoE expression, since the apoE gene is under the control of a human promoter and a primate pattern of neuronal immunoreactivity is observed (Xu et al., 1996). When subjected to transient focal ischemia and reperfusion, mice expressing the E4 isoform had significantly larger infarct volumes than mice expressing the E3 isoform (Sheng et al., 1998). This is the first animal model to address isoform-specific effects in acute CNS injury, and is consistent with clinical data suggesting that apoE might influence CNS recovery in an isoform-specific fashion.
APOLIPOPROTEIN E AND THE CNS RESPONSE TO INJURY
Evidence supports the contention that apoE may protect against oxidative injury, modulate glial activation, or have direct and indirect neurotropic properties. These mechanisms of action are not mutually exclusive, and it is certainly possible that apoE modulates the CNS response to injury at several functional levels.
One possible explanation for the effects of apoE in the CNS is that it protects against oxidative insult and the ensuing process of lipid peroxidation. This is consistent with recent observations that apoE has the ability to protect a neuronal cell line from hydrogen peroxide toxicity in isoform-specific fashion (E2 > E3 > E4) (Miyata and Smith, 1996). The possibility that apoE protects against oxidative injury also is consistent with observations that plasma lipoproteins from apoE-deficient mice are more susceptible to in vitro oxidation than those of wild-type mice, and that apoE-deficient animals express higher titers of autoantibodies directed against oxidized lipids than control animals (Palinski et al., 1994; Hayek et al., 1994). Brains from apoE-deficient animals also have increased levels of 3-nitrotyrosine, a marker of protein nitration by peroxynitrite, as compared with controls (Matthews and Beal., 1996). All of these results suggest that apoE may play a role in modulating oxidative stress. Presumably, apoE either could act directly as a free radical scavenger, or sequester peroxidized lipids to prevent the propagation of lipid peroxidation.
Another potential mechanism by which apoE may exert its protective effects is by modulating the glial response to inflammation. Apolipoprotein E is known to have immunomodulatory effects in vitro, which include suppression of lymphocyte proliferation and immunoglobulin synthesis after mitogenic stimulation (Pepe and Curtiss, 1986; Macy et al., 1983; Avila et al., 1982; Curtiss and Edgington, 1981; Edgington and Curtiss, 1981; Hui et al., 1980; Curtis and Edgington, 1978). In our laboratory, we have examined the possibility that apoE, which is the primary apolipoprotein produced within the CNS, may modulate the CNS inflammatory response as well. We have demonstrated that apoE suppresses glial secretion of inflammatory cytokines in a specific and dose-dependent fashion (Laskowitz et al., 1997a). We also have observed that apoE inhibits microglial synthesis of nitric oxide and inflammatory cytokines in an isoform-specific fashion (Laskowitz and others, unpublished data). These results are consistent with the observation that apoE modulates microglial activation after exposure to secreted amyloid precursor protein in an isoform-specific manner (Barger and Harmon, 1997). Thus, it is possible that apoE suppresses microglial activation and subsequent release of cytotoxins associated with acute CNS injury. There is increasing evidence that inflammation may contribute to the pathogenesis of chronic neurodegenerative diseases such as Alzheimer's disease, and also may exacerbate secondary neuronal injury after acute CNS insult (Arvin et al., 1996; Rogers et al., 1996; Aisen, 1996; Garcia et al., 1994). Thus, if apoE modulated the CNS inflammatory response in an isoform-specific fashion, it might play an important role in both acute and chronic CNS disease.
A third possible mechanism by which apoE might alter recovery in neurologic disease is through a direct neurotrophic effect on injured neurons. This is consistent with apoE-enhanced survival of a neuronal cell line after exposure to hydrogen peroxide, and also with observations that apoE promotes neurite outgrowth in an isoform-specific fashion (Miyata and Smith, 1996; Bellosta, et al., 1995; Holzman et al., 1995; Nathan et al., 1994). In addition to its direct neurotrophic effects, apoE has been demonstrated to bind to and potentiate the neurotrophic effects of growth factors such as ciliary neurotrophic factor (Gutman et al., 1997). Similarly, apoE binds to components of the extracellular matrix, such as laminin, to enhance neuronal adhesion and growth (Huang et al., 1995).
CONCLUSION
In summary, there is increasing evidence that apoE is a biologically important mediator of the CNS response to injury, and that a differential effect may exist between apoE isoforms. Following the original association between the APOE4 allele and increased risk of developing sporadic and late-onset familial Alzheimer's disease, recent clinical observations support the possibility that apoE may play an isoform-specific role in recovery from acute CNS insult as well. The development of apoE-deficient mice and transgenic animal models designed to express the human apoE isoforms no doubt will facilitate our understanding of the normal biological role of apoE in vivo. Although the mechanisms by which apoE influences risk of Alzheimer's disease and recovery after acute CNS insult remains to be defined, it is likely that apoE's role in the CNS is not limited to lipid transport. A better understanding of the neurobiology of apoE promises to provide new insights into our understanding of the CNS responses to acute and chronic injury (Roses, 1995).