
Abstract
Apolipoprotein E (apoE), a 34-kD glycosylated lipid-binding protein, is expressed as three common isoforms in humans (E2, E3, or E4). Clinical evidence suggests that the apoE genotype (APOE) may be a risk factor for poor outcome after acute central nervous system injury. This was examined further in transgenic mice constructed with the human APOE3 or APOE4 gene under the control of human promoter and tissue expression elements. Presence of human apoE3 and apoE4 proteins in brains of human APOE homozygous transgenic mice was confirmed by Western blotting. APOE3 (n = 12) and APOE4 (n = 10) mice underwent 60 minutes of middle cerebral artery occlusion. After 24-hour recovery, infarct size was measured. Infarct volumes (mean ± standard deviation) were smaller in the APOE3 group (cortex: APOE3 = 18 ± 4 mm3; APOE4 = 30 ± 11 mm3, P = 0.04; subcortex: APOE3 = 12 ± 4 mm3; APOE4 = 18 ± 4 mm3, P = 0.003). Hemiparesis was less severe in APOE3 mice (P = 0.02). These data indicate that human isoform-specific effects of apoE are relevant to acute pathomechanisms of focal ischemic brain damage when examined in the mouse. APOE transgenic mice may provide an appropriate model to examine the mechanistic basis for the differential effects of human apoE isoforms in acute central nervous system injury.
Apolipoprotein E (apoE) is a 34-kD glycosylated protein that originally was described in the context of cholesterol metabolism. The most common isoform in man, E3, is secreted as a 299-amino acid protein containing a single cysteine residue at position 112. The other two common isoforms, E2 and E4, differ from E3 by cysteine—arginine interchanges; E2 contains a cysteine at position 158, and E4 contains an arginine at 112.
The apolipoprotein E4 (APOE4) allele is a major determinant of risk for sporadic and late-onset familial Alzheimer disease (Corder et al., 1993; Saunders et al., 1993; Strittmatter et al., 1993). There is accumulating evidence that the APOE4 allele also may be associated with poor outcome from various forms of acute brain injury, including intracerebral hemorrhage (Alberts et al., 1995), closed head injury (Nicoll et al., 1995; Roses and Saunders, 1995; Seliger et al., 1997; Sorbi et al., 1996), and stroke (Basun et al., 1996; Slooter et al., 1997). APOE4 also has been associated with worse neuropsychiatric deficits after cardiopulmonary bypass in humans (Newman et al, 1995).
ApoE is secreted locally by macrophages after peripheral nerve injury and by astrocytes and oligodendrocytes after central nervous system insults (Skene and Shooter, 1983; Stoll and Mueller, 1986; Stoll et al., 1989). In both gerbils and rats, apoE is expressed in selectively vulnerable hippocampal neurons after transient forebrain ischemia (Ali et al., 1996; Hall et al., 1995; Horsburgh and Nicoll, 1996a,b; Kida et al., 1995). Mice deficient for apoE have worse outcomes from focal ischemic insults than wild-type controls (Laskowitz et al., 1997b). The aforementioned studies have defined a response of apoE to ischemic stress but have not examined the role of isoform specificity. The purpose of this study was to determine whether outcome from transient focal cerebral ischemia is dependent on isoform specificity of the human APOE allele in transgenic mice.
MATERIALS AND METHODS
This study was approved by the Duke University Animal Care and Use Committee.
Transgenic mice
The methods for obtaining the APOE3 and APOE4 human transgene constructs have been reported previously (Xu et al., 1996). Briefly, cosmid libraries were constructed from lymphoblasts of humans known to be homozygous carriers for APOE3 or APOE4. Fragments containing human regulatory sequences and the coding sequences for human APOE were isolated and microinjected into single-cell embryos from APOE-deficient mice to produce transgenic mice. Animals were bred to homozygosity. The presence of human and APOE “knockout” genes was confirmed by polymerase chain reaction (Xu et al., 1996). Mode of integration and copy numbers of transgenes were analyzed by Southern blotting. Serum cholesterol concentration was determined to be normalized for both E3 and E4 mice. Presence of the human apoE protein in neurons and glia was confirmed by immunohistochemistry. For the following study, the APOE3-437 and APOE4-81 lineages were selected based on determination that both lines carry two copies of the respective transgenes (Xu et al., 1996).
Immunoblot detection of apolipoprotein E
Brain homogenates from 8-week old APOE3-437 or APOE4-81 animals were generated by Dounce homogenization of tissue in ice-cold homogenization buffer (0.25 mol/L sucrose, 1 mmol/L edetic acid, 10 mmol/L HEPES [pH 7.4], 0.1% ethanol, and 5 μg/mL of the protease inhibitor phenylmethylsulfonyl fluoride [Sigma, St. Louis, MO, U.S.A.]). The protein concentration was determined using the Bio-Rad Protein Assay kit (Bio-Rad Laboratories, Hercules, CA, U.S.A.). Approximately 20 μg of protein homogenate was added to IX Laemmli buffer (2% sodium dodecyl sulfate/5% [vol/vol] 2-mercaptoethanol/10% [vol/vol] glycerol/62.5 mmol/L TRIS-hydrochloride [pH 6.8] and 0.0025% bromophenol blue) and boiled for 5 minutes. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis of solubilized proteins was performed using stacking gels of 4% (wt/vol) polyacrylamide and separating gels of 10% polyacrylamide with 2% sodium dodecyl sulfate. Electrophoresed protein was transferred to polyvinylidene flouride membranes (Millipore, Bedford, MA, U.S.A.) by standard Western transfer techniques. After transfer, membranes were incubated in blocking solution (5% nonfat dried milk in TRIS-buffered saline [pH 7.6] with 0.1% Tween 20 (Pierce, Rockford, IL, U.S.A.]) at room temperature for 1 hour. Then the membrane was incubated with goat antihuman apoE (Calbiochem-NovaBiochem International, San Diego, CA, U.S.A.) at 1:15,000 dilution in blotto overnight at 4°C, and subsequently washed five times in blotto. The membrane was exposed to swine antigoat secondary antibody conjugated with horseradish peroxidase (1:5,000 dilution; Boerhinger-Mannheim, Indianapolis, IN, U.S.A.) for 1 hour at room temperature, then washed in blotto. Horseradish peroxidase was visualized with an enhanced chemiluminescence detection kit (Life Science, Buckinghamshire, England, U.K.) and exposed to Hyperfilm ECL (Life Science). As a standard for apoE, 1 ng of purified recombinant human recombinant apoE3 protein (Calbiochem-NovaBiochem International) was run on the same protein gels. After exposure to film, protein bands were quantified further by laser-densitometric scanning using a Personal Densitometer SI and Image QuaNT 4.2 software (Molecular Dynamics, Sunnyvale, CA, U.S.A.). Western blot analysis was repeated several times, with similar results. Parallel studies were performed as negative controls on brain homogenates derived from APOE-deficient mice (The Jackson Laboratory, Bar Harbor, ME, U.S.A.).
Focal ischemia-reperfusion model
Male APOE3-437 (n = 12) and APOE4-81 (n = 10) mice (8–10 weeks of age) were fasted for 12 to 14 hours but were allowed free access to water. Animals then were prepared for middle cerebral artery occlusion using modifications of techniques described by others (Huang et al., 1994; Zea Longa et al., 1989). With the surgical operator blinded to genetic identity, mice were anesthetized with 1.0% to 1.5% halothane in 50% oxygen/balance nitrogen delivered via snout mask and allowed to breathe spontaneously. Via a midline cervical skin incision, the right common carotid artery was identified. The external carotid artery was ligated and transected. The internal carotid artery was dissected distally until the origin of the pterygopalatine artery was visualized. After surgical preparation, a 15-minute interval was allowed for physiologic stabilization. Rectal temperature was monitored continuously and servoregulated with surface heating/cooling at 37.0°C throughout the procedure.
A 6-0 nylon monofilament was blunted at the tip in a flame and then lightly coated with silicone. The filament was inserted into the proximal external carotid artery stump and advanced ≈11 mm into the internal carotid artery to occlude the middle cerebral artery. Inspired halothane concentration was reduced to 0.9% at time of filament insertion. After 60 minutes, the filament was removed, and the skin incision was closed with suture. Ten minutes after filament removal, halothane was discontinued and the mice were allowed to awaken. The mice were placed in an oxygen-enriched environment (fraction of inspired oxygen [FiO2] = 50%) for 1 hour and then returned to their cages.
All animals were evaluated neurologically 24 hours after reperfusion. Each mouse was assigned a score of 0 to 4, where 0 = no observable neurologic deficit; 1 = failure to extend the left forepaw; 2 = circling to the left; 3 = falling to the left; and 4 = cannot walk spontaneously (Yang and Betz, 1994). Neurologic examination was performed by one observer blinded to group assignment.
After neurologic evaluation, animals were anesthetized with halothane and decapitated. The brain was removed and frozen at −20°C. Using a cryotome, 20-μm thick coronal sections were taken at 320-μm intervals over the rostral—caudal extent of the infarct. The sections were dried and stained with hematoxylin and eosin.
Infarct volume was measured by digitally sampling stained sections with a video camera controlled by an image analyzer (M2 Turnkey System, Imaging Research, Inc., St. Catharines, Ontario, Canada). The image of each section was stored as a 1024 × 1024 matrix of calibrated pixel units. The digitized image then was displayed on a video screen. With the observer blinded to experimental condition, infarct borders in both cortex and subcortex were outlined individually (corpus callosum excluded) using an operator-controlled cursor. The area of infarct (mm2) was determined automatically by counting pixels contained within the outlined regions of interest. Infarct volumes (mm3) were computed as running sums of infarct area multiplied by the known interval (e.g., 320 μm) between sections over the extent of the infarct expressed as an orthogonal projection.
Measurement of physiologic parameters
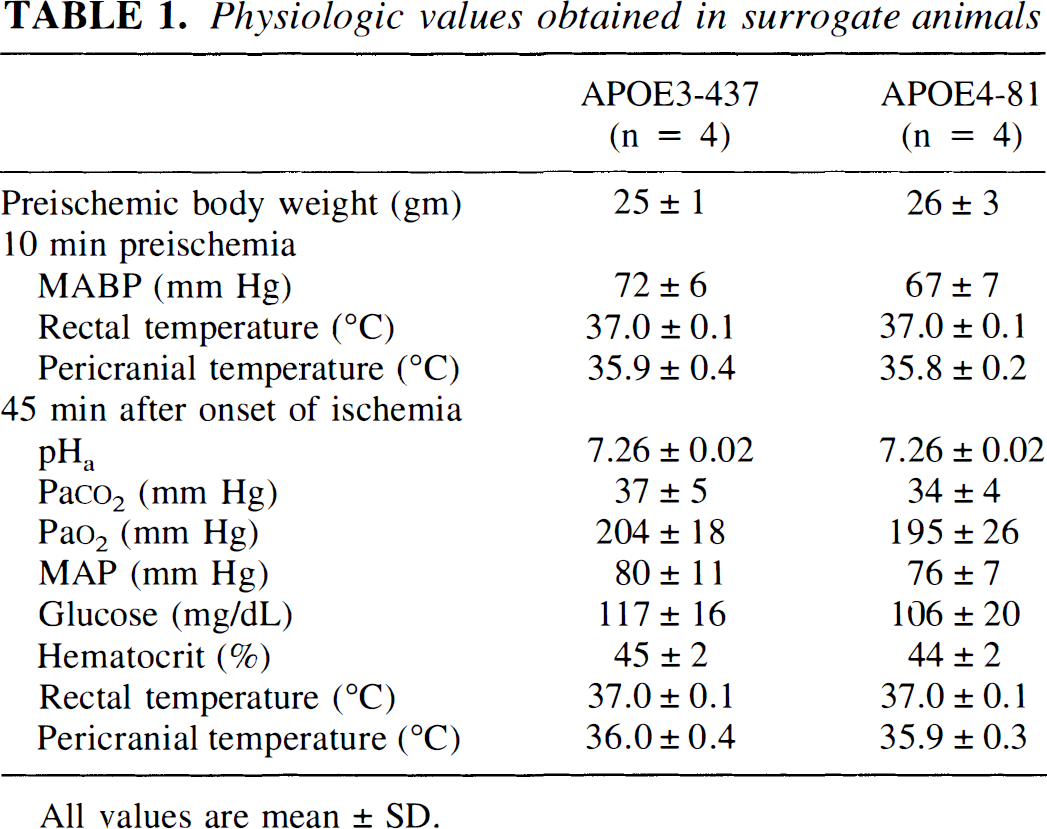
Because the blood volume required for analytic assays was great enough to cause hypovolemia, a parallel study was performed to define the probable physiologic state of mice subjected to focal ischemia. Both APOE3-437 and APOE4-81 mice were examined (n = 4 per group). These animals underwent a surgical procedure and middle cerebral artery occlusion insult identical to the aforementioned one. In addition, a pericranial needle thermistor was placed adjacent to the skull, beneath the temporalis. Servoregulation of body temperature was performed via the rectal probe. Therefore, only pericranial temperature was monitored. A right femoral arterial catheter was placed to monitor blood pressure and withdraw blood. Physiologic values were measured 10 minutes before and 45 minutes after onset of ischemia. The animals then were killed by an overdose of halothane anesthesia.
Statistical analysis
Infarct volumes and physiologic values were compared between groups with the Student's t test. Infarct volumes were correlated with neurologic grades by the Spearman rank correlation coefficient, whereas neurologic grades were compared between groups by the Mann-Whitney U statistic. Parametric values are expressed as mean ± standard deviation (SD). Significance was assumed if P < 0.05.
RESULTS
Western blotting with antihuman apoE allowed detection of apoE in both transgenic strains (Fig. 1). For tissue from the APOE3-437 and APOE4-81 strains, a single band of immunopositive protein was observed (optical density = 3048 and 5772 units, respectively) per 20 μg of brain homogenate. Optical density for human apoE in the E3-437 line was approximately 50% less than the APOE4-81 value. No immunostaining was observed in the APOE-deficient controls.

Western blot analysis of human apolipoprotein E (apoE) in nonischemic mouse brain. Brain homogenates (20 μg) from apoE-deficient (–/–), APOE3-437 (E3), and APOE4-81 (E4) transgenic animals were electrophoresed under reducing conditions and then immunoblotted. Apolipoprotein E was detected using a primary antibody against human apoE. The last lane depicts a 1-ng standard of purified recombinant human apoE3 migrating as a 34-kD protein.
No mice in either group died as a result of 60 minutes of middle cerebral artery occlusion. Hemiparesis was less severe in APOE3-437 mice (P = 0.02; Fig. 2). Infarct volumes were smaller in the APOE3 group (cortex: APOE3-437 = 18 ± 4 mm3; APOE4-81 = 30 ± 11 mm3, P = 0.04; subcortex: APOE3-437 = 12 ± 4 mm3; APOE4-81 = 18 ± 4 mm3, P = 0.003). Mean total infarct volume was 37.5% smaller in the APOE3-437 versus APOE4-81 mice (P = 0.02; Fig. 3). Correlation between neurologic score and infarct size was significant (P = 0.02).
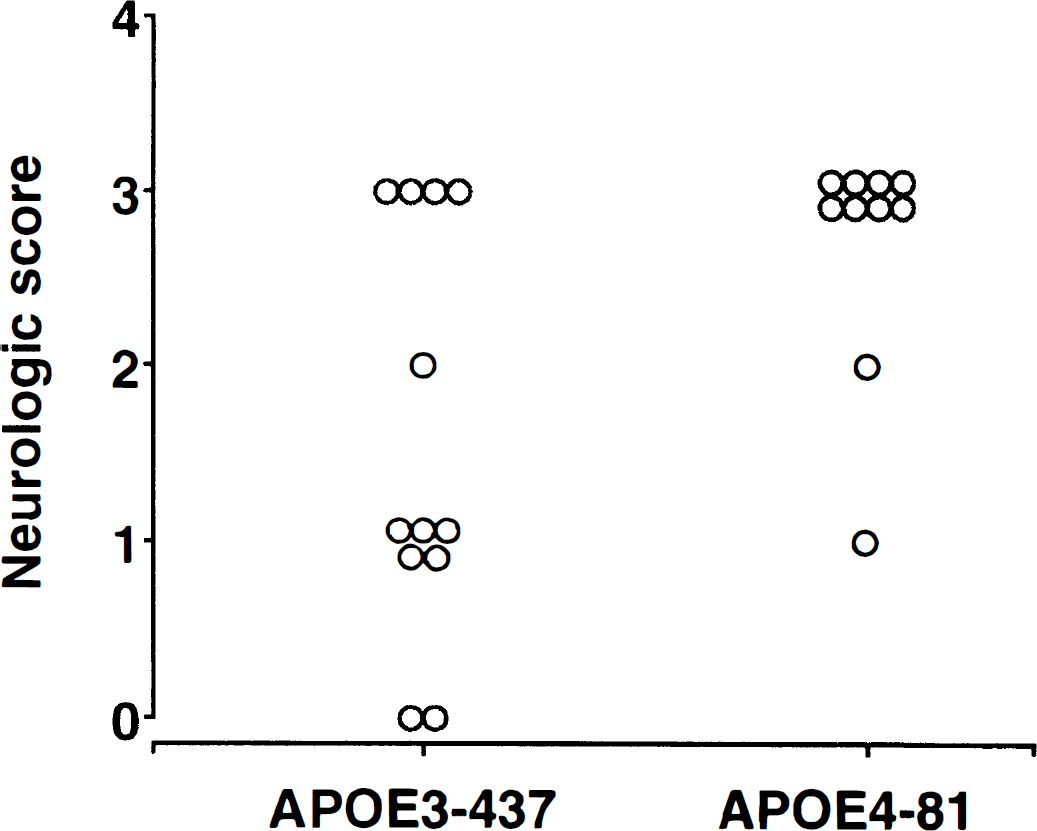
Individual neurologic scores (0 = no deficit; 4 = cannot walk spontaneously) for APOE3-437 (n = 12) vs. APOE4-81 (n = 10) mice subjected to 60 minutes of middle cerebral artery occlusion and a 24-hour recovery interval. APOE3-437 mice exhibited less severe deficits (P = 0.02).
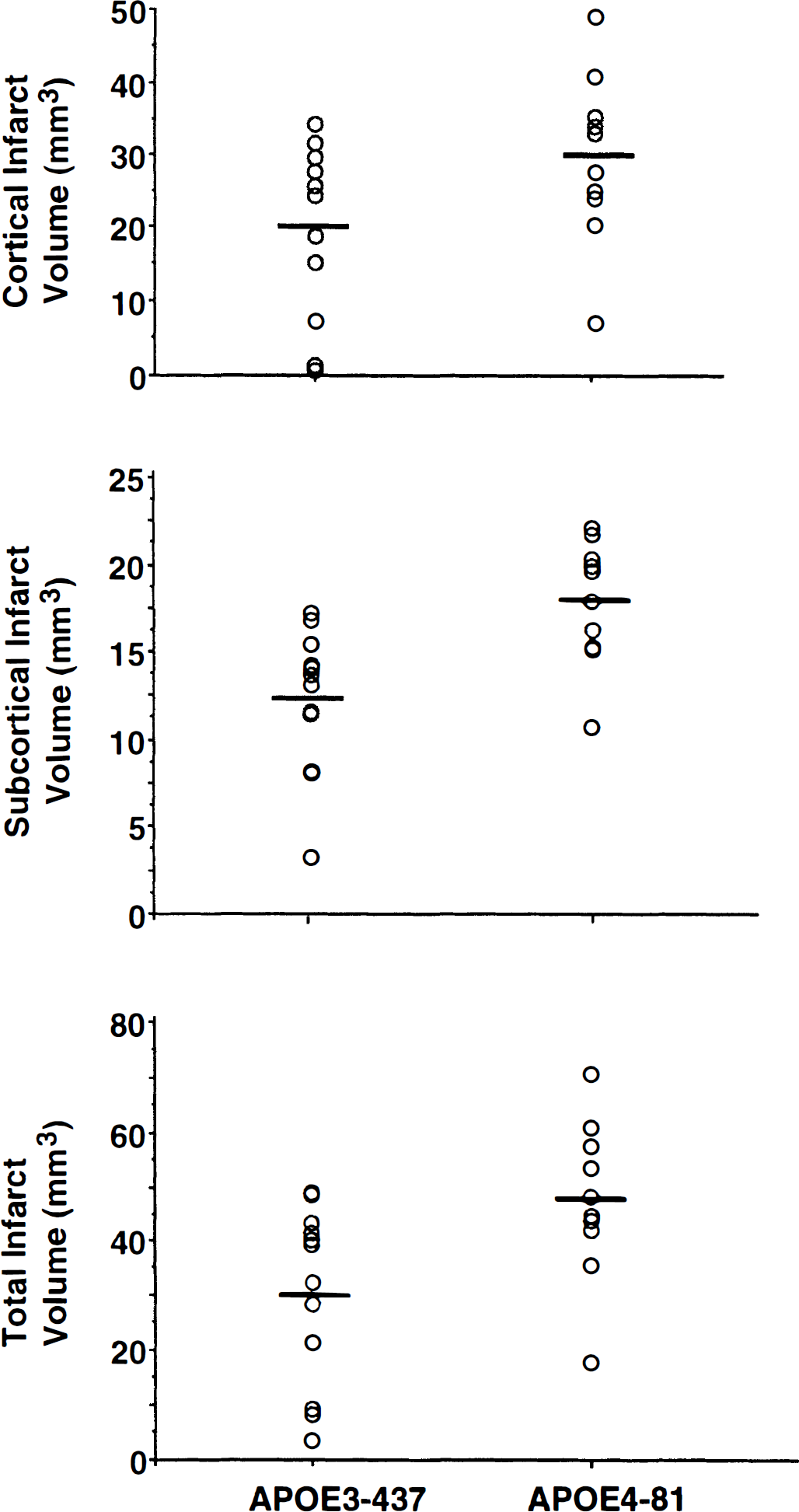
Cortical, subcortical, and total infarct volumes for individual APOE3-437 (n = 12) vs. APOE4-81 (n = 10) mice subjected to 60 minutes of middle cerebral artery occlusion and a 24-hour recovery interval. Horizontal bars depict mean values for each group. All mice survived the recovery interval. Infarct volume was greater in the APOE4-81 mice.
There were no significant differences among experimental groups for physiologic values (as determined in surrogate animals) (Table 1). Mild pericranial hypothermia was evident in both groups despite control of rectal temperature at 37.0°C, as intended.
Physiologic values obtained in surrogate animals
All values are mean ± SD.
DISCUSSION
Interest in the neurobiology of apoE has increased since the APOE4 allele was associated with Alzheimer disease (Corder et al., 1993; Saunders et al., 1993; Strittmatter et al., 1993). Recent evidence suggests that apoE also may play a unique role in a variety of acute central nervous system insults. Independent observations have linked the presence of the APOE4 allele with increased neuropsychiatric deficit after cardiopulmonary bypass (Newman et al., 1995) and poor prognosis after closed head injury, intracranial hemorrhage, and stroke (Alberts et al., 1995; Basun et al., 1996; Mayeux et al., 1995; Nicoll et al., 1995; Roses and Saunders, 1995; Seliger et al., 1997; Slooter et al., 1997; Sorbi et al, 1996).
These provocative clinical observations suggest an isoform-specific role for apoE in modifying the central nervous system response to acute and chronic insults. However, human studies have been correlative, thereby limiting potential definition of mechanistic actions of apoE. Examination of the role of apoE in a wild-type animal model of cerebral ischemia is difficult for several reasons. Rodents are known to express only one apoE isoform. Therefore, direct examination of isoform specificity is not possible. Further, murine apoE shares only a 70% homology to the human amino acid sequence (Rajavashisth et al., 1985). Differences also exist between rodents and primates/humans in histologic patterns of normal central nervous system apoE immunoreactivity. In rodents, apoE is confined largely to astrocytes (Boyles et al., 1985), although small extracellular deposits have been described in the perivascular and periarachnoid spaces (Boyles et al., 1985; Horsburgh and Nicoll, 1996a; Kida et al., 1995). There is no evidence of apoE immunoreactivity in rodent neurons other than that observed in the postischemic state (Ali et al., 1996; Horsburgh and Nicoll, 1996b). In humans, apoE immunoreactivity normally is present in some neurons, as well as in astrocytes (Han et al., 1994a,b). Therefore, the presence of species-specific histologic expression of apoE within the central nervous system and the lack of homology with the human gene complicates the use of wild-type rodent models when attempting to understand the role of apoE in neurologic disease.
Recently, transgenic mice have been created that express human apoE3 or apoE4 isoforms but lack endogenous murine apoE (Xu et al., 1996). Serum cholesterol is normal in these mice, suggesting the presence of functional apoE protein. Moreover, these mice have been created to express apoE under the control of human promoter sequences and tissue expression elements. The result is a “humanized” pattern of expression—that is, a pattern of neuronal and glial immunoreactivity is observed. This experiment demonstrates that mice expressing the apoE4 isoform have significantly larger infarct volumes than those expressing the apoE3 isoform. Also, total infarct volume in the apoE4 animals (48 ± 14 mm3) was larger that that previously observed in wild-type C57BL/6J mice (25 ± 13 mm3) exposed to an identical insult (Laskowitz et al., 1997b). The finding that apoE4 is associated with worse outcome from an acute central nervous system insult is consistent with clinical observations suggesting that the APOE genotype influences recovery from brain injury in an allele-specific fashion.
There are limitations of our model that warrant consideration. First, it is likely that the human APOE3 and APOE4 genes were inserted at different sites in the mouse genome. The extent to which specific upstream regulatory elements interact with apoE expression in response to injury is unknown. Second, Western blot analysis in this experiment has revealed that apoE production was greater in the APOE4-81 mice relative to that in the APOE3-437 mice. Our previous work suggested that murine apoE plays a protective role in response to focal ischemia (Laskowitz et al., 1997b). Therefore, the most likely explanation for our findings is that the difference in infarct volume between these two lines is secondary to an isoform-specific effect and not a difference in total protein expression. Further work examining a dose-response effect for both apoE3 and apoE4, either by comparing ischemic outcome in transgenic mice with different numbers of gene copies (expressing correspondingly different amounts of protein) or by direct intraventricular infusion of recombinant human apoE isoforms in apoE-deficient animals would help resolve this issue. Third, these transgenic animals had been back-bred to a common genetic background for six generations, which ensured a greater than 98% homology. However, subtle genetic differences still may exist between the APOE3-437 and APOE4-81 lines. Others have reported substantial interstrain sensitivities to either ischemic or kainic acid lesions (Barone et al., 1993; Schauwaecker and Steward, 1997). The magnitude of such an effect in our study is not known.
It is unlikely that the increased infarct volumes observed in the APOE4 transgenic mice were due to neuroanatomic differences between these two murine lines because no differences in the gross or histologic brain morphology have been observed in transgenic or apoE-deficient mice at the ages used in this experiment (Masliah et al., 1995; Xu et al., 1996). It also is unlikely that the observed differences in infarct volumes were due to microvascular disease, given the young age of the animals. No hemodynamically significant lesions have been observed, even in apoE-deficient mice within this age range, despite marked hypercholesterolemia (Plump et al., 1992; Reddick et al., 1994; Zhang et al., 1992). Direct cerebral blood flow measurements will be required, however, to ensure that the volume of tissue at risk for infarction was similar between the APOE3 and APOE4 groups.
Although there is mounting evidence that apoE plays a role in acute and chronic neurologic disease, its function in injured brain remains poorly defined. There is preliminary evidence suggesting several potential mechanisms of action that are not necessarily mutually exclusive. One possible role of apoE is protection against oxidative insults and the ensuing process of lipid peroxidation, as suggested by a recent report that apoE protected a neuronal cell line from hydrogen peroxide toxicity (Miyata and Smith, 1996). This mechanism also is consistent with observations that plasma lipoproteins from apoE-deficient mice are more susceptible to in vitro oxidation than those of wild-type mice (Hayek et al., 1994). Further, apoE-deficient animals express higher titers of autoantibodies directed against oxidized lipid than control animals (Palinski et al., 1994). Presumably, apoE could act as a free radical scavenger, stabilizing lipid membranes against oxidative insult. Alternatively, apoE may sequester peroxidized lipids to prevent propagation of lipid peroxidation. It also has been suggested that apoE plays a role in membrane remodeling as a lipid carrier after neuronal injury (Poirier et al., 1991).
Another mechanism by which apoE might ameliorate effects of focal ischemia is by down-regulation of the inflammatory response. Apolipoprotein E is known to have immunomodulatory properties in vitro, including suppression of lymphocyte proliferation and immunoglobulin synthesis (Curtiss and Edgington, 1976; Pepe and Curtiss, 1986). Apolipoprotein E might play a unique role in the brain, where it is the primary apolipoprotein produced. This is supported by recent experiments in cell culture that demonstrate that apoE suppresses glial secretion of tumor necrosis factor-α (Laskowitz et al., 1997a). We also recently observed that apoE inhibits microglial synthesis of nitric oxide and inflammatory cytokines in an isoform-specific fashion (Laskowitz et. al., in press). Inflammation after ischemia may exacerbate brain injury by contributing to edema, blood—brain barrier breakdown, and release of reactive oxygen species by activated microglia.
A third possibility is that apoE exerts a neurotrophic effect on injured neurons. This is consistent with in vitro observations that apoE promotes survival and neurite outgrowth in an isoform-specific fashion (Bellosta et al., 1995; Holzman et al., 1995; Nathan et al., 1994). In addition to these direct neurotrophic effects, apoE has been demonstrated to bind to and potentiate the neurotrophic effects of ciliary neurotrophic factor (Gutman et al., 1997). Similarly, apoE binds to heparinized components of the extracellular matrix, such as laminin, to enhance neuronal adhesion and growth (Huang et al., 1995).
Transgenic mice expressing the human apoE4 isoform have larger infarct volumes after 60 minutes of focal ischemia than human apoE3 transgenic mice. Apolipoprotein E appears to play an isoform-specific role in the brain, modulating the effects of ischemia. This is consistent with clinical observations that apoE plays an isoform-specific role in outcome after other forms of acute central nervous system insults, including intracranial hemorrhage, closed head injury, and stroke, and after cardiopulmonary bypass. The mechanisms by which apoE modulates the response to injury remain unclear, but transgenic models of human apoE expression probably will help to define brain metabolism of this multifunctional protein.