
Abstract
Recent evidence suggests that apolipoprotein E (ApoE) plays a role in neurologic disease. This experiment compared the neurologic and histologic outcome of ApoE-deficient mutant and wild-type mice subjected to a 60- or 90-minute episode of middle cerebral artery filament occlusion and a recovery interval of 24 hours. With 60 minutes of ischemia, there was no mortality. Apolipoprotein E-deficient mice had larger infarcts (cortex: ApoE deficient = 20 mm3 ± 12, wild-type = 9 ± 7 mm3, P = 0.03; subcortex: ApoE deficient = 22 ± 7 mm3, wild-type = 16 ± 7 mm3, P = 0.07). Hemiparesis was less severe in wild-type animals (P = 0.02). After 90 minutes of ischemia, mortality in ApoE-deficient mice (n = 10) was 40% versus 0% in wild-type mice (n = 10; P = 0.09). Intraparenchymal hemorrhage was found in 3 of the 4 dead mice. No difference in cortical (ApoE deficient = 37 ± 8 mm3; wild-type = 31 ± 18 mm3; P = 0.49) or subcortical (ApoE deficient = 30 ± 11 mm3; wild-type = 32 ± 18 mm3; P = 0.78) infarct volumes was present among survivors. ApoE-deficient mice had a prolonged activated partial thromboplastin time and increased fibrinogen concentration. This data supports the hypothesis that apolipoprotein E plays a role in the pathophysiology of ischemic brain damage.
Keywords
Apolipoprotein E (ApoE) is a 34-kd glycosylated protein that was originally described in the context of cholesterol metabolism. Apolipoprotein E is locally secreted by macrophages after peripheral nerve injury and by astrocytes and oligodendrocytes after CNS injury, suggesting a role in response to neural injury or repair (Skene and Shooter, 1983; Stoll and Mueller, 1986; Stoll et al., 1989). The most common isoform in man, E3, is secreted as a 299 amino acid protein containing a single cysteine residue at position 112. The other two common isoforms E2 and E4 differ from E3 by cysteine-arginine interchanges; E2 contains a cysteine at position 158, and E4 contains an arginine at 112. The E4 allele is a major determinant of risk for sporadic and late-onset familial Alzheimer's disease (Strittmatter et al., 1993).
There is accumulating evidence that the ApoE4 allele may also be associated with poor outcome from various forms of acute brain injury including intracerebral hemorrhage and closed-head trauma (Alberts et al., 1995; Sorbi et al., 1996). Further, ApoE4 has been associated with worsened neuropsychiatric deficits after cardiopulmonary bypass in humans suggesting an isoform-specific response to ischemia (Newman et al., 1995). In the gerbil, ApoE is selectively expressed in vulnerable areas of the hippocampus after a period of forebrain ischemia (Hall et al., 1995). Further support that ApoE accumulates in postischemic neurons in the rat was provided by others (Horsburgh and Nicoll, 1996a;Horsburgh and Nicoll, 1996b;Kida et al., 1995). We now show that ApoE-deficient mice are more sensitive to focal ischemia and reperfusion than age- and gender-matched wild-type control animals.
MATERIALS AND METHODS
This study was approved by the Duke University Animal Care and Use Committee. Homologous recombination in embryonic stem cells (129-derived E14Tg2a ES) was used to produce targeted knockout mutations of the ApoE locus (Peidrahita et al., 1992). The ApoE-deficient animals used in this experiment had been back crossed six times to the C57BL/6J mouse and purchased from The Jackson Laboratory (Bar Harbor, ME, U.S.A.). Wild-type animals were age and gender matched C57-BL/6J mice (The Jackson Laboratory).
Focal ischemia-reperfusion model
Animals were prepared for middle cerebral artery occlusion (MCAO) using modifications of techniques described by others (Huang et al., 1994; Zea Longa et al., 1989). Male mice (8 to 10 weeks of age) were fasted from food for 12 to 14 hours but allowed free access to water. With the surgical operator blinded to genetic identity, mice were anesthetized with 1 to 2% halothane in 50% O2/balance N2 delivered via snout mask and allowed to breathe spontaneously. Via a midline cervical skin incision, the right common carotid artery was identified. The external carotid artery was ligated and transected. The internal carotid artery was dissected distally until the origin of the pterygopalatine artery was visualized. After surgical preparation, a 15-minute interval was allowed for physiological stabilization. Rectal temperature was continuously monitored and servoregulated with surface heating/cooling at 37°C throughout the procedure.
A 6-0 nylon monofilament was blunted at the tip in a flame and then lightly coated with silicone. The filament was inserted into the proximal external carotid artery stump and advanced ≈11 mm into the internal carotid artery to occlude the middle cerebral artery. Inspired halothane concentration was reduced to 0.9% at time of filament insertion. After a pre-established ischemic interval (see below), the filament was removed and the skin incision closed with suture. Ten minutes after filament removal, halothane was discontinued and the mice were allowed to awaken. The mice were placed in an oxygen-enriched environment (FIO2 = 50%) for 1 hour and then returned to their cages.
All animals were evaluated neurologically 24 hours after reperfusion. Each mouse was assigned a score of 0 to 4 where 0 = no observable neurologic deficit; 1 = failure to extend the left forepaw; 2 = circling to the left; 3 = falling to the left; and 4 = cannot walk spontaneously (Yang et al., 1994). Neurological examination was performed by one observer blinded to group assignment.
After neurological evaluation, animals were anesthetized with halothane and decapitated. The brain was removed and frozen at −20°C. Using a cryotome, 20-μm thick coronal sections were taken at 320-μm intervals over the rostral-caudal extent of the infarct. The sections were dried and stained with hematoxylin and eosin.
Infarct volume was measured by digitally sampling stained sections with a Sony CCD Model XC-77 video camera controlled by an image analyzer (M2 Turnkey System, Imaging Research, Inc., St. Catharines, Ontario). The image of each section was stored as a 1024 × 1024 matrix of calibrated pixel units. The digitized image was then displayed on a video screen. With the observer blinded to experimental condition, infarct borders in both cortex and subcortex were individually outlined (corpus callosum excluded) using an operator-controlled cursor. The area of infarct (mm2) was determined automatically by counting pixels contained within the outlined regions of interest. Infarct volumes (mm3) were computed as running sums of infarct area multiplied by the known interval (e.g., 320 μm) between sections over the extent of the infarct expressed as an orthogonal projection.
Two independent experiments were performed with the above stated protocol: Experiment 1: ApoE-deficient and wild-type mice (n = 10 per group) underwent 60 minutes of MCAO. Experiment 2: ApoE-deficient and wild-type mice (n = 10 per group) underwent 90 minutes of MCAO.
Measurement of physiological parameters
Because the blood volume required for analytic assays was great enough to cause hypovolemia, a parallel study was performed to define the probable physiologic state of mice studied in Experiments 1 and 2. Both ApoE-deficient and wild-type mice were examined (n = 5 per group). These animals underwent a surgical procedure and MCAO insult identical to that described above. In addition, a pericranial needle thermistor was placed adjacent to the skull beneath the temporalis. Servoregulation of body temperature was performed via the rectal probe. Therefore pericranial temperature was only monitored. A right femoral arterial catheter was placed to monitor blood pressure and withdraw blood. Blood chemistries were measured at 10 minutes before and 45 minutes after onset of ischemia. The animals were then killed by an overdose of halothane anesthesia.
Measurement of coagulation parameters
Four control (C57-BL/6J) and four ApoE-deficient mice were administered ketamine 30 mg/kg, xylazine 5.2 mg/kg, and acepromazine 1 mg/kg intraperitoneally. Via direct cardiac puncture, 0.9 mL of whole blood was aspirated and mixed with 100 μL of 0.1 mol/L sodium citrate. Prothrombin time, activated partial thromboplastin time, and the fibrinogen concentration were determined by standard methods in accordance with the National Committee on Clinical Laboratory Standards (Clauss, 1957; Langdell et al., 1957; Quick et al., 1935). All assays were performed on an MDA 180 clot detection analyzer (Organon Teknika, Durham, NC, U.S.A.).
Statistical analysis
Physiologic values, coagulation values, and infarct volumes were compared between groups with the Student's t-test. Mortality rates were compared with Fisher's exact test (two-sided). Infarct volumes were correlated with neurologic grades by the Spearman rank correlation coefficient while neurologic grades were compared between groups by the Mann Whitney U statistic. Parametric values are expressed as mean ± SD.
RESULTS
In Experiment 1, no mice in either group died as a result of 60 minutes of MCAO. Total infarct volume was 64% greater in ApoE-deficient mice (Fig. 1). Apolipoprotein E-deficient mice had significantly larger cortical infarct volumes (20 mm3 ± 12 versus 9 ± 7 mm3, P = 0.03). Although a trend was present, ApoE genotype did not significantly alter subcortical infarct volume (ApoE deficient = 22 ± 7 mm3; wild-type = 16 ± 7 mm3, P = 0.07). Increased neurologic deficit was observed in the ApoE-deficient mice (P = 0.02; Fig. 2). Correlation between neurologic score and infarct size was significant (P = 0.02).
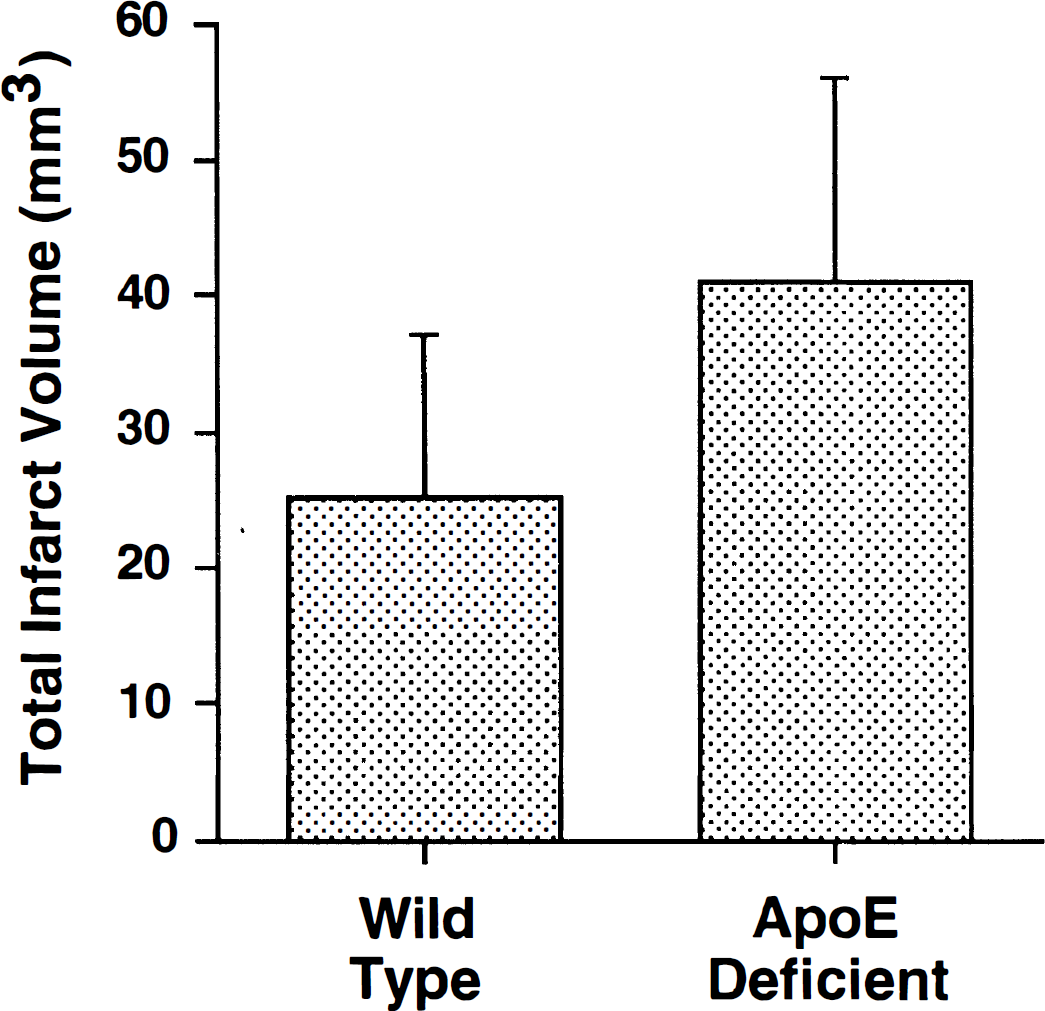
Total infarct volume for apolipoprotein E-(ApoE) deficient mice (n = 10) versus wild-type controls (n = 10) subjected to 60 minutes of middle cerebral artery occlusion and a 24-hour recovery interval (mean ± SD). All mice survived the recovery interval. Infarct volume was greater in the ApoE-deficient mice (P = 0.02).
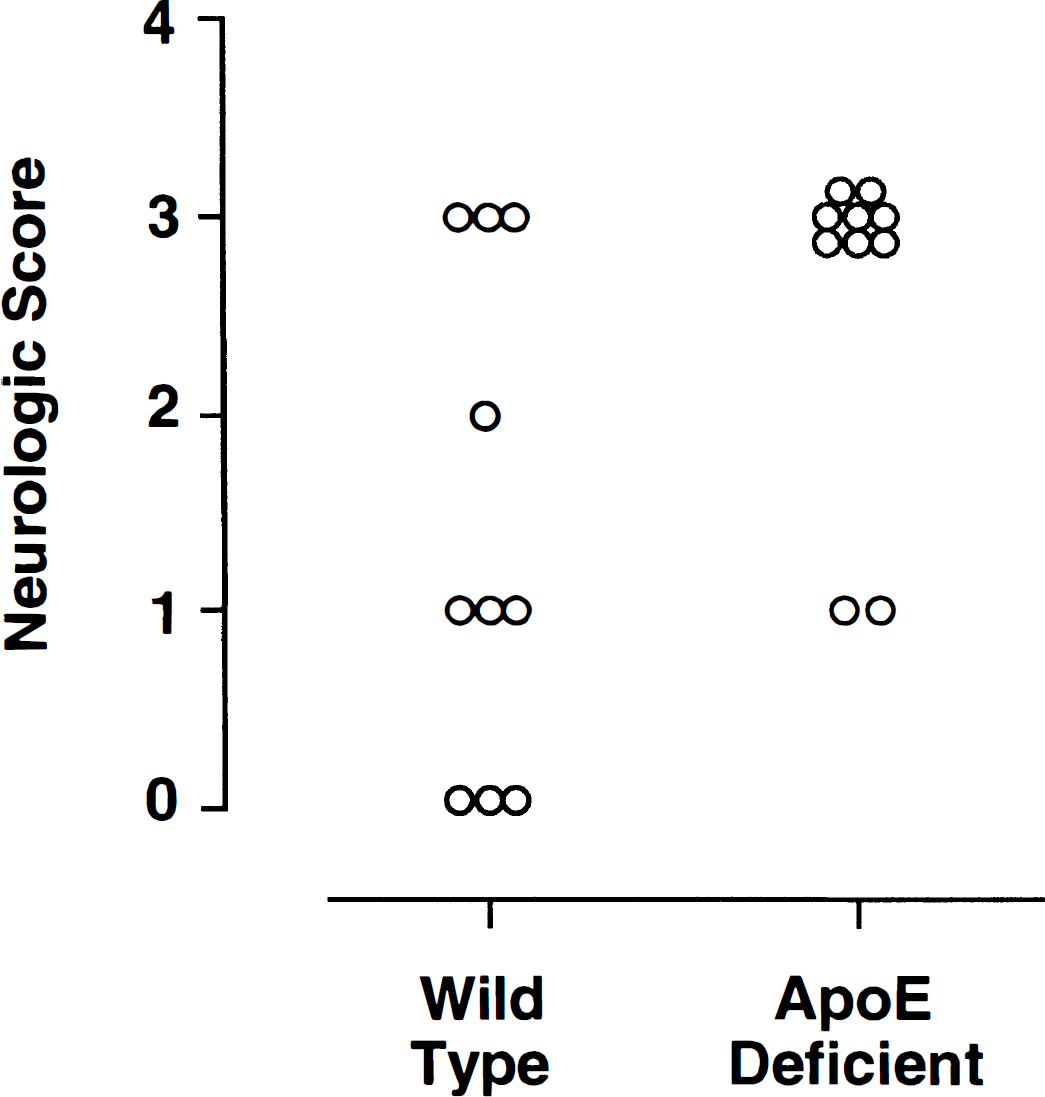
Individual neurologic scores for ApoE-deficient mice (n = 10) versus wild-type controls (n = 10) subjected to 60 minutes of middle cerebral artery occlusion and a 24-hour recovery interval. Wild-type mice showed less severe deficits (P = 0.02).
In Experiment 2, 40% of ApoE-deficient and no wild-type mice died after 90 minutes ischemia (P = 0.09). Deaths typically occurred 12 to 16 hours after reperfusion. All 4 of the ApoE-deficient mice that died had large infarcts, 3 of which had parenchymal hemorrhages with intraventricular extension. Hemorrhages were absent in the wild-type group. Cortical (ApoE deficient = 37 ± 8 mm3; wild-type = 31 ± 18mm3; P = 0.49) and subcortical (ApoE deficient = 30 ± 11 mm3; wild-type = 32 ± 18 mm3; p = 0.78) infarct volumes were similar among rats surviving 24 hours of reperfusion. Among survivors there was no difference in neurologic function (P = 0.36). Total infarct volume correlated with infarct size (P = 0.01).
Most physiological values were similar between groups (Table 1). Mild pericranial hypothermia was present in both groups despite control of rectal temperature. There was increased hypercarbia (P = 0.04) and a trend toward greater acidosis (P = 0.08) in the ApoE-deficient mice after 45 minutes of ischemia as compared with wild-type animals.
Physiologic values obtained in surrogate animals
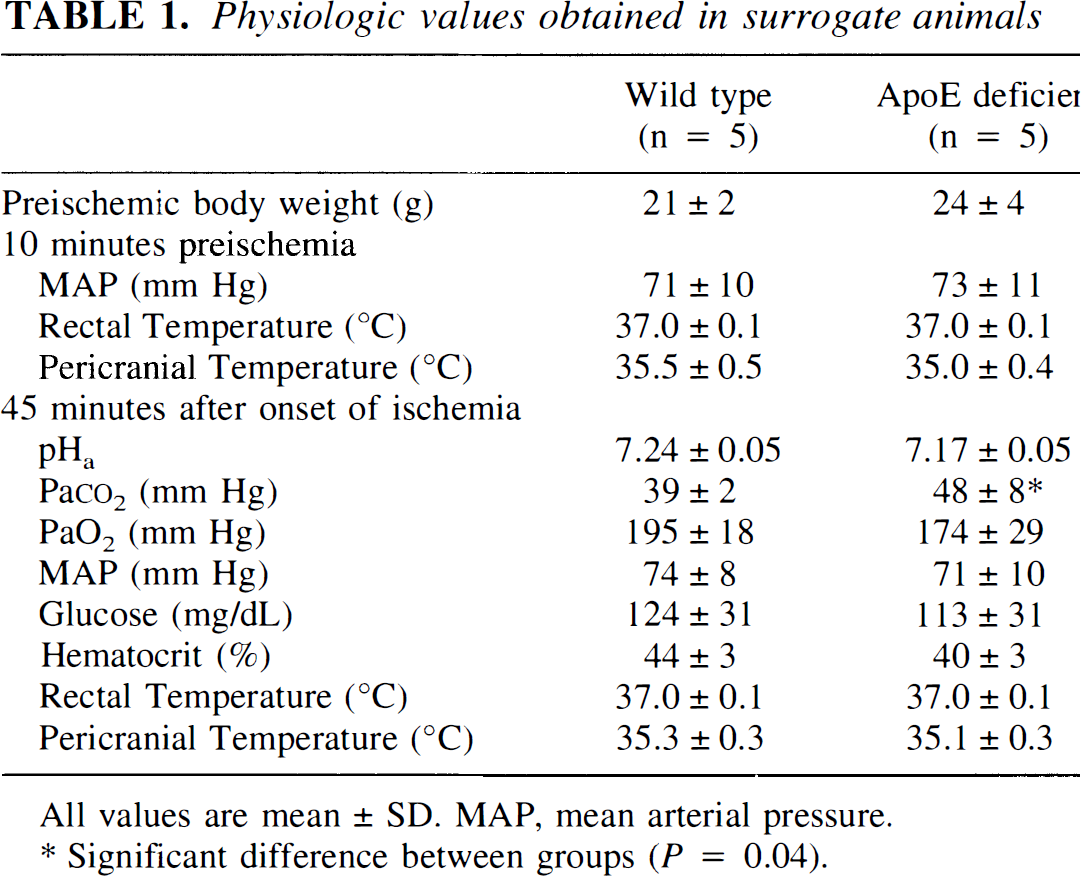
All values are mean ± SD, MAP, mean arterial pressure.
Significant difference between groups (P = 0.04).
There was no difference in prothrombin time between ApoE-deficient (13.0 ± 1.4 seconds) and wild-type (12.6 ± 0.9 seconds) mice. There was a prolongation of the activated partial thromboplastin time (64 ± 2 second versus 34 ± 5 second, P < 0.01). There was also a significant elevation of the fibrinogen concentration in the ApoE-deficient animals (132 ± 18 mg/dL versus 74 ± 11 mg/dL, P < 0.05).
DISCUSSION
Apolipoprotein E is the primary apolipoprotein produced intra-axially, where it is secreted by astrocytes and oligodendrocytes. Its functional role in the CNS remains to be elucidated, but upregulation of ApoE synthesis by astrocytes and oligodendrocytes after injury suggests that it might play a role in neuronal injury or repair. The current experiment has shown that ApoE-deficient mice have decreased resistance to focal cerebral ischemia when compared with wild-type animals.
A potential confound in these experiments is the fact that ApoE-deficient animals are markedly hypercholesterolemic and therefore are prone to develop atherosclerotic disease (Plump et al., 1992; Reddick et al., 1994; Zhang et al., 1992). Using strains identical to those examined in this experiment, Xu et al. found ApoE-deficient mice to have serum cholesterol concentrations of 756 ± 189 mg/dL while wild-type mice had concentrations of 98 ± 17 mg/dL (Xu et al., 1996). At 3 to 5 months of age, ApoE-deficient animals develop early evidence of preatherosclerotic lesions, including small foci of foam cells in the coronary sinus, and fatty streaks in the proximal aorta. These lesions advance with age, and by 9 to 12 months, ApoE-deficient animals will have lesions distributed throughout the vascular tree, including the abdominal aorta, carotid, and iliac arteries (Plump et al., 1992; Reddick et al., 1994; Zhang et al., 1992). In theory, vascular disease could have impaired collateral flow and indirectly exacerbated the effect of MCAO. This effect was minimized by the use of young (8- to 10-weeks old) adults in which no hemodynamically significant vascular lesions have been reported. Nevertheless, comparison of ApoE-deficient mice against similarly hypercholesterolemic low-density lipoprotein-deficient mice in a focal ischemia model might provide additional evidence that the effects we observed were attributable to phenomena occurring at the cellular level.
The mouse is known to have only one allele for ApoE. The DNA sequence has been defined and a 78% homology to human ApoE has been identified (Rajavashisth et al., 1985). The predicted secondary structure of the mouse protein is nearly identical to the human, including the sequence active at the receptor binding site. The mouse protein correlates best with the human ApoE4 isoform. However, direct examination of differential human isoform effects on ischemic pathophysiology will require mice carrying transgenes for the different human isoforms (Xu et al., 1996).
Concern can be raised regarding the genetic similarity between the ApoE-deficient and wild-type mice and potential for CNS abnormalities in the ApoE-deficient mutants. A sixth-generation back cross of ApoE-deficient animals against the C57BL/6J mouse should assure approximately 98% homology. Study of animals with an increased number of inbred generations would reduce the remaining difference. However, the relevance of this to the different responses to MCAO is unknown. There is evidence to support lack of neuroanatomic anomalies in young ApoE-deficient animals. Masliah et al. examined age-related changes of ApoE-deficient mutants (Masliah et al., 1995). Two-month old mice (within the age range used in our experiment) showed no morphological changes when examined for alterations in gross histology, synaptic density, or neuronal cytoskeletal architecture. However, in older mice (4 months), ultrastructural analysis revealed extensive dendritic vacuolization and disruption of the cytoskeleton. These changes were attributed to absence of active processes normally provided by ApoE rather than to developmental anomalies. Others have observed reduced brain choline acetyltransferase activity in the hippocampus and frontal cortex of 6-month old, ApoE-deficient mice. This was correlated with deficits in working but not reference memory (Gordon et al., 1995). Such effects have not been examined in younger animals.
One possible mechanism by which ApoE ameliorated the effects of focal ischemia is down regulation of the CNS inflammatory response. In addition to its role in cholesterol transport and its putative role in neurological disease, ApoE is known to have immunomodulatory functions in vitro. For example, ApoE decreases lymphocyte proliferation after mitogenic challenge, and reduces pokeweed-mitogen stimulated immunoglobulin synthesis of B lymphocytes (Curtiss and Edgington, 1976; Pepe and Curtiss, 1986). The in vivo functional significance of these immunomodulatory properties remains to be established. In theory, ApoE might play a unique role in the CNS where it is the primary apolipoprotein produced. Focal brain ischemia is associated with an inflammatory response which peaks in 48 to 72 hours (Garcia et al., 1994). Inflammation after ischemia may exacerbate brain injury by contributing to brain edema, blood-brain barrier breakdown, and release of reactive oxygen species by activated microglia (Gordon et al., 1990; Quagliarello et al., 1991; Rothwell and Relton, 1993). Inflammation in the CNS is largely mediated by cytokines such as TNFα and Il-1β. There is preliminary evidence that inhibiting the actions of these inflammatory molecules may reduce ischemic injury (Garcia et al., 1995). We have recently observed that ApoE suppresses glial secretion of the inflammatory cytokine TNFα without any effect on cell viability (Laskowitz et al., 1997). Thus, it is possible that ApoE down regulates the CNS inflammatory response, protecting neurons from secondary injury.
Another possible mechanism by which ApoE might protect against ischemic insults is by acting as an anti-oxidant. After a period of ischemia, reperfusion contributes to injury via the production of free radicals and subsequent lipid peroxidation (Globus et al., 1995; Kil et al., 1996). Apolipoprotein E has the ability to protect neurons from oxidative injury, as has been demonstrated by its ability to protect neurons in cell culture from hydrogen peroxide toxicity (Miyata and Smith, 1996). This is also consistent with the observation that plasma lipoproteins from ApoE-deficient mice are more susceptible to in vitro oxidation than observed in wild-type mice (Hayek et al., 1994). This might also explain the isoform-specific differences that have been observed in neurological disease, as the E3 isoform is a more effective at protecting cells from oxidative death than E4 (Hayek et al., 1994).
The finding that several of the ApoE-deficient mice exposed to 90 minutes of ischemia suffered an intraventricular hemorrhage was unexpected. Although ApoE has never been identified in playing a role in hemostasis, this observation led us to evaluate the clotting cascade in these animals. Apolipoprotein E seems to affect the intrinsic clotting pathway, as evidence by a significantly prolonged activated partial thromboplastin time in the ApoE-deficient mice as compared with wild-type animals. This is of particular interest, given the recent finding that the E4 allele is associated with poor outcome after intracerebral hemorrhage (Alberts et al., 1995). The ApoE-deficient strain is associated with a 10% incidence of lethal hydrocephalus (personal communication, Linda L. Washburn, The Jackson Laboratory, November 1996). It can be speculated that this is caused by spontaneous intraventricular hemorrhage with resultant obstructive hydrocephalus. The role that ApoE plays in clotting, and the possibility of any functionally relevant isoform-specific effects remains to be investigated. It is also unclear why the ApoE-deficient animals had increased serum fibrinogen. Apolipoprotein E has been found to show immunosuppressive effects (Curtiss and Edgington, 1976; Pepe and Curtiss, 1986). Because fibrinogen is an acute-phase reactant, it is possible that this reflects chronic low-grade inflammation in the ApoE-deficient animals.
In summary, after 60 minutes of focal ischemia and 24 hours of reperfusion, ApoE-deficient mice were found to have larger infarcts and worse neurologic outcome than matched controls. This is consistent with the hypothesis that in addition to its well-recognized role in cholesterol transport, ApoE may modify the CNS response to injury. Apolipoprotein E deficiency (Type III hyperlipoproteinemia) is a rare disorder in humans (Ghiselli et al., 1981; Mabuchi et al., 1989; Schaefer et al., 1986). Therefore, the ApoE-deficient murine construct may have limited direct application. However, evidence that ApoE is involved in the pathophysiology of stroke supports further work examining ApoE isoform-specific effects on ischemic outcome. Such work could have large therapeutic and prognostic implications in cerebrovascular disease.