
Abstract
Excitatory amino acids can modify the tone of cerebral vessels and permeability of the blood-brain barrier (BBB) by acting directly on endothelial cells of cerebral vessels or indirectly by activating receptors expressed on other brain cells. In this study we examined whether rat or human cerebromicrovascular endothelial cells (CEC) express ionotropic and metabotropic glutamate receptors. Glutamate and the glutamate receptor agonists N-methyl-d-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-isoxazole-4-propionic acid (AMPA), and kainate failed to increase [Ca2+]i in either rat or human microvascular and capillary CEC but elicited robust responses in primary rat cortical neurons, as measured by fura-2 fluorescence. The absence of NMDA and AMPA receptors in rat and human CEC was further confirmed by the lack of immunocytochemical staining of cells by antibodies specific for the AMPA receptor subunits GluR1, GluR2/3, and GluR4 and the NMDA receptor subunits NR1, NR2A, and NR2B. We failed to detect mRNA expression of the AMPA receptor subunits GluR1 to GluR4 or the NMDA receptor subunits NR11XX, NR10XX, and NR2A to NR2C in both freshly isolated rat and human microvessels and cultured CEC using reverse transcriptase polymerase chain reaction (RT-PCR). Cultured rat CEC expressed mRNA for KA1 or KA2 and GluR5 subunits. Primary rat cortical neurons were found to express GluR1 to GluR3 and NR1, NR2A, and NR2B by both immunocytochemistry and RT-PCR and KA1, KA2, GluR5, GluR6, and GluR7 by RT-PCR. Moreover, the metabotropic glutamate receptor agonist 1-amino-cyclopentyl-1S, 3R-dicorboxylate (1S,3R-trans-ACPD), while eliciting both inositol trisphosphate and [Ca2+]i increases and inhibiting forskolin-stimulated cyclic AMP in cortical neurons, was unable to induce either of these responses in rat or human CEC. These results strongly suggest that both rat and human CEC do not express functional glutamate receptors. Therefore, excitatory amino acid-induced changes in the cerebral microvascular tone and BBB permeability must be affected indirectly, most likely by mediators released from the adjacent glutamate-responsive cells.
Keywords
Whereas the roles of glutamate in regulating physiological responses (Collingridge and Singer, 1990) and mediating excitotoxic injury (Meldrum and Garthwaite, 1990; Small and Buchan, 1996) to neurons have been established, the possible involvement of glutamate in modulating cerebrovascular functions is less clear. Numerous reports have described various modes of vasoactive action of glutamate agonists or antagonists in different segments of the cerebral vasculature in vivo and on isolated vessels (Fukuda et al., 1983; Cavazutti et al., 1987; Busija and Leffler, 1989; Perkins et al., 1989; Faraci and Breese, 1993; Wendling et al., 1994). Studies using isolated cerebral microvessels reported that bovine and ovine cerebral microvessels lack binding sites for the N-methyl-
Due to the anatomical proximity of glutamate-releasing neurons to the cerebral capillaries and microvessels, the presence of glutamate receptors on cerebromicrovascular endothelial cells (CEC) could significantly affect vasomotor responses of the cerebral microvascular bed or the function of the BBB. Brain capillary and microvascular endothelial cells have been shown to express receptors for other neurotransmitters including noradrenaline (Bacic et al., 1992), serotonin (Cohen et al., 1995), acetylcholine (Linville et al., 1995), and dopamine (Bacic et al., 1991) as well as for the vasoactive mediators histamine (Stanimirovic et al., 1994a), adenosine (Stanimirovic et al., 1994a), endothelins (Stanimirovic et al., 1994b), bradykinin (Stanimirovic et al., 1996b), and angiotensins (Stanimirovic et al., 1996a). These neurotransmitters/mediators have also been shown to modulate both vascular tone (Luscher, 1990) and BBB permeability (Guillot and Audus, 1991; Stanimirovic et al., 1996a) by acting on endothelium-expressed receptors.
The purpose of this study was to determine if CEC express glutamate receptors on their membranes and are therefore the primary site of action for modulation of BBB permeability and/or vascular tone by glutamate in the cerebromicrovascular bed. We studied the presence of functional glutamate receptors in both freshly isolated rat and human capillaries and microvessels and cultured rat and human CEC by measuring glutamate-induced changes in [Ca2+]i, inositol trisphosphate (IP3), and cyclic AMP and the expression of glutamate receptor subunits by immunocytochemistry and reverse transcriptase polymerase chain reaction (RT-PCR). Our studies provide evidence that neither rat nor human CEC express functional glutamate receptors. Therefore, it appears that any microvessel vasomotor responses and/or BBB permeability changes effected by glutamate are endothelium independent and are likely elicited by glutamate actions on surrounding cells.
MATERIALS AND METHODS
Chemicals and reagents
All culture media and fetal bovine serum were obtained from GibcoBRL (Gaithersburg, MD, U.S.A.). Endothelial cell growth supplement, ITS Premix, human fibronectin, and rat tail type IV collagen were purchased from Collaborative Biomedical Products (Bedford, MA, U.S.A.). Human serum and glutamate were obtained from Sigma Chemical Co. (St. Louis, MO, U.S.A.). Mouse melanoma cells, Cloudman S91 (clone M-3), were obtained from American Type Culture Collection (Rockville, MD, U.S.A.). NMDA, α-amino-3-hydroxy-5-methyl-isoxazole-4-propionic acid (AMPA), kainate, 1-amino-cyclopentyl-15 3R-dicorboxylate (1S,3R-trans-ACPD), and MK-801 were purchased from Research Biochemicals International (Natick, MA, U.S.A.). Fura-2 acetoxymethyl ester (fura 2-AM) was from Molecular Probes (Eugene, OR, U.S.A.). TriReagent was obtained from Molecular Research Center (Cincinnati, OH, U.S.A.).
Freshly isolated cerebral microvessels
Rat brain microvessels and human brain capillaries or resistance microvessels were isolated using a modification(s) of the procedures of Diglio et al. (1982) and Gerhart et al. (1988), respectively. Rat brain microvessels were isolated from cortical brain tissue of four to six adult rats. Human brain capillaries and microvessels were obtained from small samples of human temporal lobe excised surgically from patients treated for idiopathic epilepsy.
In both cases, the brain samples were cleaned of pia matter and associated surface vessels, and cortical gray matter was dissected. The tissue was minced with scissors and homogenized in a glass-teflon homogenizer. The homogenate was passed once through 350-μm and twice through 112-μm mesh nylon nets (Nitex; Tetko, Elmsford, NY, U.S.A.) fitted in Millipore filter holders. Resistance microvessels were dislodged from the 112-μm mesh and collected in cold medium M199 (containing Earle's salts, 25 mmol/L N-2-hydroxy-ethylpiperazine-N′-2-ethanesulfonic acid, 4.35 g/L sodium bicarbonate, and 3 mmol/L
Cerebral endothelial cell cultures
Freshly isolated brain microvessels were enzymatically dissociated to obtain endothelial cell cultures as follows: The resistance vessels (112 to 350 μm) and the capillaries and smaller microvessels (>20 μm) derived from human brain samples were separately dissociated by exposure to 1 mg/mL type IV collagenase for 15 minutes at 37°C, centrifuged at 500 g for 5 minutes, resuspended in growth medium containing 65% medium M199, 10% fetal calf serum, 5% human serum, 20% murine melanoma cell (mouse melanoma, Cloudman S91, clone M-3, melanin-producing cells)-conditioned medium, 5 μg/mL insulin, 5 μg/mL transferrin, 5 ng/mL selenium, and 10 μg/mL endothelial cell growth supplement, seeded in 0.5% gelatin-coated plastic tissue culture dishes, and maintained at 37°C in an atmosphere of 5% CO2 in air. Microvessels of rat brain retained on the 20-μm mesh were dissociated by a brief (10-min) exposure to type I collagenase (1 mg/mL, 37°C) and seeded in rat tail collagen-coated tissue culture flasks in medium M199 supplemented with 1% basal medium Eagle's amino acids, 1% basal medium Eagle's vitamins, antibiotic/antimycotic mixture (100 units/mL penicillin G, 100 mg/mL streptomycin sulfate, 0.25 mg/mL amphotericin B), 1% glucose, 0.05% peptone, and 20% fetal bovine serum. Identified single endothelial cells or small groups of endothelial cells emerging from the microvessels 3 to 4 days after the seedings were isolated using cloning rings (Bellco Glass, Vineland, NJ, U.S.A.), and two to three of these cloned colonies were pooled and further passaged. Cells obtained from three different isolations (passages 3 to 8) were used for the experiments in this study. The CEC cultures obtained from both rat brain microvessels and human brain resistance microvessels and capillaries were routinely characterized using indirect immunocytochemical staining for von Willbrandt factor (Factor VIII)-related antigen, incorporation of acetylated low-density lipoprotein conjugated to fluorescent dye, and binding of fluorescently labeled lectins Ricinus communis agglutinin I and concanavalin A, as described and published previously (Stanimirovic et al., 1995, 1996a). More than 95% of the cells derived by the described procedures were positive for these endothelial cell markers and free of glial contamination as ascertained by the absence of staining for glial fibrillary acidic protein. Both capillary and resistance microvessel-derived human CEC were highly enriched in the BBB-specific enzyme γ-glutamytranspeptidase (Stanimirovic et al., 1996a,b).
Rat cortical neurons
Rat cortical neurons were cultured by a previously described method (Durkin et al., 1997). For RT-PCR and the determination of IP3 production, the cells were plated on poly-
[Ca2+]i measurement
To determine [Ca2+]i; human and rat CEC were cultured in complete medium on 24-mm glass coverslips coated with 10 μg/mL human fibronectin and 0.5% rat tail collagen, respectively, for 1 to 4 days, while primary fetal rat cortical neurons were grown for 15 days on poly-
Inositol trisphosphate determination
To determine inositol phosphate formation, confluent rat CEC, human CEC, and primary cultures of rat cortical neurons grown in 35-mm petri dishes were prelabeled with [3H]myo-inositol (2.5 mCi/mL) for 16 to 18 hours in serum- and inositolfree medium M199. Unbound [3H]myo-inositol was removed by washing in M199, and the cells were then exposed to glutamate and/or glutamate receptor agonists/antagonists in M199 in the presence of 20 mmol/L LiCl (Berridge et al., 1982) for 15 minutes. The reaction was stopped by replacing the medium with cold 0.3 mol/L trichloroacetic acid, and the cells were scraped, briefly sonicated, and sedimented by centrifugation. Aliquots of the supernatants were treated three times with anhydrous diethyl ether, the aqueous phases were separated by centrifugation, and the remaining ether was evaporated under nitrogen. The samples were neutralized with 6.25 mol/L sodium tetraborate and applied to a 1-mL Dowex-AG 1×8-formate form anion exchange column. Inositol phosphate fractions were eluted from the columns and quantified as previously described (Stanimirovic et al., 1994b). The IP3 fraction determined by this technique represents a mixture of IP3 isomers.
Cyclic AMP determination
To determine levels of cyclic AMP in rat and human CEC and rat cortical neurons, cells grown in 24-well tissue culture plates were treated with the metabotropic glutamate receptor agonist 1S,3R-trans-ACPD, alone or in combination with forskolin, in phosphate-buffered saline (PBS) containing 0.2% bovine serum albumin and 1 mmol/L 3-isobutyl-1-methylxanthine for 10 min. The reaction was stopped by removing the reaction mixture and adding 65% (vol/vol) ice-cold ethanol. The ethanol extraction was repeated twice, and the combined extracts were collected into test tubes and dried in a vacuum oven at 80°C. The cyclic AMP levels were determined using a commercial cyclic AMP enzyme immunoassay kit (Biotrak; Amersham). The assay is based upon the competition between unlabeled cyclic AMP and a fixed quantity of peroxidaselabeled cyclic AMP for a limited number of binding sites on a cyclic AMP-specific antibody.
Immunohistochemical detection of glutamate receptor subunits
Cells grown on glass coverslips were washed briefly in PBS and then immersed in Lana's fixative [4% paraformaldehyde and 14% (vol/vol) saturated picric acid in 0.25 mol/L NaH2PO4, pH 6.9] for 1 hour at room temperature. The cells were then rinsed in PBS, permeabilized for 5 minutes in 0.25% Triton X-100 in PBS, and then rinsed 2 × 5 min with PBS. Coverslips were inverted onto droplets of 5% normal donkey serum in 1% bovine serum albumin (Sigma) in PBS for 40 minutes at room temperature to block nonspecific binding sites. The cells were then rinsed for 5 minutes in PBS and incubated with primary antibodies against the NMDA receptor subunits NR1, NR2A, and NR2B and the AMPA receptor subunits GluR1, GluR2/3, and GluR4 for 1 hour at room temperature. All antibodies were purchased from Upstate Biotechnology (Lake Placid, NY, U.S.A.). Antibodies were used at a concentration of 10 or 2 μg/mL (for GluR2/3) diluted in 1% bovine serum albumin/PBS. Control experiments were performed in which the primary antibodies were omitted. Following incubation with primary antibody, cells were washed 3 × 5 min in PBS and incubated with a CY3-conjugated donkey anti-rabbit IgG (secondary antibody), diluted 1:500 (Jackson, Bio/Can Scientific, Mississauga, Ontario, Canada) in 1% bovine serum albumin/PBS for 1 hour at room temperature. Coverslips were rinsed and mounted in Vectashield (Vector Laboratories, Burlingame, CA, U.S.A.), viewed on an Olympus photomicroscope, and photographed using Ilford XP2 400 ASA film.
RT-PCR detection of glutamate receptor subunits
Reverse transcriptase polymerase chain reaction was used to determine the mRNA expression profile of the glutamate NMDA receptor subunits NR1, NR2A, NR2B, and NR2C, the AMPA receptor subunits GluR1, GluR2, GluR3, and GluR4, and the kainate receptor subunits KA1 or KA2, GluR5, GluR6, and GluR7 in freshly isolated rat and human brain microvessels, cultured rat and human CEC, and cultured rat neurons. Total RNA was isolated from each culture dish [total cell number, −1.5 × 105 for human CEC, −5 × 105 for rat CEC, and −7.5 × 105 for rat neurons including a 20 to 35% glial component] using TriReagent. First-strand cDNA was made in 20 μl, using Moloney murine leukemia virus RT (Perkin Elmer Cetus, NJ, U.S.A.) and primed with random hexamers. For the freshly isolated human microvessel preparations, 10 μg of total RNA, digested with DNase I (Promega Corp., Madison, WI, U.S.A.), was reverse transcribed with avian myeloblastosis virus RT (Seikagaku America, Ijamsville, MD, U.S.A.) in 50 μl and primed with oligo-dT (Perkin Elmer Cetus). First-strand cDNA (20-μL cultures, 4 μL fresh) was then used for PCR amplification of NMDA, AMPA, and kainate receptor subunits. All PCR reactions were performed in a final volume of 100 μL using the Perkin Elmer PCR kit, AmpliTaq DNA polymerase, and 50 pmol oligonucleotide primers. Following PCR amplification, the product was electrophoresed on a 1.5% agarose gel and visualized by staining with ethidium bromide. Serial dilutions of copies of full-length clones of the various glutamate subunits were used as templates in the PCR reactions to assess the amplification efficiency of each of each reaction.
The oligonucleotide primers (Sheng et al., 1994) used to amplify NR1 receptor subunits were designed to flank the NR1's exon 5, resulting in two different-sized products (235 and 172 bp) corresponding to mRNAs containing (NR11XX) or lacking (NR10XX) this exon. NR2 receptor subunits NR2A, NR2B, and NR2C were amplified using oligonucleotide primers and the cycling paradigm as described by Audinat et al. (1994) to produce products of all the same size (547 bp). To amplify the AMPA receptor subunits GluR1, GluR2, GluR3, and GluR4, the oligonucleotide primers and cycling paradigm used were as described by Lambolez et al. (1992) and produced products of all the same size (~750 bp). To amplify the kainate receptor subunits KA1 or KA2, GluR5, GluR6, and GluR7, four sets of oligonucleotide primers and the cycling paradigm described by Ruano et al. (1995) were used and produced products of 512, 208, 259, and 421 bp, respectively.
RESULTS
Effects of glutamate agonists on [Ca2+]i, IP3, and cyclic AMP in rat and human CEC and rat cortical neurons
To probe for the presence of functional glutamate receptors in cultured rat and human CEC, we measured changes in [Ca2+]i and IP3 production in response to glutamate and glutamate receptor agonists. In Mg2+-free normal buffer solution containing 30 μmol/L glycine, glutamate (100 μmol/L; data not shown), NMDA [40 μmol/L; Fig. 1A (rat) and B (human); n = 11], AMPA [40 μmol/L; Fig. 1C (rat) and D (human); n = 7], and kainate [40 μmol/L; rat and human; data not shown; n = 7] did not affect [Ca2+]i in either rat or human CEC. In all experiments, these endothelial cells were capable of responding by increases in [Ca2+]i to a subsequent addition of endothelin-1 (10 nmol/L), angiotensin II (200 nmol/L), or bradykinin (20 μmol/L) (Fig. 1A to D, F). Rat and human CEC exposed to very high concentrations of glutamate (1 mmol/L), NMDA (400 μmol/L), and AMPA (400 μmol/L) in the presence of the desensitization blocker cyclothiazide (25 μmol/L) (data not shown) and kainate (400 μmol/L; rat; Fig. 1F) also failed to show [Ca2+]i responses (data not shown). These findings are in sharp contrast to the ability of NMDA (40 μmol/L; Fig. 1E), glutamate, AMPA, and kainate (data not shown) to produce significant increases in [Ca2+]i in rat cortical neurons (Durkin et al., 1997). The results suggest that rat and human CEC in culture lack functional ionotropic glutamate NMDA, AMPA, and kainate receptors.
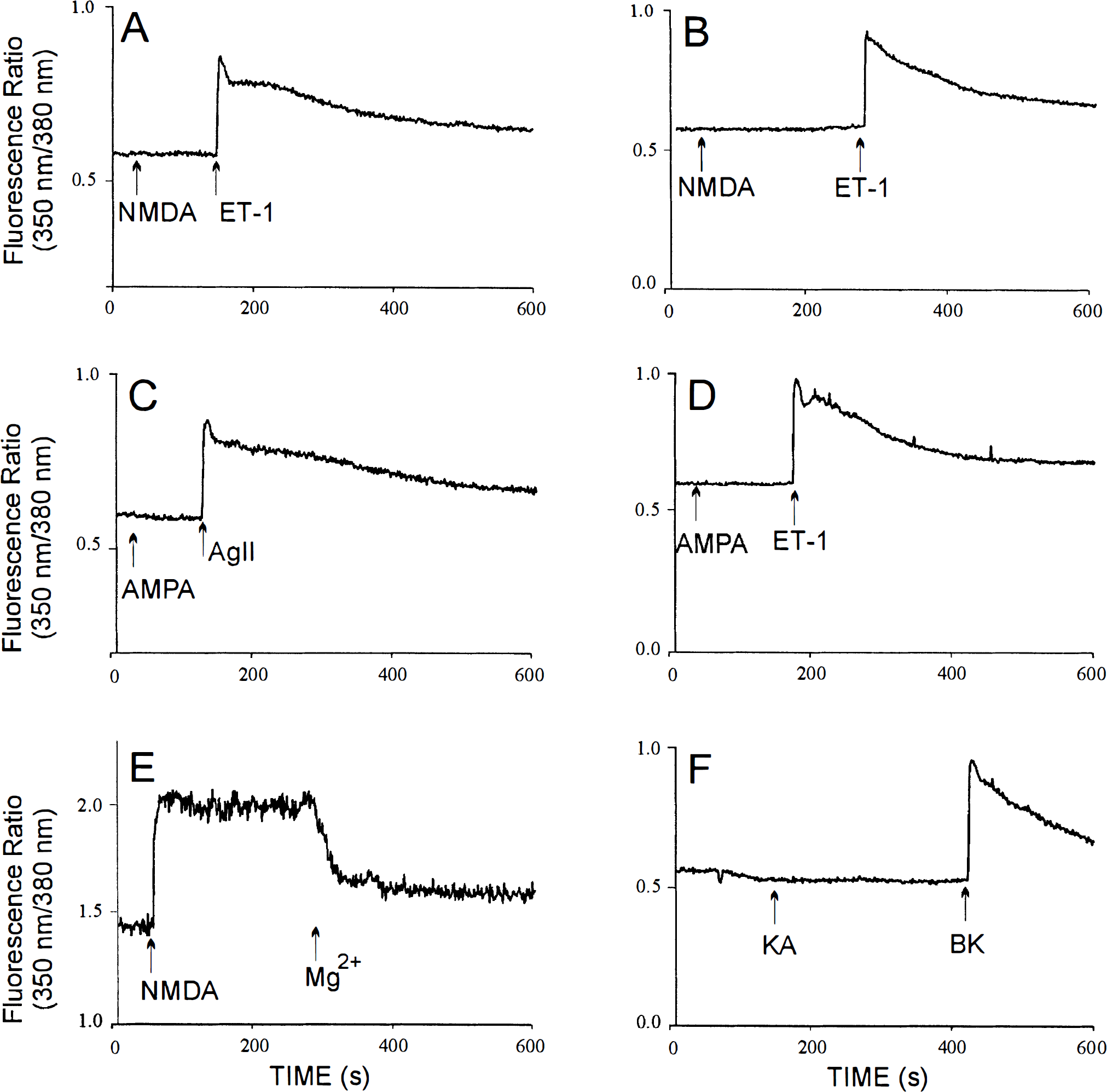
The effect of glutamate receptor agonists on [Ca2+]i in rat cerebromicrovascular endothelial cells (CEC) (
To test for metabotropic glutamate receptor expression, we determined the effects of the specific metabotropic glutamate receptor agonist 1S,3R-trans-ACPD on Ca2+ fluxes, phospholipid hydrolysis, and adenylyl cyclase activity. 1S,3R-trans-ACPD [300 μmol/L; Fig. 2A (rat) and B (human); n = 4) did not affect [Ca2+]i in either rat or human CEC. When added to either rat (Fig. 3A) or human (Fig. 3B) CEC cultures at 300 to 500 μmol/L, 1S,3R-trans-ACPD failed to induce phospholipid hydrolysis and accumulation of IP3. In the same cell cultures, the vasoactive peptides endothelin-1, previously shown to couple to phospholipase C in these cells (Stanimirovic et al., 1994b, 1996a), induced a four- to fivefold augmentation of IP3 levels [Fig. 3A (rat) and B (human)]. By contrast, in rat cortical neuronal cultures, 300 μmol/L 1S,3R-trans-ACPD induced a twofold increase in IP3 (Fig. 3C). The same concentrations of 1S,3R-trans-ACPD have been shown to trig ger a [Ca2+]i rise in cortical neuronal cultures (Durkin et al., 1997). Both endothelin-1 and angiotensin II-induced [Ca2+]i increases in rat and human CEC (Fig. 1) (Monette et al., 1996; Stanimirovic et al., 1996b) and 1S,3R-trans-ACPD-induced [Ca2+]i increases in cortical neurons (Durkin et al., 1997) were due to the release of Ca2+ from internal stores, since they were preserved in Ca2+-free medium.
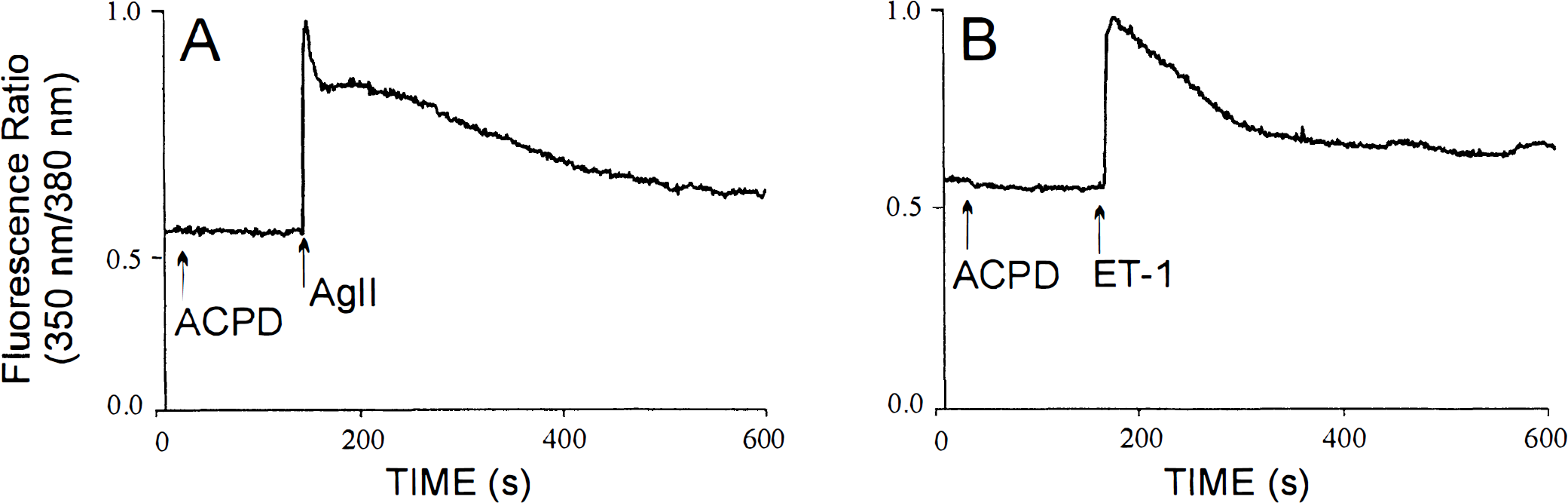
The effect of the metabotropic agonist 1S,3R-trans-ACPD on [Ca2+]i in rat (
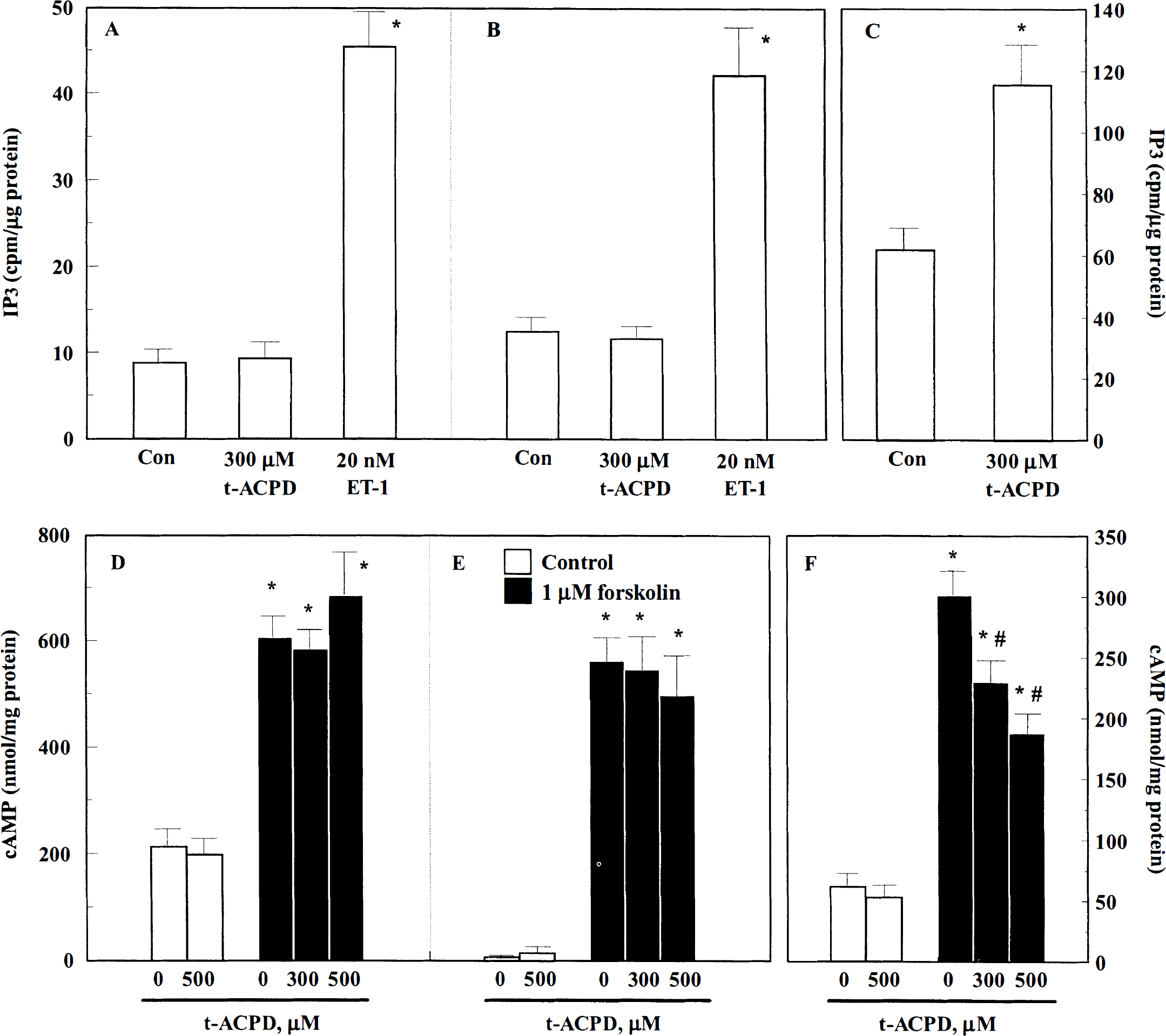
The effect of the metabotropic glutamate receptor agonist 1S,3R-trans-ACPD on IP3 (
In addition to being ineffective in activating phospholipase C, 1S,3R-trans-ACPD also failed to affect either basal or forskolin-stimulated cyclic AMP levels in rat or human CEC [Fig. 3D (human) and E (rat)]. In human CEC, previously shown to express the serotonin 5HT1D receptor (Cohen et al., 1995), 10 μmol/L sumatriptan, a selective antagonist of 5HT1D&F receptors, reduced 1 μmol/L forskolin-stimulated cyclic AMP by 40% (data not shown). However, in cultured rat neuronal cells, 1S,3R-trans-ACPD reduced forskolin-stimulated cyclic AMP (Fig. 3F) in a concentration-dependent manner, suggesting that rat neurons express mGluR2 to mGluR4 and mGluR6 to mGluR8 metabotropic glutamate receptors linked to the inhibition of adenylyl cyclase.
Collectively, these data indicate that there is an apparent lack of functional metabotropic receptor expression in rat and human CEC as determined by the absence of signal transduction upon addition of receptor agonists to CEC.
Immunocytochemical detection of glutamate receptor subunits in cerebromicrovascular endothelial cells and rat neuronal cultures
To investigate whether the lack of signal transduction in response to glutamate agonists in rat and human CEC was due to the lack of one or more glutamate receptor subunits resulting in the nonsignaling receptor(s), we used indirect immunofluorescence microscopy to detect subunits of NMDA and AMPA receptors in CEC and rat cortical neuronal cultures. Both rat and human CEC failed to display any immunopositive reaction to the glutamate receptor subunits tested. An example of the absence of immunoreactivity for the NMDA NR1 subunit, shown to be essential for establishing a functional NMDA receptor (Ishii et al., 1993), is shown in Fig. 4A. Similarly, both rat and human CEC lacked immunoreactivity for NR2A and NR2B subunits (data not shown) or AMPA GluR1, GluR2/3, and GluR4 subunits (data not shown). By contrast, cortical neurons (14 to 18 days in culture) expressed strong immunofluorescent staining for NR1, NR2A, NR2B, GluR1, and GluR2/3 (Fig. 4) subunits associated predominately with the cell bodies and were negative for the GluR4 subunit (Fig. 4B). Negative results were also seen in control cells in which the primary antibody had been omitted (Fig. 4B). The results indicate that rat and human CEC, unlike rat neuronal cultures, do not express immunoreactive NMDA and AMPA glutamate receptor subunits.
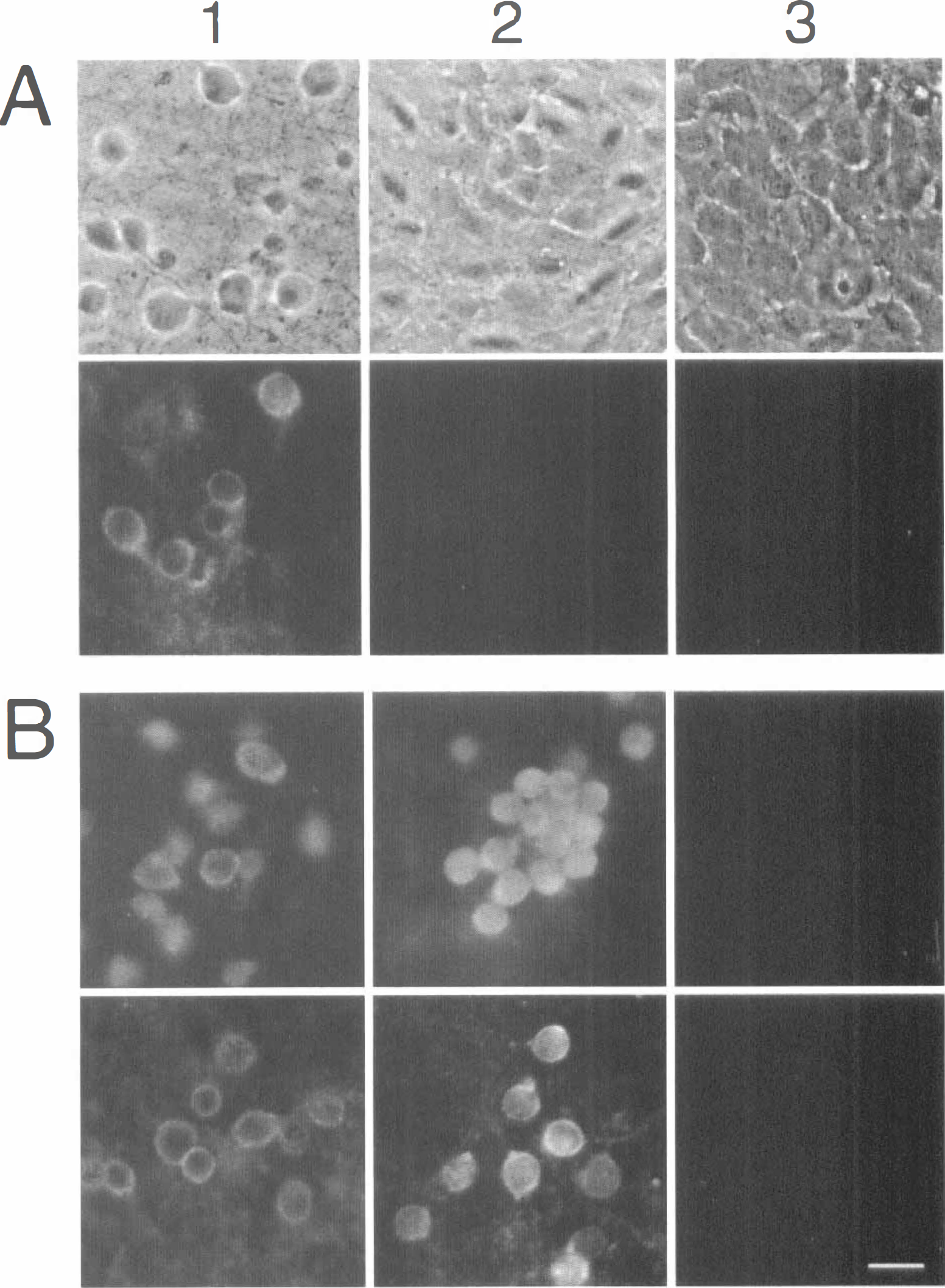
Immunocytochemical detection of glutamate receptor subunits in rat and human CEC and rat cortical neurons in culture. (
Expression of glutamate receptor subunit mRNA in rat or human cerebromicrovascular endothelial cells and rat cortical neurons
To eliminate the possibility that the lack of immunoreactive glutamate receptor subunit expression in rat or human CEC is due to the inability of the antibodies to detect “aberrant” or translationally modified proteins, we used the RT-PCR technique to assay for the presence of mRNA encoding various NMDA, AMPA, and kainate glutamate receptor subunits in rat or human CEC cultures and rat neurons. We were not able to detect mRNA for either NMDA NR11XX, NR10XX, NR2A, NR2B, or NR2C receptor subunits or AMPA GluR1, GluR2, GluR3, or GluR4 receptor subunits in any of the rat or human CEC cultures tested (Fig. 5). The lack of these glutamate receptor subunits was confirmed in both human (Fig. 5) and rat (not shown) CEC cultures derived from either resistance microvessels (112 to 350 μmol/L) or capillaries (20 to 112 μmol/L). All of the NMDA and AMPA receptor subunit mRNAs were also absent in primary cultures of CEC (data not shown), eliminating the possibility that the lack of glutamate receptor subunit mRNAs was due to cell passaging and dedifferentiation in vitro. To address the contingency of receptors being lost due to the enzymatic dissociation of microvessels and culturing conditions, we analyzed freshly isolated microvessels prior to dissociation and plating. As in the cultures, in freshly isolated preparations, no message for NMDA or AMPA receptor subunits was detected (Fig. 5).
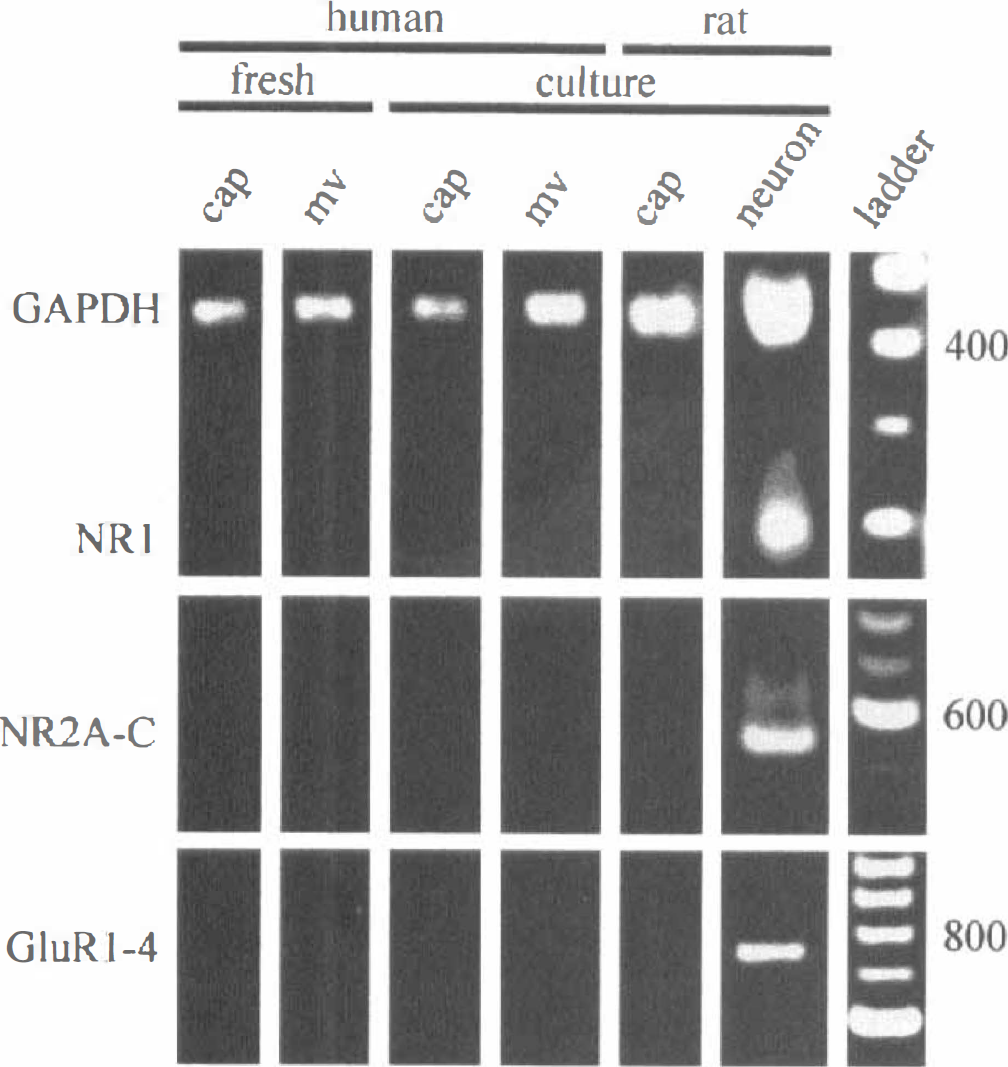
RT-PCR products from mRNA extracted from freshly isolated human capillaries (cap; 20 to 53 μmol/L) and microvessels (mv; 112 to 350 μmol/L), human capillary and microvessel, and rat capillary CEC in culture and fetal rat cortical neurons in culture using primers specific for GAPDH, NR1, NR2A, NR2B, NR2C, GluR1, GluR2, GluR3, and GluR4 receptor subunits. The DNA size marker was 100 bp ladder. The results presented are representative of at least four experiments in different cell isolations. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
The sensitivity of the primer sets was assessed using a dilution series of the full-length clones for the various subunits. The sensitivity of the primer sets was such that the GluR1 to GluR4 primer set detected as few as 103 copies, the NR1 primer set detected as few as 104 copies, and the NR2A to NR2C primer set detected as few as 103 copies (data not shown). To establish whether our inability to detect NMDA and AMPA receptor subunit message in the CEC cultures was due to extracting insufficient quantities of RNA from these preparations, each preparation was probed with a primer set specific for a ubiquitously expressed enzyme, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), using primers GGAGATTGTTGCCATCAACGAC and ATGAGCCCTTCCACAATGCCAAAG. Each preparation probed was positive for GAPDH, suggesting that there was sufficient RNA extracted from the cultures (Fig. 5, top).
Primary rat cortical neuronal cultures were probed for glutamate receptor subunit mRNAs as positive controls. The lack of NMDA and AMPA receptor expression observed in the CEC cultures was in stark contrast to RT-PCR assays performed on the neuronal cultures, in which both AMPA and NMDA receptor subunits were detected (Fig. 5). Although the NR11xx splice variants were detected when probing message that had been pooled from four or more dishes of neurons (data not shown), the predominant splice variants expressed in the rat cortical neurons were those lacking the N-terminal NR10XX (Fig. 5, top). The mRNA encoding NR2 (Fig. 5, middle) and GluR1 to GluR3 (Fig. 5, bottom) subunits was also abundantly expressed in rat neuronal cultures. Control reactions without RT product were always negative (data not shown). The results obtained using RT-PCR are in agreement with the data obtained by immunocytochemistry showing expression of NMDA receptor NR1 and NR2 subunits or AMPA receptor GluR1 to GluR3 subunits in rat cortical neurons but not in rat or human CEC.
When probing for the presence of kainate receptor subunit mRNAs in rat CEC cultures and neurons, we detected mRNA for KA1 or KA2 and GluR5 (Fig. 6) but not GluR6 or GluR7 (data not shown) in both microvascular and capillary CEC. By comparison, KA1 or KA2, GluR5 (Fig. 6), GluR6, and GluR7 (data not shown) subunit mRNA were all detected in rat cortical cultures. The specificity of the primers for rat kainate receptor subunits prevented probing human CEC, but the failure to observe any functional responses to very high concentrations of kainate or AMPA in either the rat or the human microvascular or capillary CEC (Fig. 1) suggests that the KA1, KA2, and GluR5 subunit mRNA expression is either not translated or is posttranslationally modified to render nonfunctional receptors.
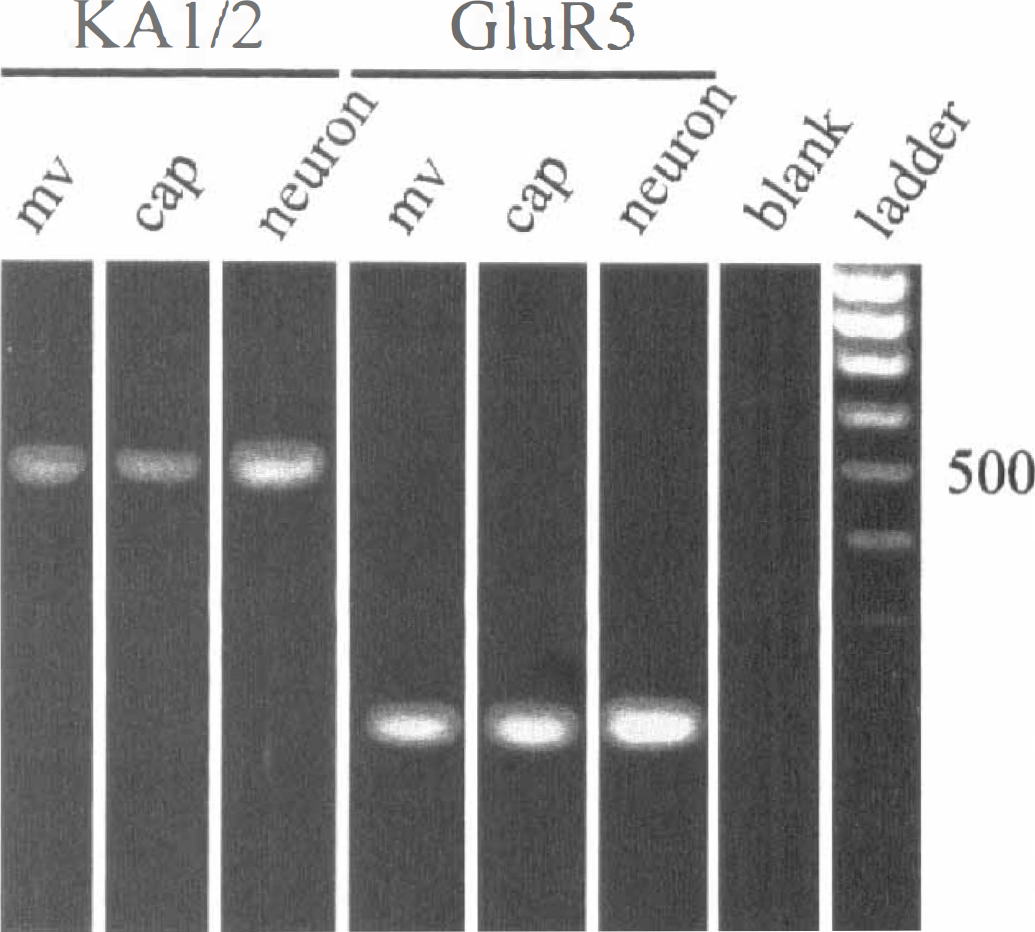
RT-PCR products from mRNA extracted from freshly isolated rat capillaries (cap; 20 to 53 μmol/L) and microvessels (mv; 112 to 350 μmol/L) and rat capillary CEC in culture and fetal rat cortical neurons in culture using primers specific for KA1 and KA2 and GluR5 receptor subunits. The DNA size marker was 100 bp ladder. The results presented are representative of at least four experiments in different cell isolations. See text for abbreviations.
DISCUSSION
This study provides evidence that endothelial cells of cerebral microvessels and capillaries in rat or human do not express functional glutamate receptors. This conclusion is corroborated by the following experimental findings: (a) Neither rat nor human CEC responded to ionotropic glutamate receptor agonists by increasing [Ca2+]i; (b) none of the NMDA or AMPA receptor subunits tested (i.e., NR1, NR2A to NR2C, and GluR1 to GluR4) was detected by either immunocytochemistry or RT-PCR; (c) the metabotropic glutamate receptor agonist 1S,3R-trans-ACPD was incapable of stimulating phospholipid hydrolysis or Ca2+ release from internal stores or inhibiting adenylyl cyclase activity in CEC; and (d) even though the mRNA for KA1 or KA2 and GluR5 was detected in rat CEC, kainate did not elicit measurable Ca2+ increases in either rat or human CEC at very high concentrations. We also demonstrated that the lack of NMDA and AMPA receptor expression in cultured CEC was not due to the enzymatic dissociation and culturing conditions, since mRNAs for NMDA and AMPA receptor subunits were also not detected in freshly isolated microvessels. Moreover, our inability to reveal glutamate receptors or glutamate-induced signaling responses in CEC was not caused by the inadequacy of the methodological approaches used, since in parallel experiments we demonstrated glutamate receptor agonist-induced [Ca2+]i increases, activation of phospholipid turnover, and inhibition of adenylyl cyclase along with the expression of both protein and mRNAs for NMDA and AMPA glutamate receptor subunits in rat neurons in culture. To our knowledge, this is the first study assessing the expression of glutamate receptors in cerebral capillary and microvessel endothelial cells in culture, in isolation from other cellular elements forming the microvascular bed.
The finding that cerebral microvascular endothelial cells lack functional glutamate receptors has important ramifications. Previous studies using isolated cerebral arteries and microvessels have sparked a significant controversy as to what roles endothelial cell glutamate receptors may play in regulating vasomotor cerebrovascular responses. Some studies have shown that both glutamate and NMDA (Busija and Leffler, 1989; Faraci and Breese, 1993) and the noncompetitive NMDA receptor antagonists ketamine (Fukuda et al., 1983; Cavazutti et al., 1987; Wendling et al., 1994) and MK-801 (Perkins et al., 1989) are potent vasodilators of the cerebral vasculature. However, while glutamate and NMD A dilated pial arterioles in situ (Busija and Leffler, 1989; Faraci and Breese, 1993), they had no effect on isolated cerebral vessels in vitro (Fujiwara et al., 1975; Swan et al., 1988; Hardebo et al., 1989; Takayasu and Dacey, 1989; Faraci and Breese, 1993; Wendling et al., 1996), indicating that this vasomotor response was not induced by direct action of glutamate on vascular cellular elements. In support of this view, it has been suggested that the NMDA-induced dilation of cerebral vessels is mediated by nitric oxide released as a result of neuronal activation (Faraci and Breese, 1993; Meng et al., 1995), since it could be inhibited by the nitric oxide synthase inhibitor NG-nitro-
The finding that capillary CEC do not express functional glutamate receptors has implications beyond those concerned with the regulation of vascular tone in the microvascular bed. Capillary cerebral endothelial cells form the BBB, and glutamate released by neurons in the vicinity of cerebral capillaries during cerebral ischemia or seizures may significantly affect BBB integrity and the formation of vasogenic brain edema. The studies of Koenig et al. (1992) using an in vivo model of vasogenic brain edema following cryogenic injury and an in vitro model of isolated cerebral microvessels have suggested that glutamate regulates the breakdown of the BBB by triggering the activation of the ornithine decarboxylase/polyamine cascade in cerebral endothelial cells. Furthermore, the same group implied that the activation of the polyamine cascade and BBB breakdown were mediated by a direct action of glutamate on endothelial NMDA receptors, since it was inhibited by the competitive NMDA receptor antagonist 2-amino-5-phosphonopentanoic acid and because their capillary preparations were reportedly devoid of glia, neurons, synaptosomes, and synaptic complexes (Koenig et al., 1992). These studies were based largely on indirect evidence of glutamate being able to activate a polyamine metabolic cascade known to be stimulated by various other diffusible biochemical stimuli including arachidonic acid (Chan et al., 1983; Pappius and Wolfe, 1983), prostaglandins (Pappius and Wolfe, 1983), diacylglycerol (Politi et al., 1985), and free oxygen radicals (Chan et al., 1984). Therefore, these results are difficult to reconcile with the evidence presented in this study of the absence of NMDA and other glutamate receptors in both cultured endothelial cells and freshly isolated microvessels. The discrepancy between the two studies may be explained by a possible non-receptor-mediated action of glutamate or NMDA on CEC or by the unlikely presence of “atypical” NMDA receptors in CEC that we were not able to detect by the methods used. Nevertheless, based on our data, it appears highly unlikely that glutamate is capable of affecting BBB opening by acting directly on cerebral capillary endothelial cells. Therefore, it appears that glutamate involvement in BBB opening during pathological events that stimulate glutamate release such as stroke, brain trauma, and epilepsy is largely caused by the paracrine effects of secondary mediators stimulated and released by surrounding glutamate receptor-expressing cells. The nature of these secondary mediators is presently unknown but may include compounds such as reactive oxygen species, eicosanoids, polyamines, and nitric oxide, all of which have been shown to induce BBB opening in vivo and/or in vitro (Black, 1995).
In conclusion, our results demonstrate the absence of functional glutamate receptor expression in rat and human microvascular and capillary CEC and suggest that glutamate, even at the high extracellular concentrations found in cerebral ischemia (Hagberg et al., 1987), is unlikely to affect microvessel tone or BBB permeability by acting directly on endothelial cells. Therefore, the neuroprotective effects of glutamate receptor antagonists developed for the clinical treatment of epilepsy, stroke, and head injury (Muir and Lees, 1995) are apparently not mediated through sites of action on cerebral microvessels.
Footnotes
Abbreviations used
Acknowledgments
The authors thank Ms. Rita Ball and Mr. Roger Tremblay for performing isolations of rat and human CEC and rat cortical neurons, respectively; Mr. Zvi Cohen for supplying the cDNA made from RNA extracted from the freshly isolated human microvessels; Dr. Suzanne Zukin for the NR1b (NR1100) clone, Dr. Shigetada Nakanishi for the NR2A and C clones, and Dr. Stephen Heinemann for the NR2B, GluR1, GluR2, and GluR4 clones.