
Abstract
Using homozygous human apolipoprotein E2 (apoE2) (2/2)-, apoE3 (3/3)-, or apoE4 (4/4)-knock-in (KI) mice, we have shown that delayed infarct expansion and reactive astrocytosis after permanent middle cerebral artery occlusion (pMCAO) were markedly exacerbated in 4/4-KI mice as compared with 2/2- or 3/3-KI mice. Here, we probed the putative causal relationship between enhanced astrocytic activation and exacerbation of brain damage in 4/4-KI mice using arundic acid (ONO-2506, Ono Pharmaceutical Co. Ltd), which is known to oppose astrocytic activation through its inhibitory action on S100B synthesis. In all of the KI mice, administration of arundic acid (10 mg/kg day, intraperitoneal, started immediately after pMCAO) induced significant amelioration of brain damage at 5 days after pMCAO in terms of infarct volumes (results expressed as the mean infarct volume (mm3) ±1s.d. in 2/2-, 3/3-, or 4/4-KI mice in the vehicle groups: 16±2, 15±2, or 22±2; in the arundic acid groups: 11±2 (P<0.001), 11±2 (P<0.001), or 12±2 (P<0.001), as compared with the vehicle groups), neurologic deficits, and S100/glial fibrillary acidic protein burden in the peri-infarct area. The beneficial effects of arundic acid were most pronounced in 4/4-KI mice, wherein delayed infarct expansion together with deterioration of neurologic deficits was almost completely mitigated. The above results support the notion that the apoE4 isoform exacerbates brain damage during the subacute phase of pMCAO through augmentation of astrocytic activation. Thus, pharmacological modulation of astrocytic activation may confer a novel therapeutic strategy for ischemic brain damage, particularly in APOE ɛ4 carriers.
Introduction
Since the establishment of the apolipoprotein E (APOE) ɛ4 allele (APOE, Apoe, and apoE denote the human gene, mouse gene, and protein, respectively) as a salient risk factor for both late onset and sporadic Alzheimer's disease (Corder et al, 1993), the presence of the APOE ɛ4 allele has been shown to worsen the outcome of other central nervous system (CNS) disorders such as cerebral trauma (Friedman et al, 1999; Nicoll et al, 1995; Sorbi et al, 1995; Teasdale et al, 1997), intracerebral hemorrhage (Alberts et al, 1995), and subarachnoid hemorrhage (Leung et al, 2002). Apolipoprotein E is mainly produced by astrocytes in the CNS (Fujita et al, 1999), and it has been shown to modulate the endogenous CNS inflammatory response in an isoform-specific manner (Barger and Harmon, 1997; Laskowitz et al, 2001; Lynch et al, 2001, 2003). While an association between the APOE ɛ4 allele and exacerbation of cerebral infarction has been supported by some clinical studies (Kim et al, 2003; Margaglione et al, 1998; McCarron et al, 1999), contradictory results have also been reported (Basun et al, 1996; Frikke-Schmidt et al, 2001; Zhu et al, 2000). Based on a reduced frequency of the APOE ɛ4 allele in patients who had survived a previous stroke (Basun et al, 1996), Laskowitz et al (1998) surmised that APOE genotype may not alter risk for stroke, but it may alter recovery once it occurs. This view is bolstered by results of experimental studies using genetically engineered mice, where the apoE4 isoform was shown to retard recovery from a variety of cerebral insults (Buttini et al, 1999, 2000; Horsburgh et al, 2000; Mori et al, 2003, 2004b; Sheng et al, 1998; White et al, 2001).
In particular, our previous studies using homozygous human apoE2 (2/2)-, apoE3 (3/3)-, or apoE4 (4/4)-knock-in (KI) mice demonstrated that the apoE4 isoform acts to exacerbate brain damage in an isoform-specific manner (apoE4 >apoE3= apoE2=wild-type background strain), not only in the acute phase (<24 h) (Mori et al, 2003) but also in the subacute phase (1 to 7 days) after permanent middle cerebral occlusion (pMCAO) (Mori et al, 2004b). Of particular importance is our finding that, in 4/4-KI mice, the infarct volume showed a progressive increase (delayed infarct expansion) from 1 to 5 days after pMCAO, and was accompanied by a further worsening of neurologic deficits.
In the above study (Mori et al, 2004b), 4/4-KI mice exhibited a progressive increase in glial fibrillary acidic protein (GFAP) burden in the peri-infarct area through 7 days after pMCAO that was significantly correlated with the progression of delayed infarct expansion. In 2/2- and 3/3-KI mice, by contrast, the increase in GFAP burden in the peri-infarct area reached a plateau at 3 days after pMCAO, and was not accompanied by any significant increase in the infarct volume until 7 days. Given the above observations, we hypothesized that a causal relationship may exist between enhanced astrocytic activation and exacerbation of brain damage in 4/4-KI mice. This is consonant with the past literature showing that apoE modulates glial activation in an isoform-specific manner (Barger and Harmon, 1997; Laskowitz et al, 2001; Lynch et al, 2001, 2003), and that endogenous CNS inflammatory response leads to exacerbation of brain damage (Benveniste, 1995; Griffin et al, 1998; Kreutzberg, 1996; Ridet et al, 1997; Wyss-Coray and Mucke, 2002).
To examine the above hypothesis, we use arundic acid (ONO-2506, Ono Pharmaceutical Co. Ltd., Osaka, Japan) in the present study for the following reasons. First, S100B plays a cardinal role in the ‘vicious cytokine cycle’ incurred by glial activation (Griffin et al, 1998; Mrak et al, 1995; Van Eldik and Griffin, 1994), and arundic acid has been shown to exert an inhibitory effect on astrocytic S100B synthesis in vitro (Shinagawa et al, 1999). Second, with regard to cerebral ischemia, administration of arundic acid in the rat pMCAO model has been shown to significantly inhibit augmented expression of S100B and GFAP in the peri-infarct area, an effect that was accompanied by both mitigation of delayed infarct expansion and neurologic deficits (Tateishi et al, 2002).
In the present study, 2/2-, 3/3-, or 4/4-KI mice subjected to pMCAO were randomly allocated to experimental groups that were treated with arundic acid or vehicle. These mice were subsequently examined for alterations in infarct volumes and neurologic deficits as well as S100/GFAP burden in the peri-infarct area during the acute (<24 h after pMCAO) and the subacute phases of ischemia. In 4/4-KI mice, the tissue levels of S100B in the peri-infarct area, which were determined by the sandwich enzyme-linked immunosorbent assay method at 3 days after pMCAO, were compared between the arundic acid and the vehicle groups. Our results show that both delayed infarct expansion and enhanced astrocytic activation in the peri-infarct area through 5 days after pMCAO were virtually abolished in 4/4-KI mice by administration of arundic acid, bolstering the view that astrocytic activation is augmented by the apoE4 isoform and acts to exacerbate brain damage.
Materials and methods
Apolipoprotein E-Knock-In Mice
Ten-week-old weight-matched male 2/2-, 3/3-, or 4/4-KI mice were used in this study. In 4/4-KI mice, a portion of Apoe has been replaced by a transgene consisting of human apoE4 cDNA through homologous recombination in embryonic stem cells such that human apoE proteins are expressed under the endogenous regulatory apoE promoter region (Hamanaka et al, 2000). Both 2/2- and 3/3-KI mice were produced using the same strategy as above except that the transgenes carried apoE2 or apoE3 cDNA in place of apoE4 cDNA. All of the KI mice used in this study were fully backcrossed onto the C57BL/6N background. The nucleotide sequences of the transgene apoE cDNAs were confirmed by sequencing cDNAs prepared from liver polyA+ RNAs of the three homozygous strains. Homozygosity was confirmed in each line of KI mice using allele-specific oligonucleotide primers and polymerase chain reaction analysis. These KI mice entirely lack mouse apoE. Expression levels of human apoE in neurons and in astrocytes, as well as the architecture of cerebral arteries including the presence or the absence of the posterior communicating artery, were comparable among the three lines of KI mice (Mori et al, 2003, 2004b).
Surgical Procedures and Neurologic Evaluation
The procedures followed were in accordance with the guidelines of the Animal Use Ethics Committee of the Saitama Medical Center/School and NIH guidelines (DHHS publication No. [NIH] 85-23, revised 1985). All efforts were made to minimize animal suffering and to reduce the number of animals used. Animals were housed in a virus-free barrier facility under a 12/12 h light—dark cycle, with ad libitum access to food and water. All of the KI mice were subjected to fasting overnight (12 h) with free access to water before surgical procedures. Anesthesia was induced and maintained with halothane (1.5% to 2% and 0.5%, respectively) in a mixture of 70% nitrous oxide and 30% oxygen with spontaneous ventilation. As repeated blood withdrawal is likely to affect the outcome of pMCAO, all parameters (PaO2, PaCO2, pH, MABP, and blood glucose) were examined in separate sets of the arundic acid and the vehicle groups for 2/2-, 3/3-, or 4/4-KI mice (n=6 per each line of KI mice for each group, total n=36) under halothane anesthesia with spontaneous ventilation as described above. The femoral artery was cannulated to monitor MABP and to collect a blood sample. The above-mentioned physiologic parameters were recorded at 15 mins before (except for PaO2, PaCO2, and pH) and 30 mins after pMCAO. Rectal temperature was monitored throughout the operative procedure using a rectal probe, and normothermia was maintained with a homeothermic blanket control unit that was preset to 37°C. The distal segment of the middle cerebral artery (MCA) crossing over the rhinal fissure was exposed for induction of pMCAO as described elsewhere (Mori et al, 2004b). Briefly, a 1-cm skin incision was made approximately midway between the left outer canthus and anterior pinna. The temporalis muscle was incised and retracted to expose the squamous portion of the temporal bone. Under a surgical microscope, a burr hole (2 mm in diameter) was made by an electrical drill over the junction of the zygomatic process and the temporal bone. The dura mater was opened with a fine curved needle to expose the MCA. The left common carotid artery (CCA) was ligated with 8-0 silk, and then a 2-mm segment of the MCA was electrocauterized. The coagulated MCA segment was then transected with microscissors. Thereafter, the burr hole was covered with the temporalis muscle. After the skin was approximated, the wound was infiltrated with lidocaine. After halothane was discontinued, mice were returned to their cages and allowed free access to food and water. Evaluation of neurologic deficits was performed at 24-h intervals after pMCAO until euthanasia as follows: score 0, no neurologic deficit; score 1, forelimb flexion; score 2, decreased resistance to lateral push and forelimb flexion without circling; score 3, same behavior as grade 2, with circling, and score 4, inability to walk spontaneously. A single investigator, who was masked to the treatment and the animal genotype, performed the neurologic evaluation every 24 h after pMCAO until the animals were euthanized.
Experimental Groups
Arundic acid [ONO-2506, (R)-(−)-2-propyloctanoic acid] is a novel agent developed by Ono Pharmaceutical Co. Ltd. The compound's general pharmacological action on cultured astrocytes or cocultures of neurons and astrocytes has been described elsewhere (Tateishi et al, 2002). A total of 120 2/2, 3/3, or 4/4-KI mice were subjected to pMCAO and were assigned either to the arundic acid or to the vehicle groups to be euthanized at 1 or 5 days after pMCAO (n=10 for each line of KI mice for each group per day). Intraperitoneal administration of arundic acid (at a daily dose of 10 mg/kg) or vehicle (saline) was started immediately after pMCAO and repeated every 24 h until the animals were euthanized. In the previous study using the rat pMCAO model, the beneficial effects of arundic acid in terms of amelioration of infarct volumes and neurologic deficits were dose-dependent, and its optimal dose was determined to be 10 mg/kg (intravenous bolus administration, once a day). Because the pharmacodynamics of the agent were similar between the rat and the mouse regardless of the route of administration (intravenous or intraperitoneal) (unpublished data), the present study used the above dose of arundic acid administered by intraperitoneal injection.
Determination of Brain Damage
At 1 and 5 days after pMCAO, mice were reanesthetized as described above, and euthanized by transcardial perfusion of 200 mL of 10 U/mL heparin in saline followed by 200 mL of 4% paraformaldehyde in 0.1 mol/L (pH 7.4) phosphate-buffered saline (PBS). The brain was removed and fixed in the same fixative as above at 4°C for 48 h. Then, the bilateral cerebral hemispheres were embedded in paraffin with 48 h of processing. Serial sections (5-μm in thickness) of the cerebral hemispheres at six predetermined coronal planes separated by 1-mm intervals were sequentially labeled as sections 1 to 6 and stained with hematoxylin and eosin (H&E) or cresyl violet. The infarct area in each section was measured using a computer-based image analyzer (Scion Image beta 4.02 for Windows, Scion Corporation, Frederic, MD, USA). To exclude the effects of brain edema, the infarct area was corrected by the ratio of the whole area of the ipsilateral hemisphere to that of the contralateral hemisphere. Since the interval between sections was 1 mm, the infarct volume (mm3) was calculated as the running sum of corrected infarct area in all six slices. Measurements were performed in a masked manner by a single investigator.
Immunohistochemistry
Additional sections adjacent to the coronal brain slice at the level of the anterior commissure (section No. 3) were used for immunohistochemistry. Detection of S100 and GFAP was performed according to the manufacturer's protocol using a Vectastain ABC Elite kit (Vector Laboratories, Burlingame, CA, USA) coupled with the diaminobenzidine reaction. Rabbit polyclonal anti-S100 and anti-GFAP antibodies (ready to use and diluted 1:1000, respectively; incubated at 4°C overnight, DAKO, Carpinteria, CA, USA) were used as primary antibodies; hence, the designation of ‘S100’ was used to describe the corresponding results. Phosphate-buffered saline (0.1 mol/L (pH 7.4)) or normal rabbit serum (isotype control) was used instead of primary antibody or ABC reagent as a negative control.
Image Analysis
Images were acquired using an Olympus BX60 microscope with an attached digital camera system (DP-50, Olympus, Tokyo, Japan), and the digital image was routed into a Windows PC for quantitative analysis using SimplePCI software (Compix, Inc. Imaging Systems, Cranberry Township, PA, USA). As shown in the inset of Figures 5 and 8, the regions of interest (ROI) within the cortex abutting the outer border of the infarct were subdivided into five different subfields (604800 pixels per field), from which images were captured via a × 20 objective lens with a threshold optical density that discriminated staining from background. Each ROI was manually edited to eliminate artifacts. S100 or GFAP burden in the peri-infarct area is presented as the percentage of immunolabeled area captured (positive pixels) divided by the full area captured (total pixels). Each analysis was performed in a masked manner by a single investigator.
Effects of Arundic Acid on the Tissue Levels of S100B in the Peri-Infarct Area
In a separate experiment, 4/4-KI mice were subjected to pMCAO and randomly allocated to the arundic acid (10 mg/kg, intraperitoneal administration) or the vehicle groups to be euthanized at 3 days after pMCAO (n=10 each group). Administration of arundic acid was started immediately after pMCAO and repeated every 24 h until the animals were euthanized. At 3 days after pMCAO, mice were reanesthetized with halothane as described above, and perfused via the transcardial route with 200 mL of 10 U/mL heparin in saline. The coronal brain slice encompassing the anterior commissure (2 mm in thickness) was dissected. Under a surgical microscope, the infarct area could be discerned on account of its whitish appearance and the peri-infarct area in the vicinity of the outer border of the infarct was dissected on the cold plate. The location of tissue sampling is shown in the inset of Figure 6. The tissue levels of S100B in the peri-infarct area were measured as described elsewhere with modifications (Tateishi et al, 2002). Briefly, S100B was extracted with 0.15% SDS solution containing 2 mmol/L EDTA and measured by the sandwich enzyme-linked immunosorbent assay method using two different antibodies. Microtiter plates coated with anti-S100B antibody (diluted 1:1,000, clone: SH-B4, Sigma, St Louis, MO, USA) were incubated overnight with standard bovine S100B protein (0.01 to 600 ng/mL, Calbiochem, La Jolla, CA, USA) or samples at 4°C before the plates were further incubated with anti-S100 antibody (diluted 1:1,000; DAKO, Carpinteria, CA, USA) at 23°C to 25°C for 2 h. After the plates had been washed, horseradish peroxidase was added, and the plates were incubated at 23°C to 25°C for 2 h. After extensive washing, horseradish peroxidase substrate was finally added, and the plates were incubated for 5 mins. The resulting absorbance was measured at an optical density of 412 nm.
Statistical Analysis
Data are presented as the mean±1 or +1s.d. The Kolmogorov—Smirnov test was used to determine the normality of the data. If data were found not to be normally distributed and/or were ordinal level, the nonparametric Kruskal—Wallis H test was performed followed by post hoc testing using the Mann—Whitney U-test. If data were found to be normally distributed and were interval or ratio level, statistical analysis was performed using parametric one-way analysis of variance (ANOVA), followed by post hoc comparison of the means using Bonferroni's or Dunnett's T3 methods (determined using Levene's test for equality of the variance) or t-test for independent samples as appropriate. In instances where multiple comparisons of the means were performed using the Mann—Whitney U-test or t-test for independent samples as post hoc analyses, Bonferroni's family-wise correction (P value/number of comparisons) was used to correct for multiple testing. Bivariate correlations were performed using Pearson's product-moment correlation coefficient (Pearson's R). P-values of less than 0.05 were considered to be significant. All analyses were performed using the Statistical Package for the Social Sciences, release 11.0 (SPSS, Chicago, IL, USA).
Results
Evaluation of Physiologic Parameters During Surgery
There were no significant differences in blood gases, MABP, blood glucose, or body temperature between the arundic acid and the vehicle groups, or among the three lines of KI mice, as shown in Table 1.
Intraoperative physiologic variables
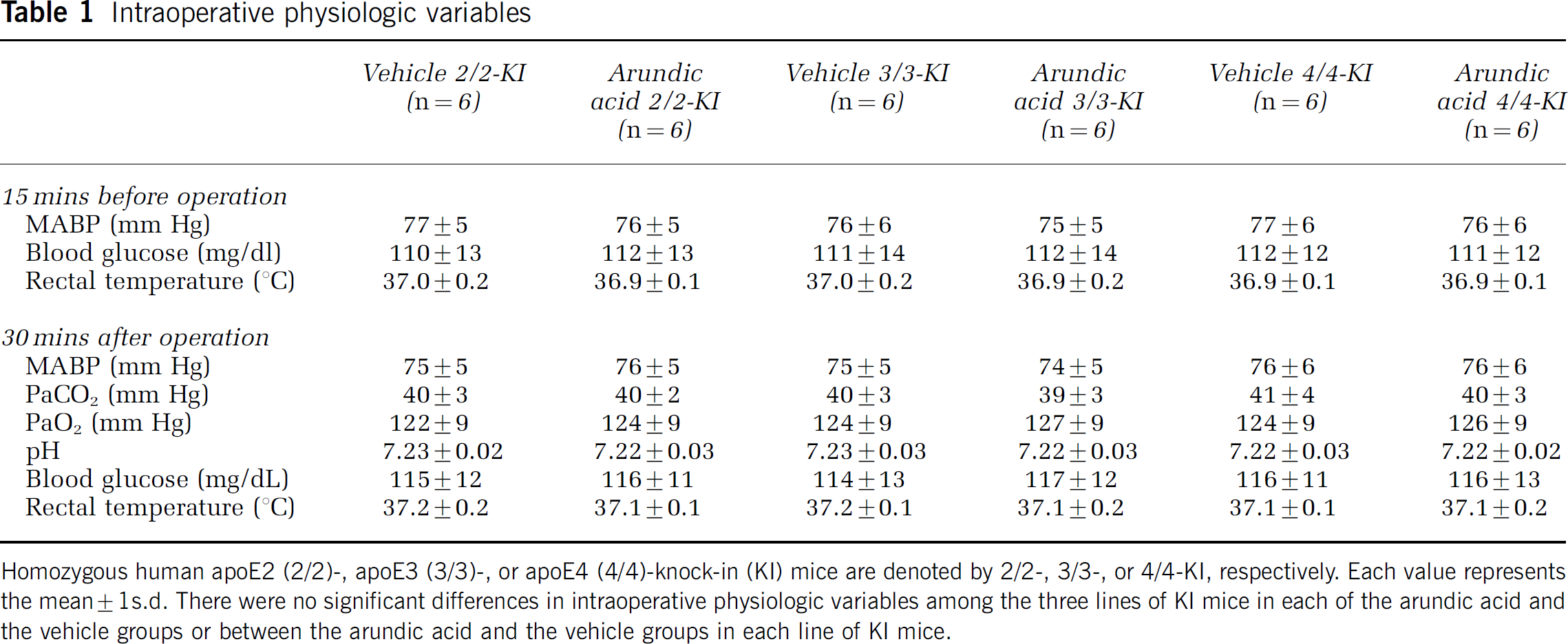
Homozygous human apoE2 (2/2)-, apoE3 (3/3)-, or apoE4 (4/4)-knock-in (KI) mice are denoted by 2/2-, 3/3-, or 4/4-KI, respectively. Each value represents the mean±1s.d. There were no significant differences in intraoperative physiologic variables among the three lines of KI mice in each of the arundic acid and the vehicle groups or between the arundic acid and the vehicle groups in each line of KI mice.
Effects of Arundic Acid on Brain Damage after Permanent Middle Cerebral Artery Occlusion in Apolipoprotein E-Knock-In Mice
At post-mortem examination, neither subarachnoid hemorrhage nor subdural/intracerebral hematoma was observed in any of the KI mice subjected to pMCAO. Mortality was not encountered before the end of the experiment. All of the KI mice consumed food and water normally.
On microscopic examination, extensive pallor, highlighted in H&E- or cresyl violet-stained specimens, was observed in the ipsilateral cortex at 1 and 5 days after pMCAO, and there was a slight extension of pallor into the adjacent white matter and the outer portion of the basal ganglia in a few 4/4-KI mice at 5 days after pMCAO. As shown in Figures 1A and 1B, infarct volumes in 4/4-KI mice were significantly larger than that in 2/2- (P<0.01 at 1 day; P<0.001 at 5 days) or 3/3-KI mice (P<0.001 at 1 and 5 days) in the vehicle groups, whereas no significant differences were detected between 2/2- and 3/3-KI mice at 1 and 5 days after pMCAO (results expressed as the mean infarct volume (mm3) ±1s.d. in 2/2-, 3/3-, or 4/4-KI mice after pMCAO in the vehicle groups: at 1 day, 14±2, 13±2, or 17±2; at 5 days, 16±2, 15±2, or 22±2). Administration of arundic acid significantly reduced infarct volumes in 2/2- (P<0.01 at 1 day; P<0.001 at 5 days), 3/3- (P<0.05 at 1 day; P<0.001 at 5 days), or 4/4-KI mice (P<0.001 at 1 and 5 days), as compared with the vehicle groups. In the arundic acid groups, no significant differences in infarct volumes were present among the three lines of KI mice at 1 or 5 days after pMCAO (results expressed as the mean infarct volume (mm3) ±1s.d. in 2/2-, 3/3-, or 4/4-KI mice after pMCAO in the arundic acid groups: at 1 day, 11±2, 11±2, or 12±1; at 5 days, 11±2, 11±2, or 12±2). The significant increase in infarct volumes that was observed at 5 days after pMCAO in the vehicle group of 4/4-KI mice (P<0.05 versus 1 day) did not occur in the arundic acid group of 4/4-KI mice, showing that administration of the agent almost completely mitigated delayed infarct expansion.
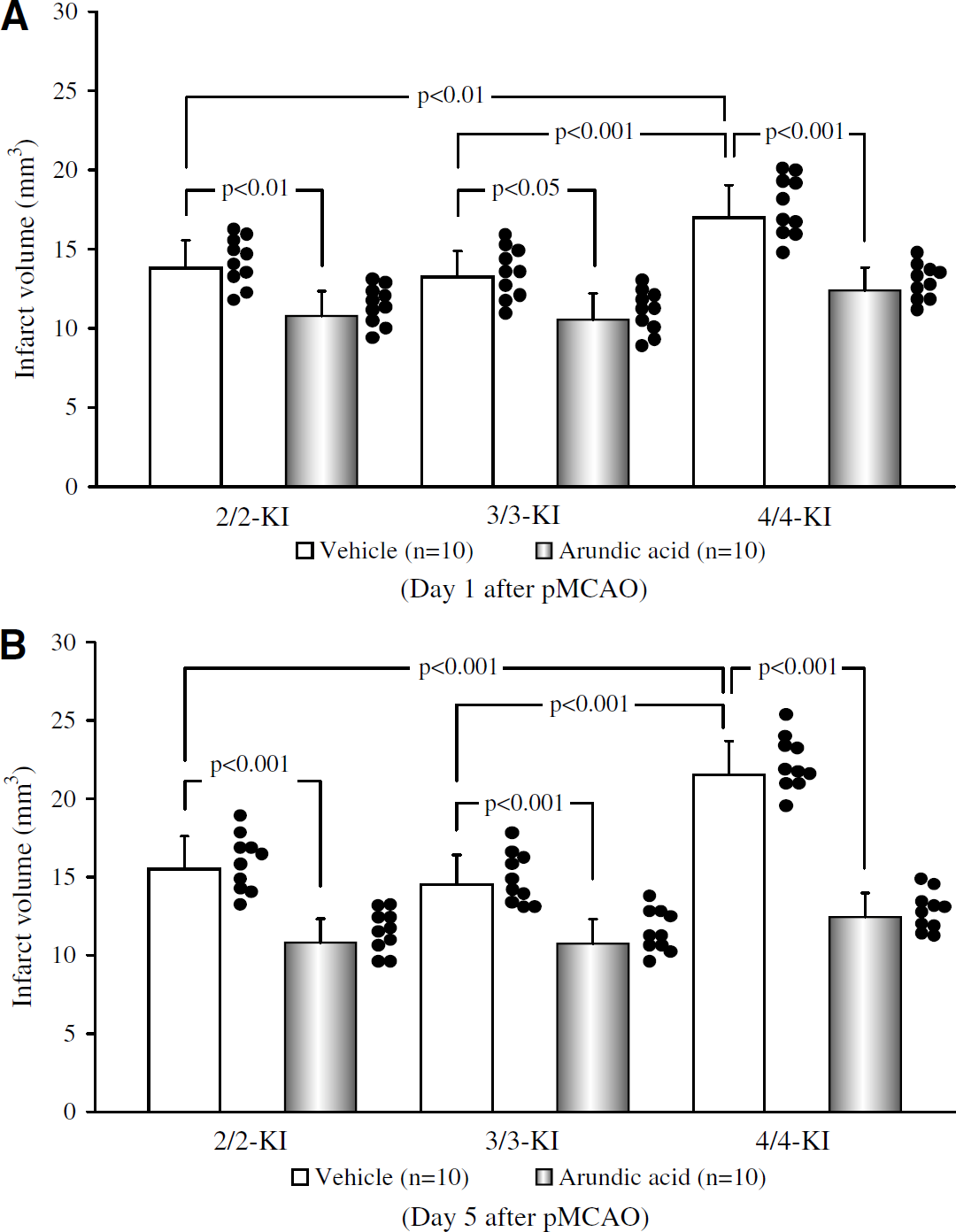
Effects of arundic acid on infarct volumes at 1 (
In each of the KI mouse groups examined, section No. 3 exhibited the largest infarct area of all the sections at 5 days after pMCAO, when infarct volumes became maximal (Figures 2A–2F). In the majority of the sections, administration of arundic acid significantly decreased infarct areas in 2/2- (P<0.01 in sections 1 to 3, 5 and 6; P<0.001 in section 4), 3/3- (P<0.01 in section 3; P=0.001 in section 4), or 4/4-KI mice (P<0.01 in section 1 and P<0.001 in sections 2 to 6), as compared with the vehicle groups (Figures 3A–3C). In the arundic acid groups, no significant differences were found in infarct areas among the three lines of KI mice in any of the section planes examined.
Hematoxylin and eosin staining in coronal brain slices at the level of the anterior commissure (section No. 3) at 5 days after permanent middle cerebral artery occlusion (pMCAO) in homozygous human apoE2 (2/2)-, apoE3 (3/3)-, or apoE4 (4/4)-knock-in (KI) mice. In the vehicle groups (
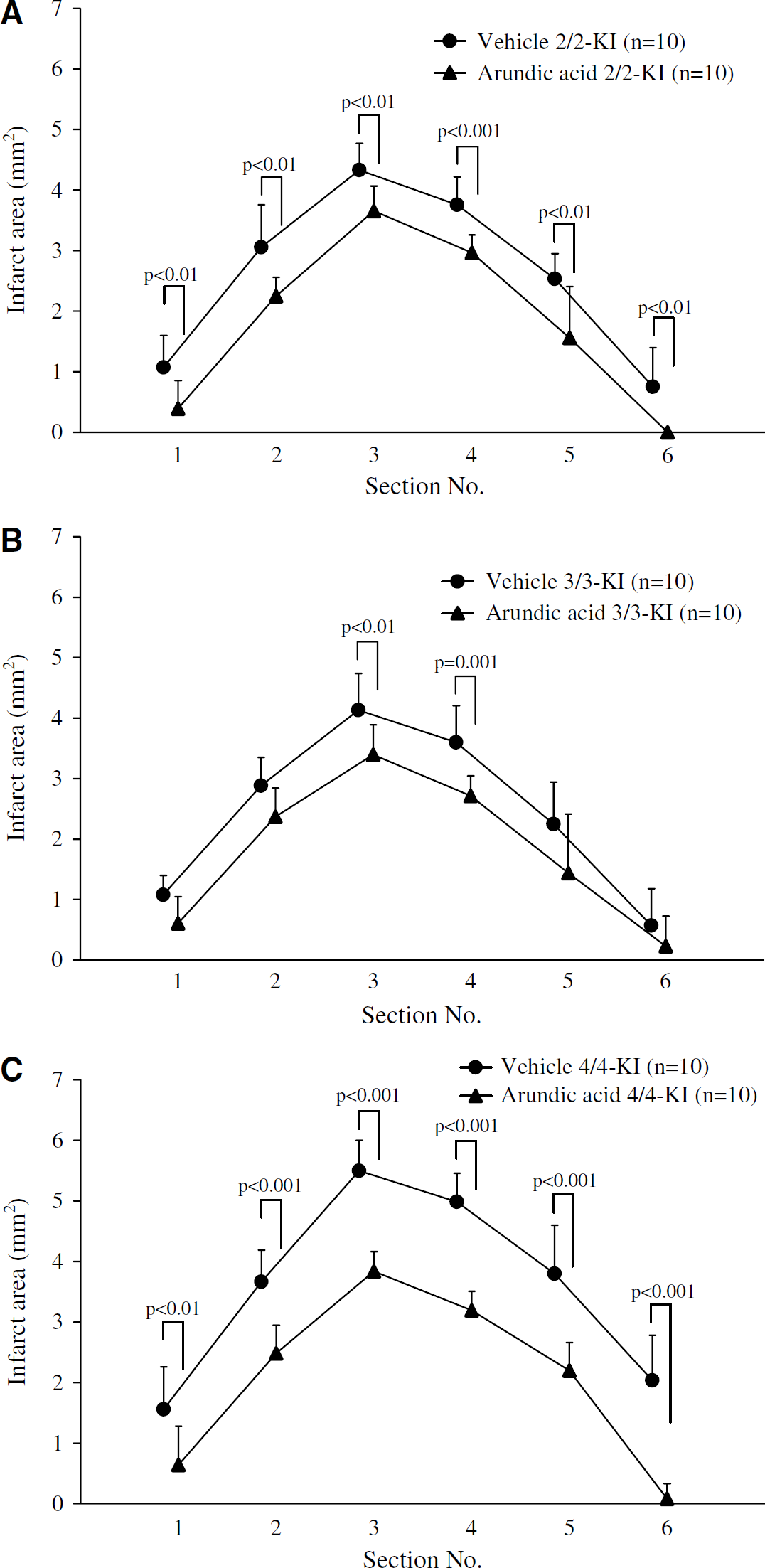
Effects of arundic acid on infarct areas in each coronal plane section at 5 days after permanent middle cerebral artery occlusion (pMCAO) in homozygous human apoE2 (2/2)-, apoE3 (3/3)-, or apoE4 (4/4)-knock-in (KI) mice (
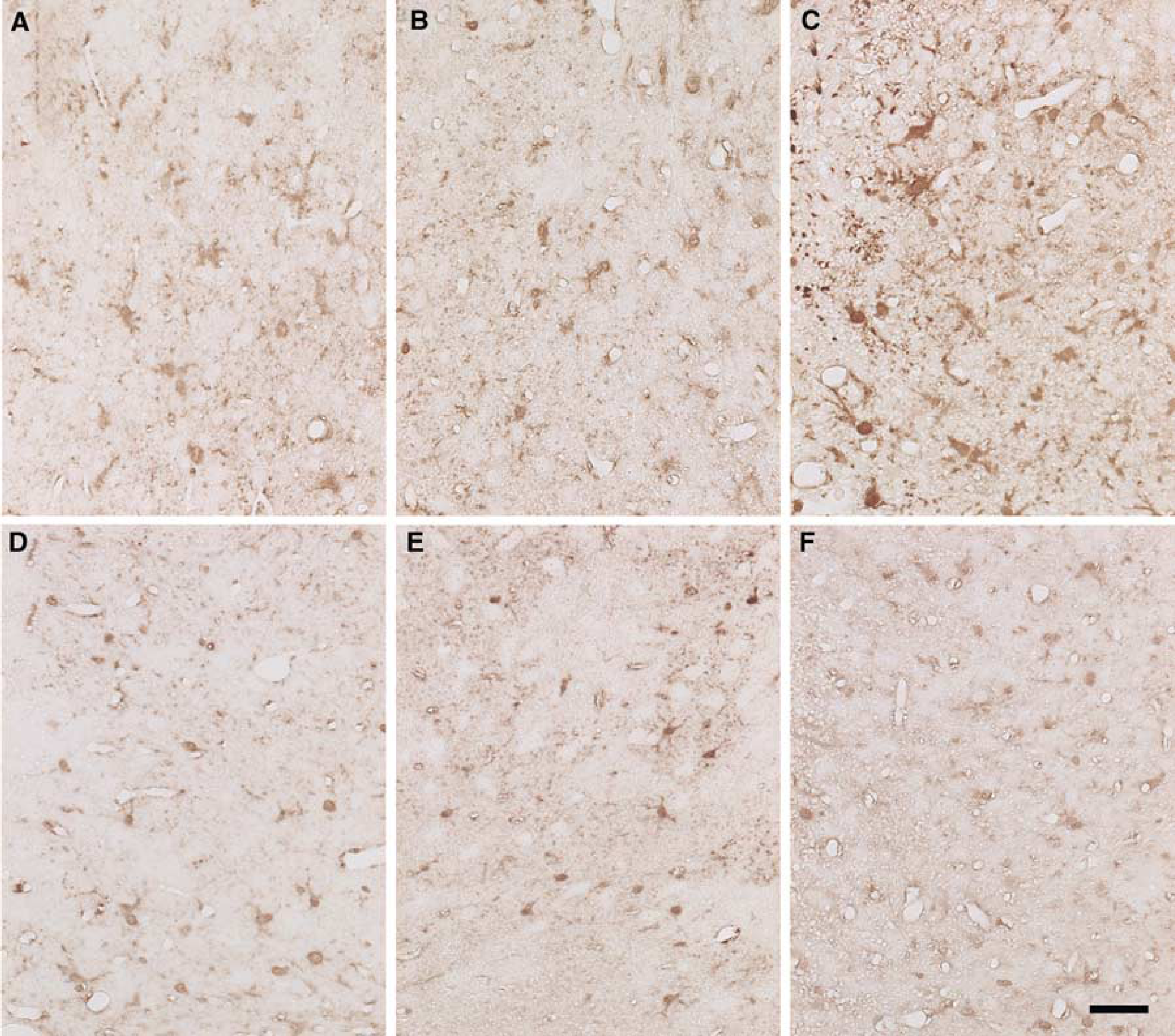
S100 immunohistochemical staining in the peri-infarct area at 5 days after permanent middle cerebral artery occlusion (pMCAO) in homozygous human apoE2 (2/2)-, apoE3 (3/3)-, or apoE4 (4/4)-knock-in (KI) mice. In the vehicle (
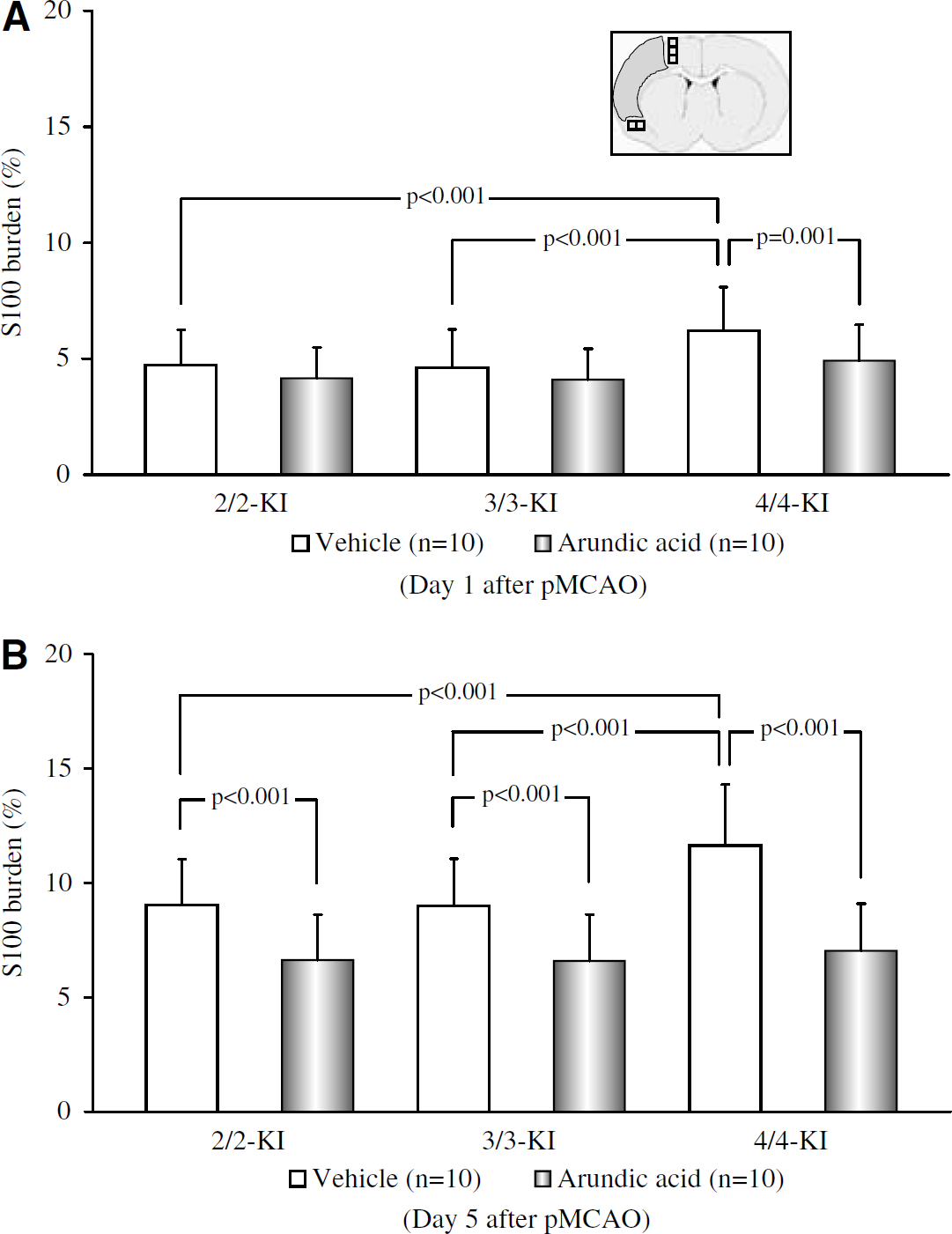
Quantitative assessment of S100 burden in the peri-infarct area at 1 (
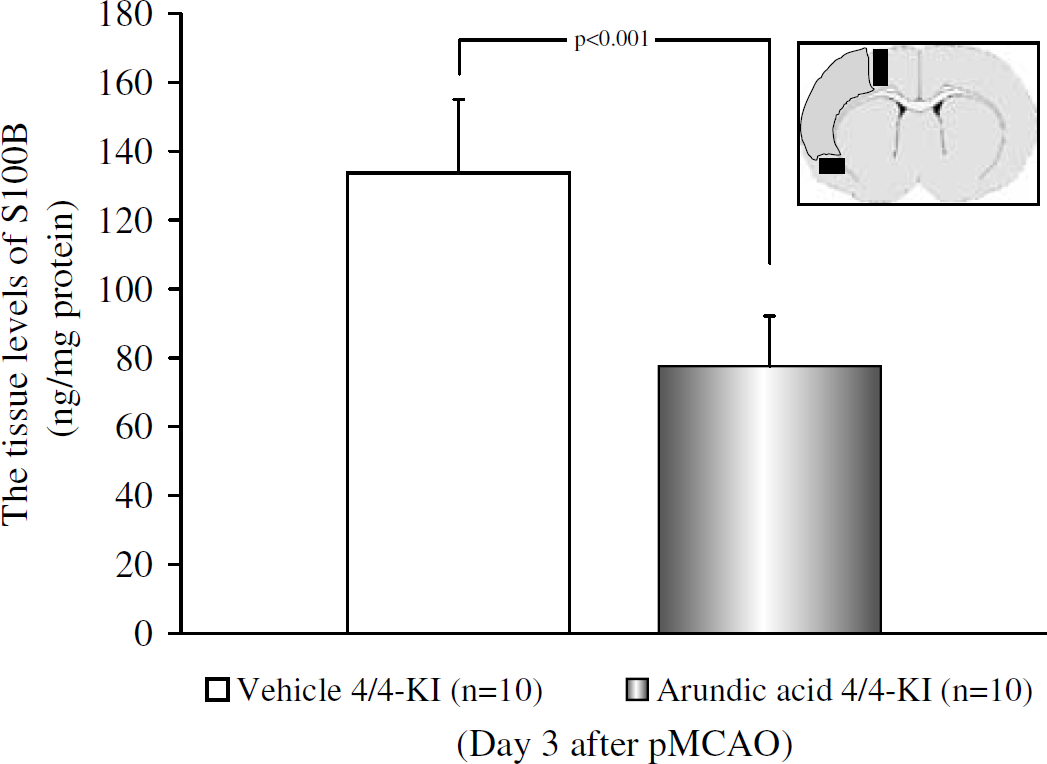
Effects of arundic acid on the tissue levels of S100B in the peri-infarct area at 3 days after permanent middle cerebral artery occlusion (pMCAO) in homozygous human apoE4(4/4)-knock-in (KI) mice in the arundic acid and the vehicle groups. Each value represents the mean+1s.d. Two closed rectangles at the level of the anterior commisure anterior commisure in the inset of the coronal brain section diagram designate the location of sampling in the peri-infarct area along the outer border of the infarct. Values obtained in the arundic acid and the vehicle groups were compared.
Effects of Arundic Acid on Neurologic Deficits after Permanent Middle Cerebral Artery Occlusion in Apolipoprotein E-Knock-In Mice
Neither atypical neurologic deficits (torsion of the neck or rolling fits) nor severe neurologic scores (score 4) were encountered in any of the KI mice examined. At every time-point examined, neurologic scores in the vehicle groups were significantly higher in 4/4-KI mice than in 2/2- (P=0.01 at 2 to 5 days; P<0.001 at 1 day) or 3/3-KI mice (P⩽0.01 at 2 to 5 days; P<0.001 at 1 day), whereas no significant differences were detected between 2/2- and 3/3-KI mice. Administration of arundic acid significantly improved neurologic deficits in 2/2- (P<0.01 at 4 and 5 days), 3/3- (P<0.01 at 3 to 5 days), or 4/4-KI mice (P<0.01 at 1 to 3 days; P⩽0.001 at 4 and 5 days), as compared with the vehicle groups (Table 2).
Effects of arundic acid on neurologic scores after permanent middle cerebral artery occlusion in the three lines of knock-in mice

Homozygous human apoE2 (2/2)-, apoE3 (3/3)-, or apoE4 (4/4)-knock-in (KI) mice are denoted by 2/2-, 3/3-, or 4/4-KI, respectively. pMCAO; permanent middle cerebral artery occlusion. Statistical analysis was performed by comparing among the three lines of KI mice in each of the arundic acid and the vehicle groups or between the arundic acid and the vehicle groups in each line of KI mice.
Versus 4/4-KI mice in the vehicle group.
Versus 2/2-KI mice in the vehicle group.
Versus 4/4-KI mice in the vehicle group.
Versus 3/3-KI mice in the vehicle group.
Versus 4/4-KI mice in the vehicle group.
Effects of Arundic Acid on S100 Burden after Permanent Middle Cerebral Artery Occlusion in Apolipoprotein E-Knock-In Mice
In all of the KI mice, S100-positive astrocytes were present in both hemispheres excluding the infarct area. From 1 to 5 days after pMCAO, S100-positive astrocytes became increasingly numerous along the outer border of the infarct, and could clearly be identified by typical morphologic features as reactive astrocytes. Specifically, at 1 day after pMCAO, astrocytic nuclei in the peri-infarct area were moderately positive for S100 in all of the experimental groups. Astrocytic processes were faintly stained, disclosing delicate structures of their processes. Numerous minute S100-positive granules, which were probably within the astrocytic processes, were dispersed between neurons. At 5 days after pMCAO, both the astrocytic nuclei and swollen processes in the peri-infarct area were moderately to strongly positive for S100 in all of the experimental groups, containing numerous minute S100-positive granules.
Among the vehicle groups, there was a clear tendency at 5 days after pMCAO for more pronounced S100 immunoreactivity in the peri-infarct area in 4/4-KI mice compared with 2/2- or 3/3-KI mice (Figures 4A–4C). When the arundic acid and the vehicle groups were compared, S100 immunoreactivity was generally reduced by administration of arundic acid in all of the KI mice, while the reduction tended to be more pronounced in 4/4-KI mice than in other KI mice (Figures 4D–4F). The above findings were in agreement with the results of image analysis as follows. In any of the KI mice treated with vehicle, S100 burden at 5 days was significantly greater than at 1 day after pMCAO (P<0.001 for 2/2-, 3/3-, or 4/4-KI mice), with a significantly larger increase in 4/4-KI mice than in 2/2- or 3/3-KI mice at both 1 and 5 days after pMCAO (P<0.001 at 1 and 5 days versus 2/2-KI mice; P<0.001 at 1 and 5 days versus 3/3-KI mice). Administration of arundic acid induced a significant decrease of S100 burden at 1 day after pMCAO in 4/4-KI mice (P=0.001 versus the vehicle groups), but not in 2/2- or 3/3-KI mice. At 5 days, however, arundic acid induced a significant decrease in S100 burden in all three lines of KI mice (P<0.001 at 5 days in 2/2-, 3/3-, or 4/4-KI mice versus the vehicle groups) (Figures 5A and 5B).
Effects of Arundic Acid on the Tissue Levels of S100B in the Peri-Infarct Area
The above immunohistochemical results were confirmed by the sandwich enzyme-linked immunosorbent assay method. These data revealed that, at 3 days after pMCAO, administration of arundic acid in the 4/4-KI mice significantly lowered the tissue levels of S100B in the peri-infarct area (results expressed as the mean S100 B levels (ng/mg protein) ±1s.d. in 4/4-KI mice at 3 days after pMCAO in the vehicle group: 134±21, in the arundic acid group: 78±15; P<0.001) (Figure 6). Further, these results confirm that S100B levels per se are reduced by arundic acid treatment in 4/4-KI mice (the antibody utilized for immunohistochemistry is polyclonal and does not discriminate against different S100 isoforms).
Effects of Arundic Acid on Glial Fibrillary Acidic Protein Burden after Permanent Middle Cerebral Artery Occlusion in Apolipoprotein E-Knock-In Mice
Reactive astrocytes, characterized by enhanced GFAP expression, hyperplasia, and gemistocytic changes, started to appear in the infarct border at 1 day after pMCAO in all of the KI mice. At 5 days after pMCAO, reactive astrocytosis was pronounced in the peri-infarct area, particularly along the infarct border. Immunoreactivity for GFAP was markedly enhanced in the plump somata and processes of astrocytes, which were interwoven to form a glial barrier in the outer boundary in the infarct border. In the vehicle groups, reactive astrocytosis was apparently more pronounced in 4/4-KI mice than in 2/2- or 3/3-KI mice at 5 days after pMCAO (Figures 7A–7C). At 5 days after pMCAO, the magnitude of reactive astrocytosis was considerably attenuated in the arundic acid groups as compared with the vehicle groups, particularly in 4/4-KI mice (Figures 7D–7F).
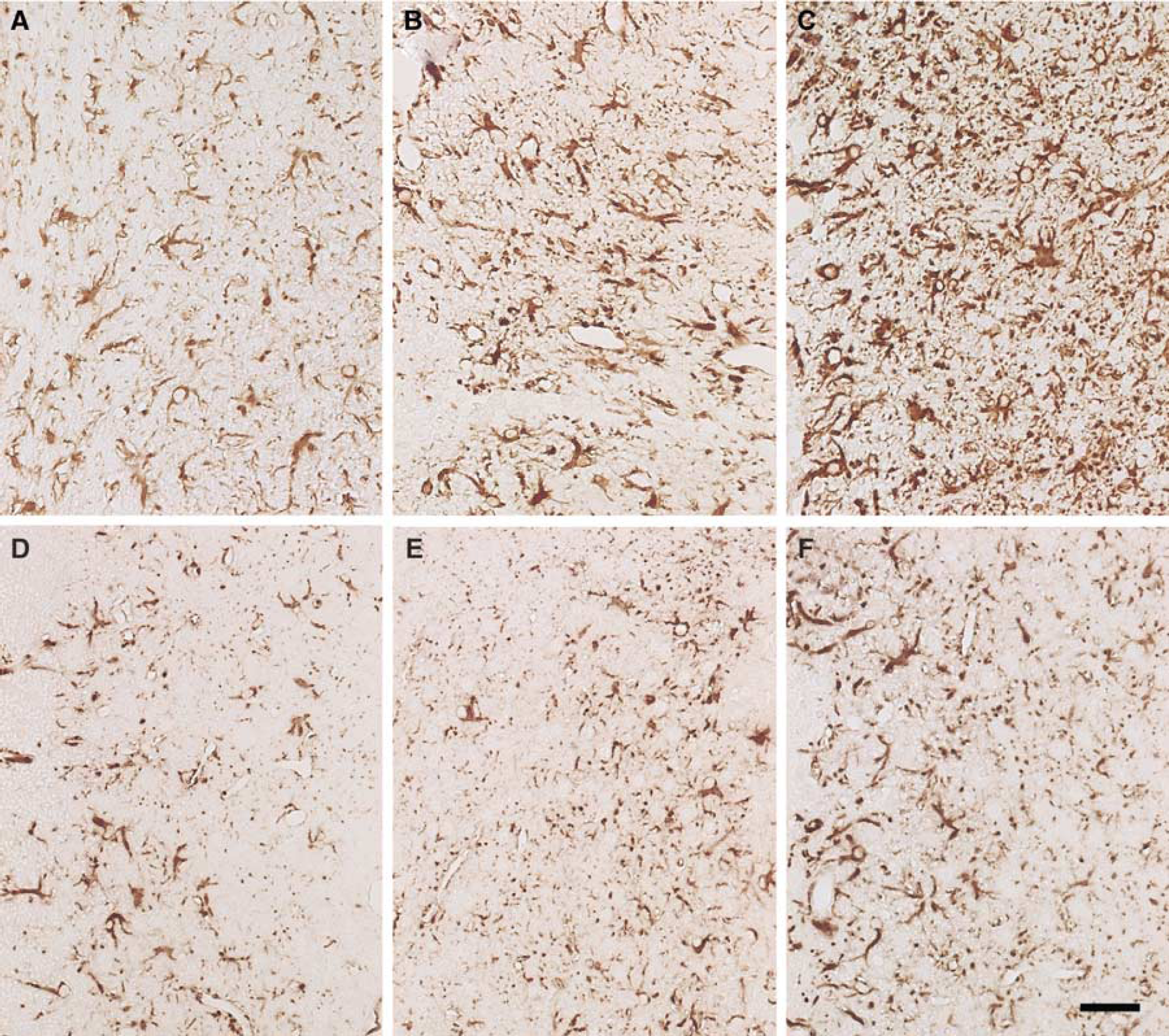
Glial fibrillary acidic protein (GFAP) immunohistochemical staining in the peri-infarct area at 5 days after permanent middle cerebral artery occlusion (pMCAO) in homozygous human apoE2 (2/2)-, apoE3 (3/3)-, or apoE4 (4/4)-knock-in (KI) mice. The vehicle (
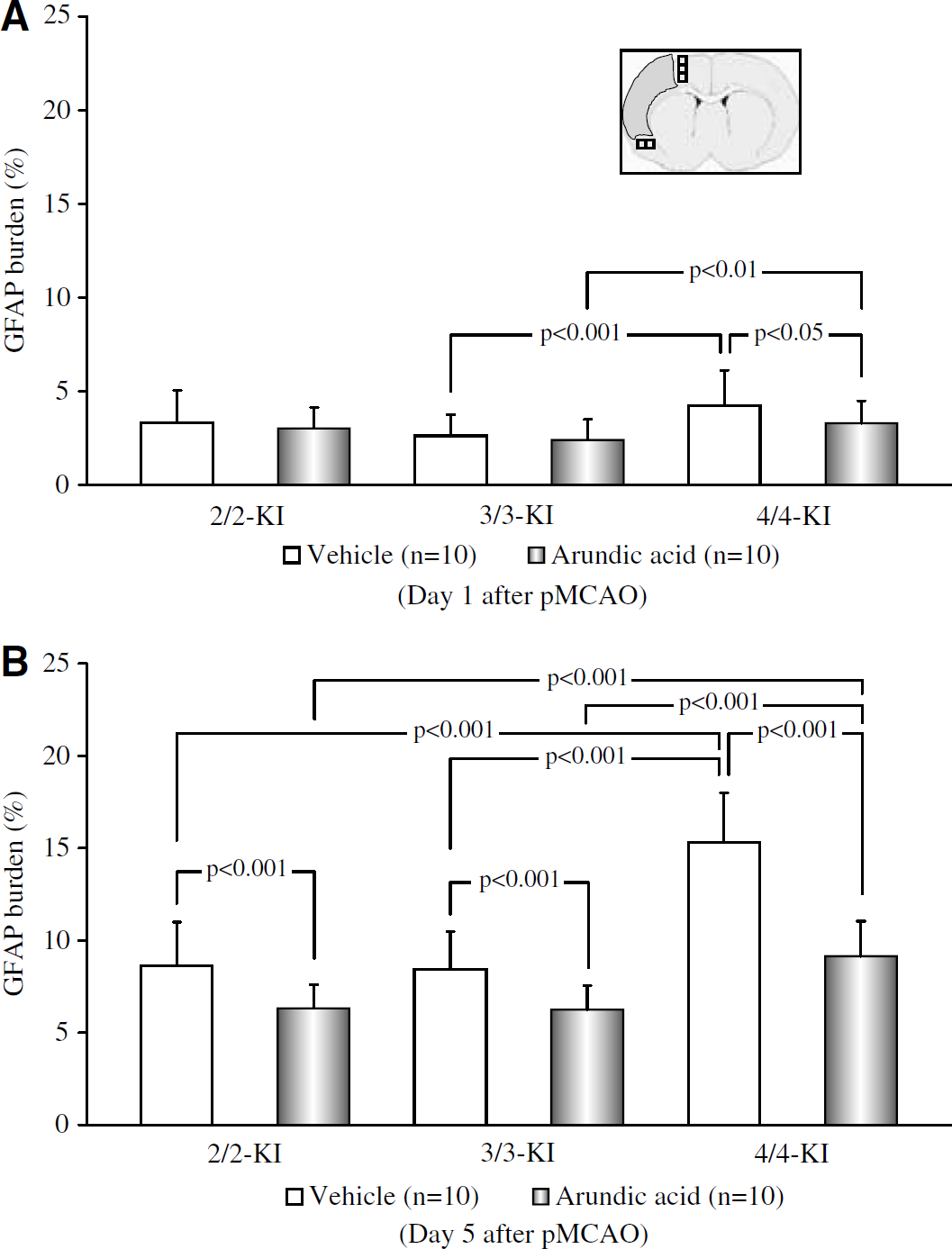
Quantitative assessment of glial fibrillary acidic protein (GFAP) burden in the peri-infarct area at 1 (
Image analysis clearly showed that, in the vehicle groups, GFAP burden at 5 days after pMCAO was significantly pronounced as compared with that at 1 day in all of the KI mice (P<0.001 for 2/2-, 3/3-, or 4/4-KI mice), whereas it was significantly greater in 4/4-KI mice than in 2/2- or 3/3-KI mice (P<0.001 at 5 days versus 2/2-KI mice; P<0.001 at 1 and 5 days versus 3/3-KI mice). Administration of arundic acid induced significant attenuation of GFAP burden at 1 or 5 days after pMCAO in all of the KI mice (P<0.05 at 1 day in 4/4-KI mice; P<0.001 at 5 days in 2/2-, 3/3-, or 4/4-KI mice versus the vehicle groups) (Figures 8A and 8B). Bivariate correlation analyses between the infarct area (as determined in section No. 3) and GFAP burden (as determined in the coronal section adjacent to section No. 3) across all days showed statistical significance in 4/4-KI mice in the vehicle group (r=0.75, P<0.001), but not in 2/2- or 3/3-KI mice in the vehicle groups or in any of the KI mice treated with arundic acid.
Discussion
In the three lines of KI mice treated here with vehicle, changes in infarct volumes, neurologic deficits, and the magnitude of astrocytic activation in the peri-infarct area as estimated by S100/GFAP burden conformed to results of our previous study (Mori et al, 2004b). Similarly, we further confirmed the detrimental effects of the apoE4 isoform in the present study, in that we found a significant difference in the infarct volume and in the magnitude of S100/GFAP burden in the peri-infarct area between 4/4-KI mice and 2/2- or 3/3-KI mice at 5 days after pMCAO. Using arundic acid, the present study primarily aimed to examine the putative causal relationship between enhanced reactive astrocytosis and exacerbation of ischemic brain damage. Arundic acid treatment resulted in protection of all of the KI mice from pMCAO-induced brain damage, with 4/4-KI mice generally enjoying the greatest magnitude of protection. Interpretation of this drug effect in a pMCAO model requires consideration as to temporal and topographical alterations in the pathogenic mechanism causing cell death. Whereas acute infarct expansion in the ischemic core up to 24 h after pMCAO is primarily because of energy failure, delayed infarct expansion in the peri-infarct area during 1 to 7 days after pMCAO has been ascribed to the occurrence of apoptotic cell death (Charriaut-Marlangue et al, 1996; Davies et al, 1998; Dirnagl et al, 1999; Griffin et al, 1998; Stoll et al, 1998; Tateishi et al, 2002; Wyss-Coray and Mucke, 2002). Therefore, the results obtained at 1 and 5 days after pMCAO are discussed separately, and the issue of possible pathogenic mechanism(s) underlying the interrelations among the apoE4 isoform, enhanced astrocytic activation, and subsequent exacerbation of brain damage are finally addressed.
Acute Effects of Arundic Acid after Permanent Middle Cerebral Artery Occlusion in Apolipoprotein E-Knock-In Mice
In the vehicle groups, infarct volumes were significantly greater in 4/4-KI mice than in 2/2- or 3/3-KI mice at 1 day after pMCAO. At this time-point, arundic acid induced significant decreases in infarct volumes in all of the experimental groups of KI mice (Figure 1A). This result differs somewhat from the results of our previous study using the rat pMCAO model (Tateishi et al, 2002), where differences in infarct volumes between the arundic acid and the vehicle groups did not reach a significant level, up to 3 days after pMCAO. This incongruity is likely attributable to differences in the models used.
The extant literature shows that S100B exerts detrimental effects through augmentation of inducible nitric oxide synthase (iNOS) and Cyclooxygenase-2 (COX-2) expression (Hu et al, 1996; Lam et al, 2001; Murphy, 2000). In accordance with these findings, suppression of astrocytic S100B synthesis by arundic acid led to a significant reduction in the expression of these enzymes, both in vitro and in vivo (Shimoda et al, 1998; Shinagawa et al, 1999). However, the increase in S100/GFAP burden in the peri-infarct area at 1 day after pMCAO was not significantly attenuated by administration of arundic acid in 2/2- or 3/3-KI mice in the present study (Figures 5A and 8A). Furthermore, the increase in brain iNOS activity after astrocytic activation reportedly peaked at 2 to 3 days after pMCAO in mice (Iadecola et al, 1995). Therefore, it seems rather unlikely that the significant brain-protective effects of arundic acid regarding infarct volumes, as observed at 1 day after pMCAO in 2/2- or 3/3-KI mice, is owing to suppression of astrocytic production of S100B or expression of iNOS/COX-2. Alternatively, it has been shown that arundic acid augments expression and activity of glial glutamate transporters (GLT-1 and GLAST) both in vitro and in vivo (Shinagawa et al, 1998, 1999). In the previous study using the rat transient MCAO model, administration of arundic acid markedly ameliorated brain damage (Tateishi et al, 1999), an effect that was associated with marked suppression of delayed elevation of extracellular glutamate levels in the peri-infarct area, presumably because of augmentation of glutamate transporter activity (Mori et al, 2004a). Therefore, it seems plausible that this mechanism of action of arundic acid is involved in the amelioration of acute infarct expansion in 2/2- or 3/3-KI mice at 1 day after pMCAO.
A similar mechanism to that described above may also operate in 4/4-KI mice. However, the increase in S100/GFAP burden in the peri-infarct area at 1 day after pMCAO was more pronounced in 4/4-KI mice than 2/2- or 3/3-KI mice, indicating an isoform-specific action of apoE4. Since arundic acid significantly inhibited both enhanced astrocytic activation and acute infarct expansion at 1 day after pMCAO in these mice, it seems likely that reinforced astrocytic activation played a significant role in the exacerbation of acute infarct expansion in 4/4-KI mice. Nonetheless, it remains to be clarified whether enhanced astrocytic activation in 4/4-KI mice led to accelerated expression of iNOS/COX-2, or to other noxious events such as a fulminant elevation of extracellular glutamate levels in the peri-infarct area.
Subacute Effects of Arundic Acid after Permanent Middle Cerebral Artery Occlusion in Apolipoprotein E-Knock-In Mice
In the vehicle groups of 2/2- or 3/3-KI mice, infarct volumes at 5 days were unaltered as compared with that at 1 day, whereas S100/GFAP burden in the peri-infarct area was significantly increased. In the vehicle group of 4/4-KI mice, both infarct volumes and S100/GFAP burden in the peri-infarct area were markedly increased at 5 days as compared with 1 day after pMCAO. Administration of arundic acid produced significant amelioration of infarct volumes, neurologic deficits, and S100/GFAP burden in the peri-infarct area at 5 days after pMCAO in all of the KI mice. The inhibitory effects of arundic acid on astrocytic S100B synthesis were confirmed by the measurement of tissue S100B levels in 4/4-KI mice at 3 days after pMCAO (Figure 6). Of particular importance, arundic acid-induced attenuation of S100/GFAP burden in the peri-infarct area and delayed infarct expansion from 1 to 5 days after pMCAO was far more pronounced in 4/4-KI mice than in 2/2- or 3/3-KI mice (Figures 1B, 5B and 8B). In all of the KI mice, the significant decrease in GFAP burden in the peri-infarct area was associated with a marked decrease in the number of hypertrophic astrocytes as is apparent from Figures 4 and 7.
The present data may be interpreted as indicating that, during the subacute phase of pMCAO, arundic acid mitigates delayed infarct expansion and deterioration of neurologic deficits through modulation of astrocytic activation, regardless of the presence or the absence of the apoE4 isoform. Given the data showing that arundic acid acts to inhibit S100B synthesis, the protective effect in 4/4-KI mice as compared with 2/2- or 3/3-KI mice is likely because of the participation of S100B in the detrimental effects of the apoE4 isoform, and this is discussed further below.
Glial Activation and the Role of S100B Induced by the Apolipoprotein E4 Isoform
S100B belongs to a multigenic family of Ca2+-binding proteins of the EF-hand type known as S100 that are differentially expressed in a large number of cell types, where they act as calcium-modulated regulatory proteins that intervene in the fine-tuning of a relatively large number of specific intracellular and extracellular activities (Donato, 1999). In the brain, S100B is selectively expressed in astrocytes and Schwann cells. Recently, there has been a surge of interest in the roles of S100 proteins, as S100/calgranulin polypeptides present at sites of inflammation were shown to bind the receptor for advanced glycation end products (RAGE) that triggers cellular activation, culminating in the generation of key proinflammatory mediators (Bucciarelli et al, 2002; Hofmann et al, 1999). It is noteworthy that recent reports documented the presence of RAGE in microglia (Yan et al, 1996), in reactive astrocytes (Sasaki et al, 2001), and at the BBB (Deane et al, 2003). In rat primary cortical astrocytes, S100B was shown to activate nuclear factor κB, iNOS promoter, and nitric oxide production (Lam et al, 2001). Thus, it is tempting to speculate that S100B-induced activation of signal transduction pathways is at least in part mediated by the binding of S100B with RAGE.
Our data provide insight into the complicated interrelations between the apoE4 isoform, glial activation, and subsequent exacerbation of brain damage. There is a growing consensus that inflammation and glial responses play an important role in both acute and chronic brain damage (Barone and Feuerstein, 1999; Griffin et al, 1998; Norenberg, 1994; Ridet et al, 1997; Rothwell, 1999; Stoll et al, 1998; Wyss-Coray and Mucke, 2002; Yankner, 1996). Brain apoE is a multifunctional molecule, with potential roles in amyloid deposition and clearance, microtubule stability, intracellular signaling, immune modulation, glucose metabolism, oxidative stress, and other cellular processes (Saunders, 2000). A unifying mechanism that would account for a role of apoE in both acute and chronic neurologic diseases would be that it suppresses glial response to brain damage in an isoform-specific fashion (Barger and Harmon, 1997; Laskowitz et al, 1997, 2001). Although it is unclear whether the detrimental effects of inflammation generally outweigh a neuroprotective mechanism (Stoll et al, 1998), astrocytic activation may play as much of a role as microgliosis in certain brain pathologies. In fact, the remarkable protective effects of arundic acid, showed here and in a previous report (Tateishi et al, 2002), indicate that augmented expression of S100B upon astrocytic activation does act to exacerbate brain damage during the subacute phase of pMCAO. This assertion is in keeping with the recent finding of Wainwright et al (2004) that S100B transgenic mice are more susceptible than wild-type or S100B knock-out mice to injury after perinatal hypoxia—ischemia.
Based on our data and the pertinent literature, we suggest that the following train of events is triggered by the apoE4 isoform: apoE4 initially acts to reinforce microglial activation (Laskowitz et al, 2001) and subsequent overexpression of microglial cytokines such as interleukin-1β boosts astrocytic activation, leading to enhanced synthesis of S100B and many other biologically active substances (Das and Potter, 1995; Ridet et al, 1997). The above substances induce further activation of astrocytes (Hu et al, 1996) as well as of microglia (Adami et al, 2001), culminating in the formation of a vicious cytokine cycle. The resultant overproduction of nitric oxide together with superoxide anion (Culcasi et al, 1994; Gunasekar et al, 1995) causes apoptosis of both neurons and astrocytes (Donato, 1999; Hu et al, 1996; Scotto et al, 1998). The remarkable inhibitory effects of arundic acid on both peri-infarct astrocytic activation and delayed infarct expansion as observed in 4/4-KI mice are suggestive of the pivotal role of S100B in perpetrating the above vicious cascade.
Conclusion
Arundic acid has been shown to act exclusively on astrocytes to suppress their activation. In genetically engineered mice subjected to permanent focal ischemia, we have shown that the agent remarkably inhibits both peri-infarct astrocytosis and delayed infarct expansion in 4/4-KI mice, indicating that these phenomena are causally related such that the detrimental effect of the apoE4 isoform resides in augmentation of glial activation. Thus, pharmacological modulation of astrocytic activation may confer a novel and useful therapeutic strategy against both acute and subacute stroke as well as a wide spectrum of chronic neurodegerative disorders, particularly in carriers of the APOE ɛ4 allele.
Footnotes
Acknowledgments
The authors thank Mr N Koyama, Ms A Kudo, Mr T Shimoda, Dr J Yamamoto, Mr S Mizote, Ms S Ono, Ms M Nakanishi, and Mr S Kawaharada for expert assistance, as well as Dr H Yamaguchi for helpful discussions.