
Abstract
The ability of intraventricular infusion of apolipoprotein E (apoE) to reduce neuronal damage after global cerebral ischemia was investigated in apoE-deficient and wild-type mice. ApoE (5 μg/mL lipid-conjugated derived from human plasma; 1 μL/h, continuous infusion) significantly reduced neuronal damage in the caudate nucleus and CA2 pyramidal cell layer by approximately 50% in apoE-deficient mice after global ischemia compared to vehicle infusion. In wild-type mice infused with apoE, there was a trend for ischemic neuronal damage to be reduced. ApoE-infused mice had a marked reduction in 4-hydroxynonenal immunoreactivity, as a marker of lipid peroxidation. The results show that the presence of apoE at or after the time of injury can be neuroprotective, possibly via an anti-oxidant mechanism.
The apolipoprotein E gene (APOE, gene; apoE, protein) exists as three different alleles in humans (∊2, ∊3 and ∊4). APOE genotype underlies a genetically determined susceptibility to the effects of several forms of acute brain damage including that caused by head injury (Nicoll et al., 1995; Teasdale et al., 1997), spontaneous intracerebral hemorrhage (Alberts et al., 1995), and repetitive traumatic brain injury in boxers (Jordan et al., 1997). The APOE ∊4 allele, carried by one third of the population, is associated with a worsened response or poorer outcome following the initial injury. ApoE is the most abundant apolipoprotein in cerebrospinal fluid (Pitas et al., 1987) and is produced within the CNS (Linton et al., 1991) primarily by astrocytes. After brain injury, apoE is markedly upregulated in astrocytes and accumulates in neurons within hours of the insult (Horsburgh et al., 1996; Horsburgh et al., 1999a) and is consistent with a role for apoE in synaptic remodelling, repair, and regeneration (Poirier, 1994). When the function of apoE is deficient or impaired, in genetically-modified apoE mice, there is an increased susceptibility of the brain to the effects of ischemic insults (Laskowitz et al., 1997b; Horsburgh et al., 1999b) and closed head injury (Lomnitski et al., 1997). We have previously shown that apoE-deficient mice have approximately a twofold increase in ischemic neuronal damage following a transient episode of global cerebral ischemia (Horsburgh et al., 1999b). We hypothesized that increasing the levels of apoE in the CSF is a possible mechanism by which to ameliorate neuronal damage associated with acute brain injury. The study determined the ability of intraventricular infusion of lipid-conjugated apoE to reduce neuronal damage after an episode of global ischemia in apoE-deficient and wild-type mice. It has been proposed that apoE can modulate oxidative damage (Miyata and Smith, 1996). The extent of 4-hydroxynonenal (4-HNE) (Esterbauer et al., 1991) immunoreactivity, a marker of lipid peroxidation, was additionally examined in apoE-deficient and wild-type mice infused with lipid-conjugated apoE compared to vehicle-infused mice.
MATERIALS AND METHODS
ApoE-deficient mice were derived at Glaxo-Wellcome from a colony produced by Dr. Maeda and colleagues (University of North Carolina, U.S.A.) as previously described (Horsburgh et al., 1999b). Briefly, homologous recombination in mouse embryonic stem cells (129J) was used to produce targeted knockout mutations of the apoE locus. Offspring were backcrossed with C57BL/6J mice (six times at Dr. Maeda's laboratory followed by further backcrossing at Glaxo-Wellcome). ApoE-deficient (n = 24) and wild-type (n = 24) mice used in this study (12 to 16 week old males) were littermates which were produced by selective matings between heterozygous apoE knockout breeding pairs. The genotype of the mice was confirmed by polymerase chain reaction analysis.
Alzet osmotic minipumps (model 1003D, delivery rate 1 μL/h) attached to a brain infusion cannula via a catheter were filled with 100 μL of artificial CSF (mmol/L: Na, 150; K, 3; Ca, 1.4; Mg, 0.8; P, 1; Cl, 155) or 100 μL of apoE (5 μg/mL in artificial CSF; derived from pooled human plasma, very-low density lipoprotein, Calbiochem, Novabiochem, U.K.). Each pump system was placed in a sealed container with sterile saline and kept at 37°C overnight to reach a steady state before implanting. Anesthesia was induced with 3% halothane in 70% N2O / 30% O2 and maintained at 2.5% for the duration of the experiment. The mice were put on a stereotactic frame and mechanically ventilated. Body temperature was strictly controlled at 37°C. A midline sagittal incision was made to expose the skull. Under sterile conditions an osmotic minipump was placed in a subcutaneous pouch under the skin of the neck and connected via a plastic catheter to the brain infusion cannula. A burr hole was drilled in the skull and the cannula placed into the right lateral ventricle at stereotactic coordinates (0.8 mm lateral, 0.2 mm from Bregma, 2.5 mm below dura). The cannula was held in place with cyanacrylate glue and secured to the skull with dental cement and the skin was sutured. The mouse was then disconnected from the frame and allowed to breath spontaneously. Halothane was delivered via a face mask and the concentration reduced to 1.5%. Thirty minutes after the implantation of the intraventricular cannula, transient global cerebral ischemia was induced as previously described (Horsburgh et al., 1999b). Both common carotid arteries were occluded for exactly 17 minutes. The animals were placed in an incubator for 2 hours after which they were returned to the animal housing and allowed to recover. Animals were killed 72 hours after carotid artery occlusion by perfusion fixation using 4% paraformaldehyde. The brains were removed and post-fixed in 4% paraformaldehyde for 2 hours and embedded in paraffin. Sections (6 μm) were cut and stained with hematoxylin and eosin. Adjacent sections were immunostained with anti-4-hydroxynonenal Michael adducts (4-HNE) (1:2000, rabbit polyclonal, Calbiochem, Novabiochem). Sections were deparaffinized, rehydrated into citrate buffer (pH 6.0), incubated in a microwave oven twice for 5 minutes for antigen enhancement and for 30 minutes in hydrogen peroxide (3%). Nonspecific sites were blocked with 0.5% bovine serum albumin and 10% normal goat serum (1 hour). After overnight incubation with anti-4HNE in blocker, the sections were incubated with biotinylated anti-rabbit IgG and processed with a Vectastain Elite kit (Vector Labs, U.K.). Color was developed using diaminobenzidine. Using a similar method, sections from apoE-deficient mice were immunostained with an antibody to apoE (1:5000; goat polyclonal, Chemicon International Ltd., U.K.).
The numbers of morphologically normal neurons and neurons showing the features of ischemic cell change in the hematoxylin and eosin stained sections (Horsburgh et al., 1999b) were counted in 3 different fields in both hemispheres of the caudate nucleus and CA2 pyramidal cell layer using a 10 mm2 grid and x400 magnification. The percentage of ischemic neurons in apoE-infused compared to vehicle-infused in apoE-deficient mice and in wild-type mice were compared using one-tailed Student's unpaired t-est. Semi-quantitative assessment of cellular 4-HNE immunoreactivity was performed at ×400 magnification. 4-Hydroxynonenal immunoreactivity was classified as follows: 0 = no cellular staining; 1 = < 35% cellular staining; 2 = 35% to 70% cellular staining; 3 = > 70% cellular staining. The extent of 4-HNE immunoreactivity in apoE-infused compared to vehicle-infused in apoE-deficient mice and in wild-type mice were compared using Student's unpaired t-test.
RESULTS
We chose to infuse a lipid-associated form of apoE (5 μLg/mL), derived from pooled human plasma of all apoE isoforms and complexed with very low density lipoprotein and injected into the CSF ensuring direct delivery of apoE to the injured brain. ApoE in the CSF (and also within the extracellular space) is normally complexed to lipid which is required for apoE to be biologically active (Pitas et al., 1987). Furthermore, the infusion of apoE was initiated before the insult and continuously thereafter to ensure that apoE would be available immediately before and after ischemic injury. We sought to determine that exogenously administered apoE was present in brain parenchyma by immunostaining sections of apoE-deficient mice with an antibody to apoE. We did find evidence that apoE immunostaining was increased in apoE-infused compared to vehicle-infused mice in brain parenchyma at the site of infusion but did not find evidence that apoE immunostaining was increased distal from this (data not shown). Histological sections were examined from all animals to ensure that the cannula delivering vehicle or apoE was placed directly in the ventricle. Transient global cerebral ischemia induced by bilateral carotid artery occlusion for a period of 17 minutes in wild-type mice is associated with selective neuronal damage. In this model, as we previously demonstrated, there is significantly greater neuronal damage in apoE-deficient mice than wild-type mice (Horsburgh et al., 1999b). A nonbiased quantitation of the number of normal and ischemic neurons was performed in the caudate nucleus. In the present study, as expected, apoE-deficient mice infused with vehicle had twice as much neuronal damage 3 days after the insult as wild-type mice infused with vehicle (47 ± 6% % versus 22 ± 5%). However, ischemic neuronal damage was significantly reduced in apoE-deficient mice infused for 3 days with apoE compared to vehicle-infused apoE-deficient mice (26 ± 6% versus 47 ± 6%; P = 0.0135, Fig. 1A). Ischemic neuronal damage in the apoE-deficient mice infused with apoE was similar to that observed in wild-type mice infused with vehicle (26% versus 22% respectively). In the wild-type mice there was also a trend toward a reduction in ischemic neuronal damage in mice infused with apoE compared with vehicle (22 ± 5% versus 12 ± 3%; P = 0.054, Fig. 1A). In another brain region, the CA2 hippocampal pyramidal cell layer, there was a significant reduction in the extent of ischemic neuronal damage in apoE-deficient mice infused with apoE compared with vehicle (5.0 ± 2.0% versus 21 ± 7%; P = 0.02, Fig. 1B). In the wild-type mice, ischemic neuronal damage was reduced in apoE-infused mice compared to vehicle-infused mice but this did not reach statistical significance (7.4 ± 2.8 versus 13.8 ± 9.3%; P = 0.25, Fig. 1B).
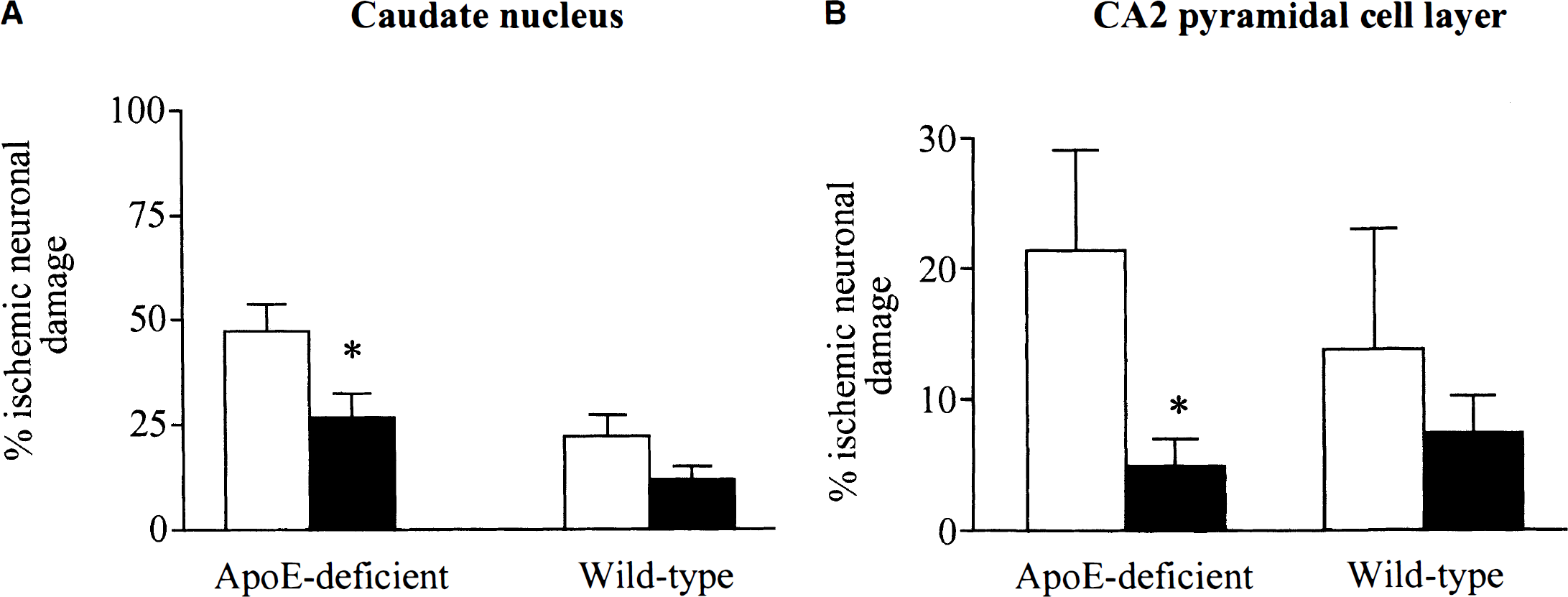
Quantification of the percentage of ischemic neuronal damage in the caudate nucleus (
4-HNE immunoreactivity, a marker of lipid peroxidation, was semiquantified in adjacent sections to those used for quantification of ischemic neuronal damage to determine whether there were alterations in lipid peroxidation after apoE-infusion. 4-HNE immunoreactivity was minimal in morphologically normal tissue. However, in areas with histological evidence of ischemic damage, 4-HNE immunoreactivity was increased in the neuropil, glia, and cytoplasm of triangular neurons. Semiquantification of 4-HNE immunoreactivity indicated there was increased 4-HNE cellular immunoreactivity in apoE-deficient compared to wild-type mice after global ischemia (2.25 ± 0.18 versus 1.46 ± 0.14; P = 0.002, Fig. 2). However, the extent of 4-HNE immunoreactivity was significantly reduced in apoE-deficient mice infused with apoE compared to vehicle-infused apoE-deficient mice (1.54 ± 0.2 versus 2.25 ± 0.18; P = 0.022, Fig. 2). In the wild-type mice there was also a trend toward a reduction in 4-HNE immunoreactivity in mice infused with apoE compared with vehicle (1.17 ± 0.20 versus 1.46 ± 0.14). The extent of ischemic neuronal damage was correlated with the extent of 4-HNE immunoreactivity in the wild-type and apoE-deficient mice infused with either vehicle or with apoE (r2 = 0.73 and r2 = 0.63; Fig. 3).
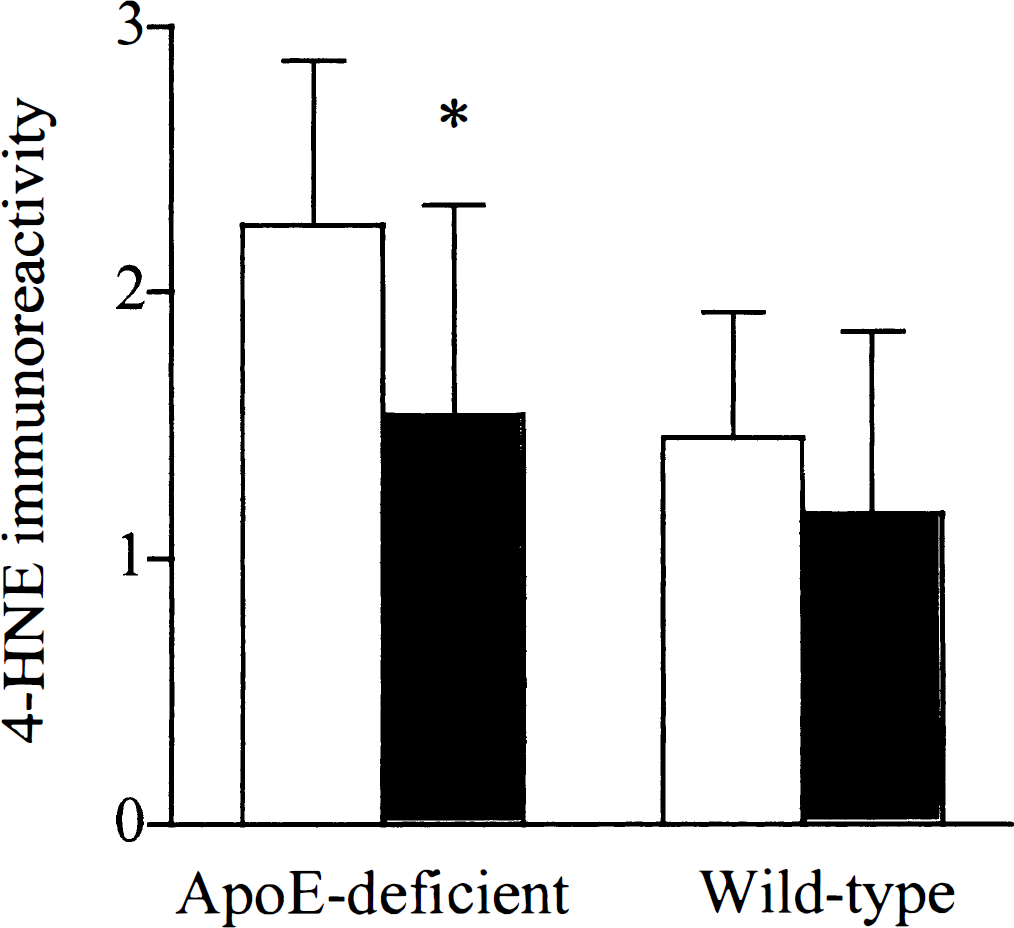
Semiquantitative assessment of 4-hydroxynonenal (4-HNE) immunoreactivity in the caudate nucleus following global ischemia of apolipoprotein E (ApoE)-deficient and wild-type mice after intraventricular infusion of apoE (very low density lipoprotein) or vehicle. Values are the mean ± SD. There is significantly less 4-HNE immunoreactivity in the apoE-deficient mice infused with apoE as compared to vehicle (P = 0.022, Student's unpaired Mest). (□) Vehicle. (▪) ApoE (5 μg/mL).
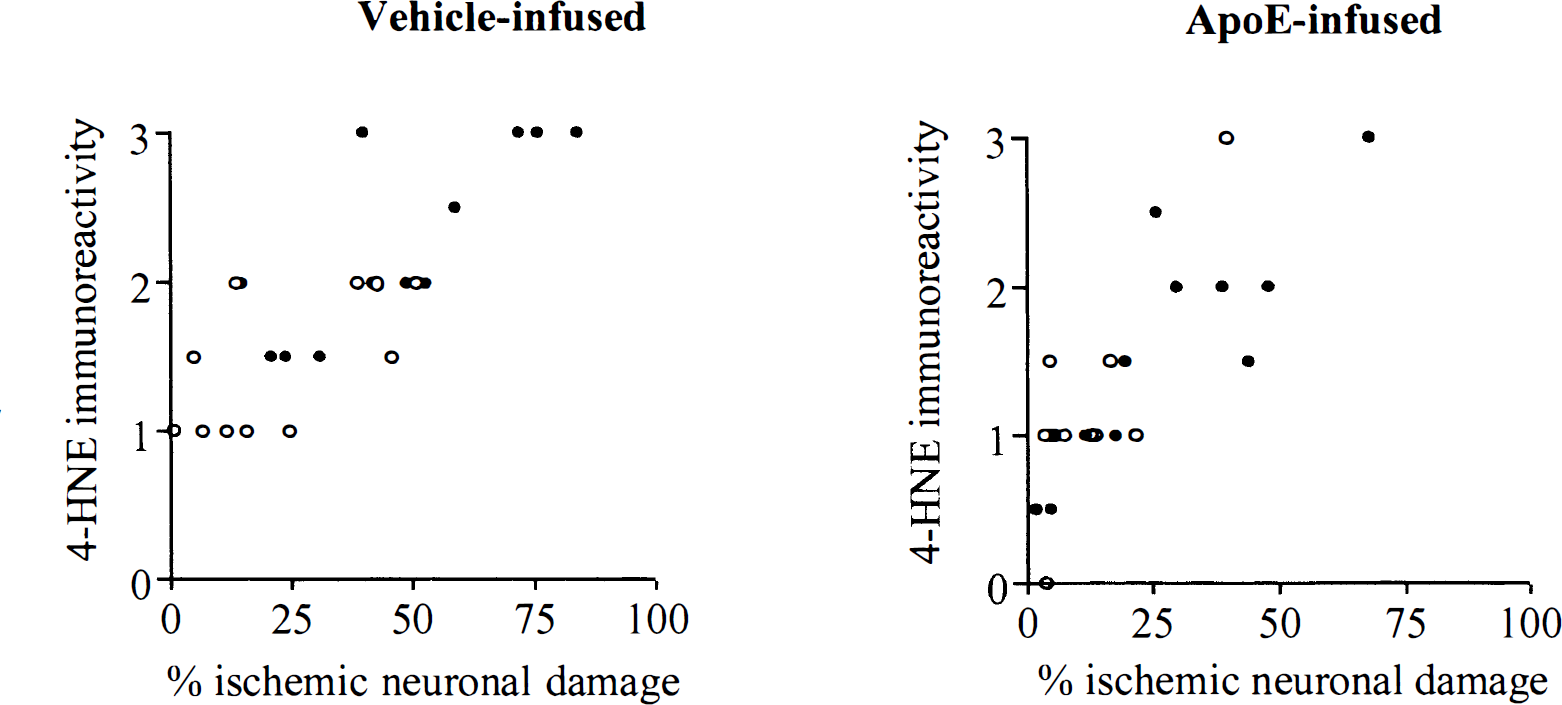
The extent of ischemic neuronal damage correlates with the extent of 4-hydroxynonenal (4-HNE) immunoreactivity in the wild-type mice and in the apolipoprotein E (ApoE)-deficient mice infused with vehicle, r2 = 0.73 (
DISCUSSION
This study demonstrates that intraventricular infusion of apoE markedly ameliorates neuronal damage with a concomitant reduction in lipid peroxidation after acute brain injury. The presence of lipid-associated apoE, throughout the postischemic period, was shown to reduce the extent of neuronal damage by approximately 50% in both apoE-deficient and wild-type mice following an episode of global ischemia. The reduction of ischemic neuronal damage paralleled a reduction in lipid peroxidation in the apoE-deficient mice infused with apoE. This demonstrates that the increased sensitivity of apoE-deficient mice to brain injury and resultant poor outcome is a consequence of reduced availability of apoE in the CNS at, or shortly after, the time of injury. The study also indicates the potential for apoE, or more realistically, an apoE mimetic to be used as a therapy for reducing neuronal damage as a result of acute brain injury.
ApoE and lipids are unable to cross the blood-brain barrier and their concentrations in CSF are a reflection of the production within the CNS (Linton et al., 1991). Intraventricular infusion of apoE ensured direct availability of apoE to the CNS. Experiments in cell cultures exposed to whole CSF demonstrate that apoE can be taken up into both neurons and astrocytes (Rebeck et al., 1998). A previous study indicated that recombinant apoE infusion without lipid could reduce the chronic neurodegeneration and cognitive impairment observed in aged apoE-deficient mice (Masliah et al., 1997). However, it is unclear whether the apoE, once infused, subsequently bound to circulating lipids in the CSF. Since we wanted to ensure that apoE, in a lipid bound form, was immediately available at the time of injury we chose apoE complexed with very low-density lipoprotein. This in vivo neuroprotective effect of apoE illustrated in the present study is consistent with biological effects of apoE in vitro in which similar physiologic concentrations of apoE have been shown to enhance neurite outgrowth of dorsal root ganglion cells (Handelmann et al., 1992). These trophic effects of apoE in vitro required the presence of lipid, very low density lipoprotein, similar to the form of apoE infused in the present study. It should be noted that it is unclear whether the very low-density lipoprotein contained within the apoE infusion contributed to the neuroprotectant effect in vivo.
The principal mechanism by which apoE exerts its influence on outcome after brain injury is unclear as yet but several have been proposed. One possible mechanism by which apoE ameliorates neuronal damage following global ischemia is by an antioxidant effect. 4-HNE immunoreactivity has been previously used to demonstrate increased lipid peroxidation after experimental traumatic brain injury (Zhang et al., 1999) and transient cerebral ischemia in rats (Urabe et al., 1998). In the present study, lipid peroxidation as indicated by 4-HNE (Esterbauer et al., 1991) immunoreactivity was markedly increased in the apoE-deficient compared to wild-type mice. However, apoE-infusion was associated with a reduction in lipid peroxidation. ApoE has also been shown to protect neurons from oxidative damage in cell culture (Miyata and Smith, 1996). Another possible mechanism by which apoE could protect against ischemic insults is by reducing inflammatory responses (Laskowitz et al., 1997a). After brain injury apoE is markedly upregulated (Poirier et al., 1991) and is increased in neurons (Horsburgh et al., 1996, 1999a) possibly via receptor-mediated uptake (Poirier, 1994). Intraneuronal apoE may protect via the transport of cholesterol and lipids to neurons for repair and remodelling (Poirier, 1994). ApoE-deficient mice have an increased sensitivity to brain injury and resultant poor outcome perhaps as a consequence of reduced availability of apoE and/or transported lipids. Intraneuronal apoE may also modulate neuronal function through an interaction with cytoskeletal proteins such as tau and microtubule associated proteins (Fleming et al., 1996).
It is unclear as yet whether patients with APOE ∊4 have a poorer outcome after brain injury (Alberts et al., 1995; Jordan et al., 1997; Nicoll et al., 1995; Teasdale et al., 1997) because of reduced levels of apoE or because of apoE isoform-specific effects. For example, reduced concentrations of apoE have been shown in the brains of patients with Alzheimer's disease with an APOE ∊4 allele (Bertrand et al., 1995). The concentration of available apoE may be the factor of key importance in the response to acute brain injury. Alternatively, the isoform of the available apoE may be of major importance as many of the studies of putative mechanisms of action of apoE have shown isoform specific differences with the apoE E4 generally having reduced efficiency compared with E3. For therapeutic purposes, in patients with a head injury, a continuous intrathecal infusion could conceivably be used to directly deliver neuroprotective agents such as an apoE mimetic.
Our data substantiate a critical role for apoE in the response of the CNS to injury and demonstrate the potential utility of apoE infusion as a therapy for the reduction of neuronal damage in individuals after acute brain injury.
Footnotes
Acknowledgments
The authors thank Michael Evans who arranged the transfer of animals from Glaxo Wellcome to the Wellcome Surgical Institute, Scott Poynter who was responsible for polymerase chain reaction genotyping at Glaxo-Wellcome, and Mrs. Janice Stewart for confirmation of the mouse genotypes at the Department of Neuropathology, Glasgow.