
Abstract
Interleukin-1 (IL-1) converting enzyme (ICE) is a cysteine protease that cleaves inactive pro-IL-1β to active IL-1β. The pro-inflammatory cytokine IL-1β is implicated as a mediator of hypoxic-ischemic (HI) brain injury, both in experimental models and in humans. ICE is a member of a family of ICE-like proteases (caspases) that mediate apoptotic cell death in diverse tissues. The authors hypothesized that in neonatal mice with a homozygous deletion of ICE (ICE-KO) the severity of brain injury elicited by a focal cerebral HI insult would be reduced, relativefto wild-type mice. Paired litters of 9- to 10-day-old ICE-KO and wild-type mice underwent right carotid ligation, followed by 70 or 120 minutes of exposure to 10% O2, In this neonatal model of transient focal cerebral ischemia followed by reperfusion, the duration of hypoxia exposure determines the duration of cerebral ischemia and the severity of tissue damage. Outcome was evaluated 5 or 21 days after lesioning; severity of injury was quantified by morphometric estimation of bilateral cortical; striatal, and dorsal hippocampal volumes. In animals that underwent the moderate HI insult (70-minute hypoxia), damage was attenuated in ICE-KO mice, when evaluated at 5 or 21 days post-lesioning. In contrast, in mice that underwent the more severe HI insult (120-minute hypoxia), injury severity was the same in both groups. Reductions in intra-HI CBF, measured by laser Doppler flowmetry, and intra- and post-HI temperatures did not differ between groups. These results show that ICE activity contributes to the progression of neonatal HI brain injury in this model. Whether these deleterious effects are mediated by proinflammatory actions of IL-lβ and/or by pro-apoptotic mechanisms is an important question for future studies.
A growing body of epidemiologic, pathologic, and experimental evidence implicates inflammatory mediators, including the pleiotropic pro-infammatory cytokine interleukin-1β (IL-1β), in the pathophysiology of perinatal and neonatal cerebral hypoxic-ischemic (HI) injury (Silverstein et al., 1997; Yoon et al., 1997a; Martin-Ancel et al., 1997). Much of this experimental work has been done in a model of focal cerebral HI induced by unilateral carotid ligation followed by timed exposure to 8% oxygen in 7-day-old rats (Rice et al., 1981). In this model the duration of hypoxia exposure determines the duration of cerebral ischemia and the severity of subsequent tissue damage (Towfighi et al., 1991). Also, cerebral ischemia is followed by resumption of normal CBF, after the end of hypoxia exposure (Mujsce et al., 1990). Thus, this model incorporates both focal cerebral ischemia and subsequent reperfusion. In 7-day-old rats, IL-1β mRNA and IL-1 bioactivity increase acutely in HI brain tissue, within hours after cerebral HI (Szaflarski et al., 1995; Hagberg et al., 1996). If IL-1β is a pivotal mediator of brain injury, then it would be predicted that functional inhibition of IL-1β could confer neuroprotection. In the neonatal model several strategies to block IL-1 activity have been tested. For example, systemic or intracerebroventricular administration of IL-1 receptor antagonist protein attenuated HI injury in 7-day-old rats (Martin et al., 1994; Hagberg et al., 1996). Another potential strategy is inhibition of enzymatic production of mature IL-1β by IL-1 converting enzyme (ICE). Systemic or intracerebroventricular administration of the general ICE-like protease (caspase) inhibitor boc-aspartyl(OMe)-fluoro-methylketone attenuated HI injury in P7 rats (Cheng et al., 1998). Despite the latter finding, the role of ICE in neonatal cerebral ischemia is unclear; although caspase-3–like activity increases after a neonatal HI insult, there is no evidence of increased ICE (caspase 1) activity after neonatal cerebral HI (Cheng et al., 1998). Factors that might account for these disparate outcomes are unknown. Genetically modified mice could represent a complementary approach to evaluating the pathophysiologic roles of IL-1β and ICE in neonatal cerebral HI.
ICE is a cysteine protease, synthesized by cells of the monocytic lineage, which cleaves inactive, pro-IL-1β to the active pro-inflammatory cytokine IL-1β (Dinarello, 1996). ICE is a member of a family of proteases, or caspases, that play distinct and complex roles as mediators of the apoptotic cell death cascade (Yuan et al., 1993; Schwartz and Milligan, 1996). Although overexpression of ICE can induce apoptosis in vitro (Miura et al., 1993), it is unclear whether ICE itself mediates apoptosis in vivo. To delineate the in vivo role of ICE, genetically modified mice deficient in a functional gene for ICE were produced (Li et al., 1995; Kuida et al., 1995). These ICE-deficient mice are morphologically normal, fertile, and capable of radiation or dexamethasone-induced apoptosis in thymocytes and ATP-induced apoptosis in macrophages (Li et al., 1995; Kuida et al., 1995). The only apoptotic defect in these mice is in Fasmediated apoptosis (Kuida et al., 1995). In contrast with caspase-3 knockout mice, in which brain development is hyperplastic and disorganized (Kuida et al., 1996), ICE-deficient mice have normal CNS development (Li et al., 1995). In vitro, macrophages from ICE-deficient mice can produce IL-1β precursors, but are incapable of producing or secreting active IL-1β (Li et al., 1995). In vivo, ICE-deficient mice are resistant to endotoxin-induced lethality (a cytokine mediated model of septic shock) and lack a circulating IL-1β response to endotoxin (Li et al., 1995). The extent to which other pro-inflammatory cytokines compensate for deficient IL-1 activity in these animals is currently unknown.
Because they do not produce mature IL-1β, ICE-deficient mice could enable direct evaluation of the hypothesis that IL-1β is a mediator of neonatal HI brain injury. The potential utility of this strategy is supported by the recent report that adult ICE-deficient mice are resistant to focal cerebral ischemic injury (Schielke et al., 1998). Yet, it is important to emphasize that results in an adult animal stroke model cannot be directly extrapolated to neonatal animals. Maturational stage may influence pathophysiologic mechanisms both of injury and of recovery. For example, in studies to evaluate the role of superoxide dysmutase in ischemic brain injury that incorporated superoxide dysmutase overexpressing mice, completely disparate results were obtained between neonatal and adult animals (Ditelberg et al., 1996; Yang et al., 1994). We hypothesized that in neonatal mice deficient in ICE, the severity of brain injury elicited by focal cerebral HI would be reduced when compared to concurrently lesioned wild-type (WT) controls.
METHODS
Cerebral hypoxia—ischemia
The neonatal rat model of unilateral carotid artery ligation followed by timed hypoxia exposure is designed to elicit ipsilateral forebrain infarction (Rice et al., 1981), and was recently adapted for use in mice (Ditelberg et al., 1996; Ferriero et al., 1996). In the neonatal rat, the minimum duration of hypoxia necessary for induction of cerebral ischemia is I to 1.5 hours (Silverstein et al., 1984; Towfighi et al., 1991), and severity of ipsilateral injury corresponds directly with duration of hypoxia exposure (Towfighi et al., 1991). In the neonatal rat, CBF decreases during hypoxia exposure, but returns to normal after the cessation of hypoxia expopure (Mujsce et al., 1990), Thus, this model incorporates both focal cerebral ischemia and reperfusion. In neonatal mice, a shorter duration of hypoxia exposure induces infarction (Ditelberg et al., 1996; Ferriero et al., 1996), Precise correlations with human brain development are difficult; however, brains of 7- to 10-day-old mice and rats are similar to third-trimester human fetuses (and thus premature human newborns), in terms of cellular proliferation, cortical organization, synapse number, neurochemical indices such as neurotransmitter synthetic enzymes, and electrophysiology (Dobbing and Sands, 1979; Romijn et al., 1991; Marret et al., 1995; McIlwain and Bachelard, 1985).
Postnatal day 9 to 10 mice, weighing 4 to 5 g, underwent right common carotid artery ligation under methoxyflurane anesthesia. We used 9- to 10-day-old mice based on preliminary experience with WT mice in which surgical mortality in WT was >50% in 7-day-old mice weighing 2 to 3 g. After 1 hour of recovery, mice were placed in 10% oxygen (balance nitrogen), in glass chambers partially immersed in a 38°C water bath (chamber air temperature 37°C). We used 10% oxygen rather than 8%, which is usually used in the rat model, based on preliminary experiments in WT mice (data not shown) in which 8% oxygen elicited inconsistent lesions and high mortality. Use of 10% oxygen yielded more uniformity of lesion severity at a given duration of hypoxia exposure and permitted titration of lesion severity. After the end of hypoxia exposure, mice were observed in a heated incubator with air temperature maintained at 37°C for 30 minutes, then they were returned to their mothers. Brief clonic seizures lasting a few seconds were observed infrequently in mice from both groups. Mice in both groups resumed normal activity by 30 minutes after the end of hypoxia, and fed well when returned to the dam. In a preliminary experiment with ICE-deficient animals, a large litter (n = 12) underwent HI lesioning and there was a high mortality rate (8 of 12 died intraoperatively or postoperatively); data from this experiment were not included. In subsequent experiments, litters were culled preoperatively to a maximum of eight, and mortality markedly decreased subsequently. To obtain samples for evaluation of histopathologic outcome, mice were decapitated and brains rapidly dissected and frozen under powdered dry ice either 5 days or 21 days later. Experimental protocols were approved by the University of Michigan Committee on Use and Care of Animals. All efforts were made to minimize animal suffering and minimize the number of animals used.
In one experiment, we evaluated intrahypoxic and post-hypoxic body temperature in both ICE-deficient (n = 7) and WT (n = 8) mice because IL-1β plays a role in thermoregulation, and changes in core temperature can alter brain injury severity (Yager et al., 1993; Saeed et al., 1993). Skin temperature was measured every 15 minutes during hypoxia and esophageal temperature at 15, 30, 60 and 120 minutes post-hypoxia (YSI thermometer 43TA with probe 554, Yellow Spring Instruments, Yellow Spring, OH, U.S.A.).
ICE-deficient and control mice
The ICE-deficient mice were generated by homologous recombination in embryonic stem cells, by investigators at BASF Corporation, (Worcester, MA, U.S.A.) (Li et al., 1995), and homozygous ICE-deficient mice (ICE -/-) were interbred since 1994. The WT (ICE +/+) mice were generated from the same chimeric founder, and also interbred among themselves since 1994. In this study heterozygotes were not used. Mice were the product of matings between either homozygous ICE-deficient (ICE -/-) or homozygous WT (ICE +/+) partners. Both strains have a mixed background of 129/Sv and C57BL/6-derived genes (S. Banerjee, personal communication, February 1998). Genetic background of both knockouts and controls is a critical issue when using genetically modified mice in cerebral ischemia research. In both adult and neonatal mice, the 129 strain is resistant to ischemia, compared to the susceptible C57BL/6 strain (Yang et al., 1997; Connolly et al., 1996; O'Donnell et al., 1996; Sheldon et al., 1997). Before we began our studies with the ICE knockouts, we first established that unilateral carotid ligation followed by exposure to 10% O2 resulted in ipsilateral cerebral infarction in the related WT mice of mixed 129/Sv and C57BL/6 background. We also verified that body ad brain size were the same in ICE-deficient and WT mice in the first month of life. We obtained ICE-deficient and WT mice born on the same dates (n = 8 to 9/group), and measured body and brain weights on day of life 31; body and brain weights of the two groups did not differ [WT (n = 8) versus ICE-deficient (n = 9), mean ± SD: body weight (g) 20.8 ± 2.5 versus 21.8 ± 2.5; brain weight (g) 0.32 ± 0.01 versus 0.32 ± 0.03].
Histopathologic outcome of neonatal cerebral hypoxia—ischemia
Histopathologic outcome was evaluated in seven independent experiments, each of which included concurrently lesioned litters of ICE-deficient and WT mice. In the first three experiments, all mice, both ICE-deficient (n = 24) and WT (n = 25), were exposed to 10% oxygen for 120 minutes, and histopathology was evaluated 5 days later. Based on initial results, the protocol was modified, and in the next four experiments, all mice, both ICE-deficient (n = 27) and WT (n = 25), were exposed to 10% oxygen for 70 minutes. Histology was evaluated both 5 days later (two paired litters, n = 11, ICE-deficient and n = 12 WT) and 3 weeks later (two paired litters, n = 16, ICE-deficient and n = 13 WT).
To evaluate tissue injury, 20-μm coronal frozen brain sections, post-fixed over paraformaldehyde vapors, were stained with cresyl violet for assessment of histopathology and morphometry. A computerized video-camera-based image analysis system (with NIH Image software) was used to measure bilateral neocortical, striatal, and hippocampal cross-sectional areas from the level of the anterior genu to the posterior genu of the corpus callosum. Measurements were obtained by an observer unaware of animal group assignment. Five brains were excluded (two WT, severe HI, 5-day survivors; one ICE-deficient, severe HI, 5-day survivor; one WT, moderate HI, 5-day survivor; and one ICE-deficient, moderate HI, 5-day survivor) because poor quality of sectioning precluded image analysis. Cortex and striatum measurements included only intact tissue; evaluation of tissue integrity was based on intensity and uniformity of cresyl violet staining. Hippocampal measurements included the entire residual tissue because it was often not possible to reliably outline intact tissue on the basis of distinct and well-demarcated differences in staining (Figs. 1 to 3). Regional volumes were estimated by summating areas and multiplying by the distance between sections.
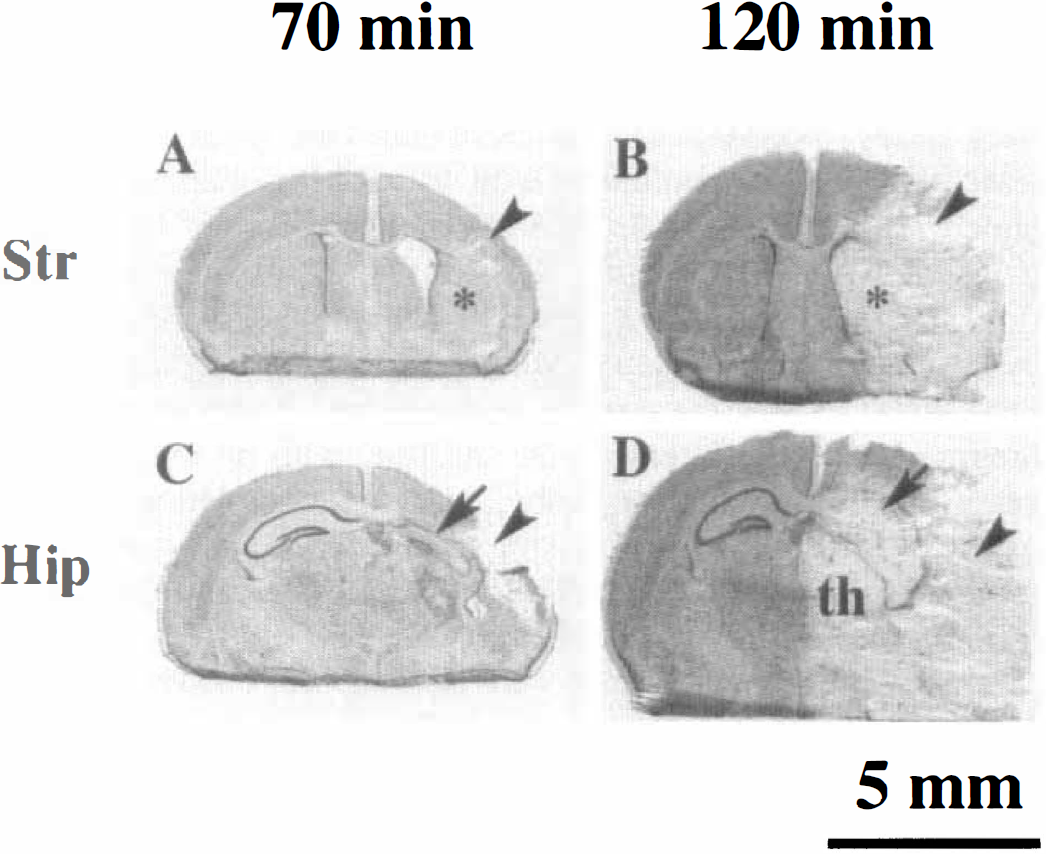
Severity of hypoxia-ischemia—induced brain damage is determined by duration of hypoxia exposure. These coronal sections, taken 5 days after lesioning, at the level of the striatum
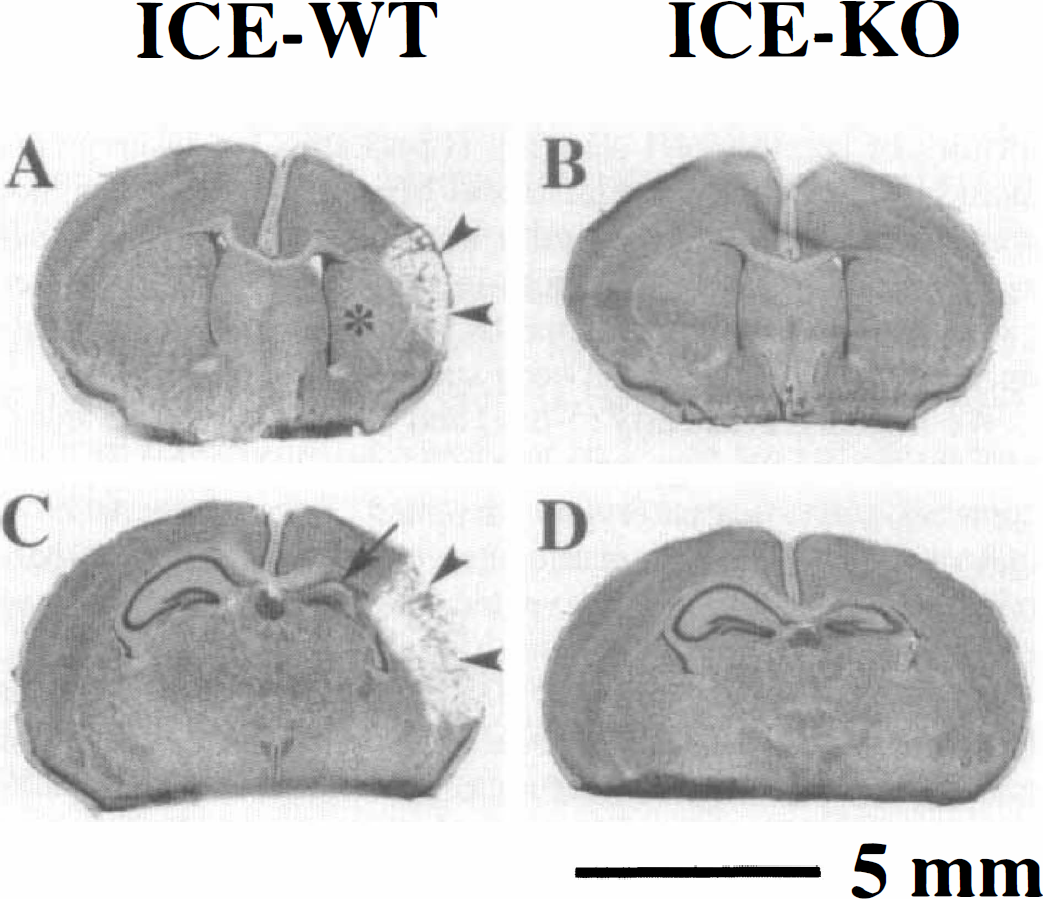
Attenuation of brain damage 5 days after neonatal hypoxic-ischemic lesioning in interleukin-1 converting enzyme (ICE)–deficient neonatal mice. These coronal sections taken at the level of striatum
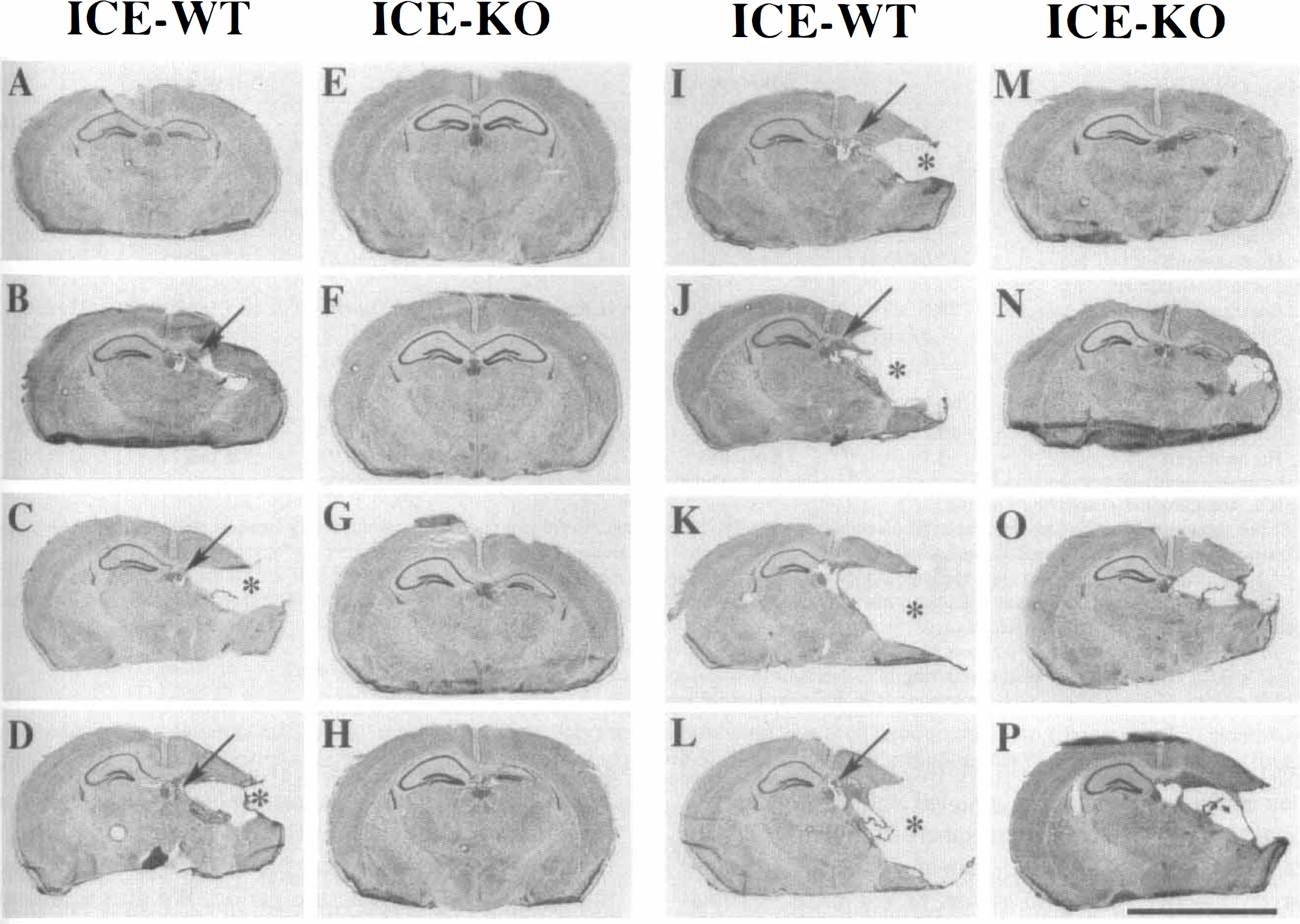
Attenuation of brain damage 21 days after neonatal hypoxic-ischemic lesioning in interleukin-1 converting enzyme (ICE)–deficient neonatal mice. These coronal sections, at the level of the dorsal hippocampus, were obtained from eight wild-type (ICE-WT, panels
We included both early (5-day) and late (21-day) time intervals for neuropathology outcome measures to confirm that any apparent neuroprotection was sustained. Recent reports have indicated that some therapeutic interventions, such as hypothermia, delayed, rather than prevented, the ultimate expression of cerebral ischemic injury (Dietrich et al., 1993; Trescher et al., 1997). This delayed neuronal death may be apoptotic rather than necrotic in nature (Du et al., 1996). Because ICE could contribute to either early necrotic or later apoptotic death pathways, we evaluated outcome at both time points.
CBF measurement
To evaluate whether there were equivalent reductions in CBF in ICE knockout and WT animals in additional experiments, we evaluated bilateral cortical blood flow by laser Doppler flowmetry (n = 8 WT, from two litters; n = 9 ICE-deficient, from two litters) at 5, 15, 30, 45 and 70 minutes after the onset of 10% oxygen exposure. To obtain reliable measurements, continuous anesthesia was required and isoflurane was used. P9-10 mice underwent right carotid ligation as described above. Then, a midline scalp incision was made so that the blood flow probe could be applied directly to the skull surface. Animals were exposed to 10% oxygen during continuous isoflurane anesthesia (4% for induction, 2% for maintenance) for 70 minutes. In these experiments, hypoxia exposure was via a snug-fitting, non-rebreathing anesthesia face mask, and body temperature was maintained with a warming mattress maintained at 37°C. The scalp incision and probe were frequently irrigated with sterile saline solution. Bilateral cortical blood flow was measured over a period of 1 minute per measurement, by laser Doppler flowmetry (Model BMP2, Vasamedics Inc. St. Paul MN, U.S.A.); the probe was positioned 2 mm posterior to bregma and 2 mm lateral to the midline, with no major blood vessel visible beneath the probe. After the completion of hypoxia exposure, the scalp incision was sutured, and mice were returned to the dam. Histopathology was analyzed 5 days after Iesioning, as described above.
Data analysis
Microcomputer-based statistical programs [Statview II (ABACUS, Berkeley, CA, U.S.A.), Systat (Systat, Inc., Evanston, IL, U.S.A.) and InStat (GraphPad Software, San Diego, CA, U.S.A.)] were used. Right- and left-sided cortical, striatal, and hippocampal volumes were compared between ICE-deficient and WT mice by unpaired, two-tailed t-tests, and by the Mann-Whitney test for nonparametric data. Percent damage, or percent reduction in regional volume, ipsilateral to carotid ligation, (i.e., mean % left-right difference in volumes [100*(L-R)/L]) was first compared independently for each of the three regions evaluated, between ICE-deficient and WT mice, by unpaired two-tailed t-tests. To evaluate regional differences in the effect of ICE on the severity of brain damage, percent damage was also compared between groups by two-way analysis of variance factoring group and region. Post-hoc testing (Scheffe test) was used to evaluate the significance of subgroup differences. To evaluate the magnitude of the protective effect of ICE deficiency on the severity of brain damage, percent protection was calculated, based on comparison of the percent damage in ICE-deficient and WT animals, 5 days after 70 minutes of HI, using the formula
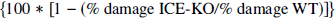
(McDonald et al., 1989). As a measure of the variability of neuroprotection values observed, the SD for %protection was calculated as
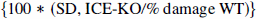
(McDonald et al., (989). Serial intra-HI and post-HI animal temperatures, and serial bilateral cortical blood flow values, were compared between groups by repeated measures analysis of variance.
RESULTS
In all WT mice exposed to 120 minutes of HI, hemispheric infarction was readily apparent 5 days later; 15 of 18 had severe ipsilateral hemispheric infarctions and 1 of 18 had bilateral infarction. In all WT mice exposed to 70 minutes of HI there was at least microscopic evidence of brain damage ipsilateral to the carotid ligation, both at 5 and 21 days post-lesioning. In this latter control group there was substantial variability in severity; abnormalities in the cortex, striatum, and hippocampus ranged from regional atrophy with preservation of anatomic landmarks to infarction (most common in lateral cortex) (Fig. 1). The mean severity of ipsilateral tissue loss in WT mice was directly related to the duration of hypoxia exposure (Table 1). Both the variability in severity of injury in WT mice that underwent the same lesioning procedure and the relationship between duration of hypoxia exposure and severity of tissue damage replicate findings in 7-day-old rats that undergo unilateral carotid ligation followed by 2 to 3 hours of exposure to 8% O2 (Rice et al., 1981; Towfighi et al., 1991). Mortality did not differ between groups overall (7 of 51 ICE-deficient; 6 of 50 WT), nor between groups exposed to either severe (6 of 24 ICE-deficient; 2 of 25 WT) or moderate hypoxia-ischemia (l of 27 ICE-deficient; 4 of 25 WT). The majority of deaths occurred in the period between post-HI recovery and the intended time of brain removal.
Regional volume measurements after moderate or severe unilateral cerebral hypoxia-ischemia in ICE-deficient and wild-type neonatal mice
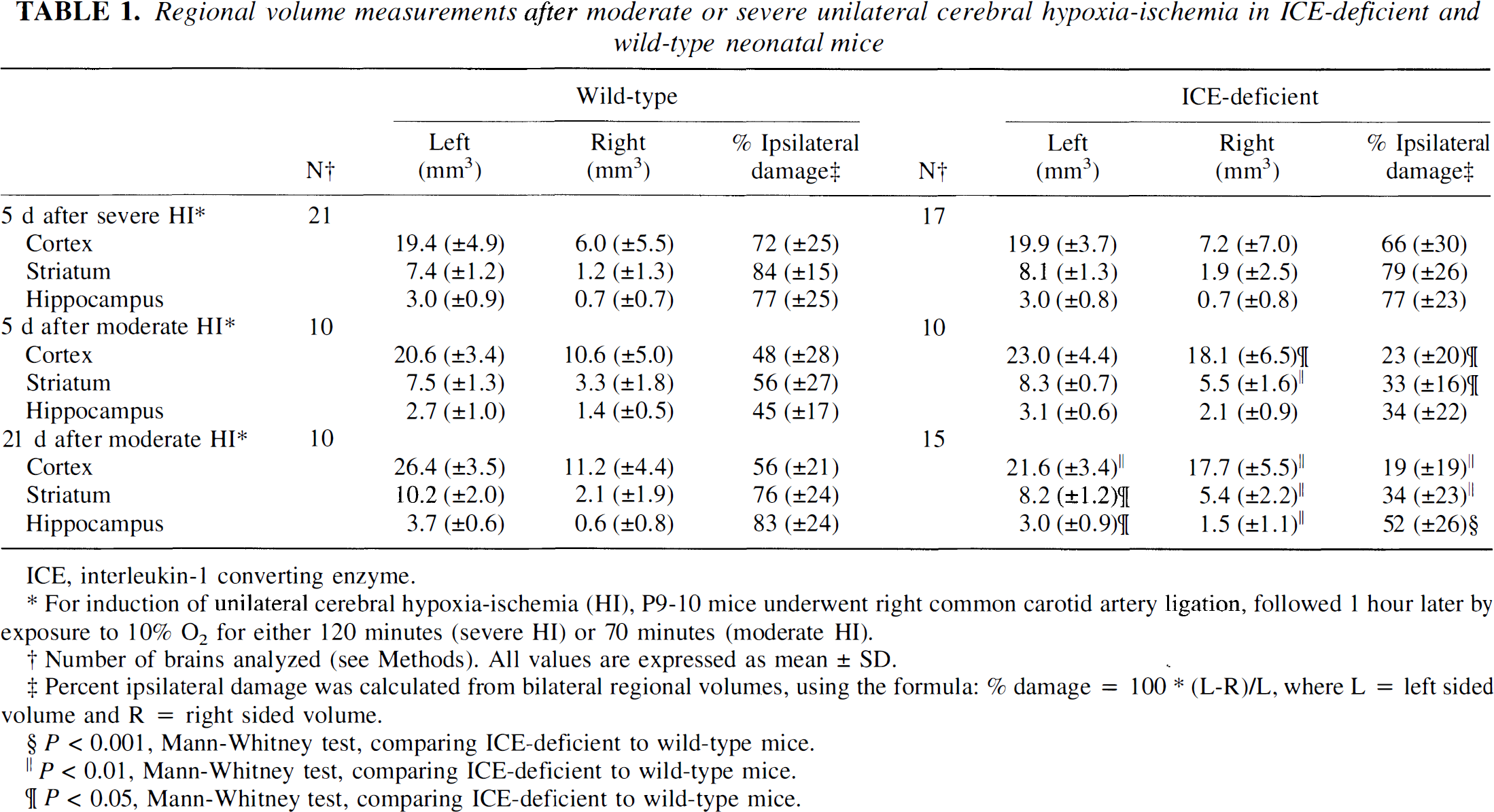
ICE, intcrleukin-1 converting enzyme.
For induction of unilateral cerebral hypoxia-ischemia (HI), P9-10 mice underwent right common carotid artery ligation, followed 1 hour later by exposure to 10% O2 for either 120 minutes (severe HI) or 70 minutes (moderate HI).
Number of brains analyzed (see Methods). All values are expressed as mean ± SD.
Percent ipsilateral damage was calculated from bilateral regional volumes, using the formula: % damage = 100 * (L-R)/L, where L = left sided volume and R = right sided volume.
P < 0.001, Mann-Whitney test, comparing ICE-deficient to wild-type mice.
P < 0.01, Mann-Whitney test, comparing ICE-deficient to wild-type mice.
P < 0.05, Mann-Whitney test, comparing ICE-deficient to wild-type mice.
In the first three experiments in paired litters of ICE-deficient and WT mice, under conditions (120 minutes of hypoxia exposure) that induced severe ipsilateral cerebral infarctions, there was no intergroup difference in the severity of cortical, striatal, or hippocampal damage evaluated at 5 days post-lesioning (Table 1).
In subsequent experiments, under conditions (70 minutes of hypoxia exposure) that induced damage of moderate severity ipsilateral to the carotid ligation in WT mice, there was attenuation of HI brain damage in ICE-deficient mice, as compared with concurrent WT controls (Table 1 and Fig. 2). In animals evaluated at 5 days after lesioning, a visible cortical scar or cavitation was observed at the time of brain removal in 8 of 10 WT mice, but in only 3 of 10 ICE-deficient mice. The incidence of cerebral infarction at 21 days after lesioning was 7 of 10 in WT mice, but only 4 of 15 in ICE-deficient mice.
Comparison of regional volume measurements confirmed this trend. At 5 days, the severity of ipsilateral cortical and striatal damage was significantly reduced in ICE-deficient mice, compared to WT controls (Table 1 and Fig. 2). At 5 days, within each group, the severity of injury was similar in all three regions evaluated. In the ICE-deficient group, there was no difference in the degree of attenuation of injury between cortex and striatum; in comparison to WT controls, the mean (± SD) percent protection, a measure of reduction in injury, in ICE-deficient mice was 53% (±41) and 40% (±29) in cortex and striatum, respectively. On histopathologic evaluation, hippocampal injury also appeared to be attenuated in the ICE-deficient mice; however, comparison of right hippocampal volumes did not show statistical significance. This finding may reflect underestimation of the extent of injury in WT controls because the method that was used included measurement of the entire residual hippocampal area and did not distinguish between intact and damaged tissue.
The attenuation of moderate HI brain injury in ICE-deficient mice persisted at 3 weeks postiesioning, and was confirmed in all three regions evaluated (Table 1 and Fig. 3). At 3 weeks after lesioning, there was no difference in the attenuation of injury associated with ICE deficiency among the three regions evaluated. In WT mice, the magnitude of ipsilateral cortical, striatal, and hippocampal damage was greater at 3 weeks than at 5 days after lesioning, based on percentage reductions in ipsilateral regional volumes (Table I). This trend likely reflects continuing growth of the intact contralateral hemisphere over 3 weeks, coupled with reduced development of the ipsilateral hemisphere during this phase of rapid brain growth, rather than a difference in injury severity between the two groups of WT mice. Analysis of regional volume measurements suggested that the contralateral hemisphere was smaller in ICE-deficient mice than in concurrent WT controls at 3 weeks post-lesioning (Table 1); although the significance of this finding is currently uncertain, it did not reflect any intrinsic difference in brain size between the two groups (see Methods).
Intra-HI skin temperatures and post-HI esophageal temperatures were measured in ICE-deficient mice and concurrent WT controls; no between-group differences were noted (Table 2).
Intra- and post-hypoxic-ischemic body temperature in ICE-deficient and wild-type mice
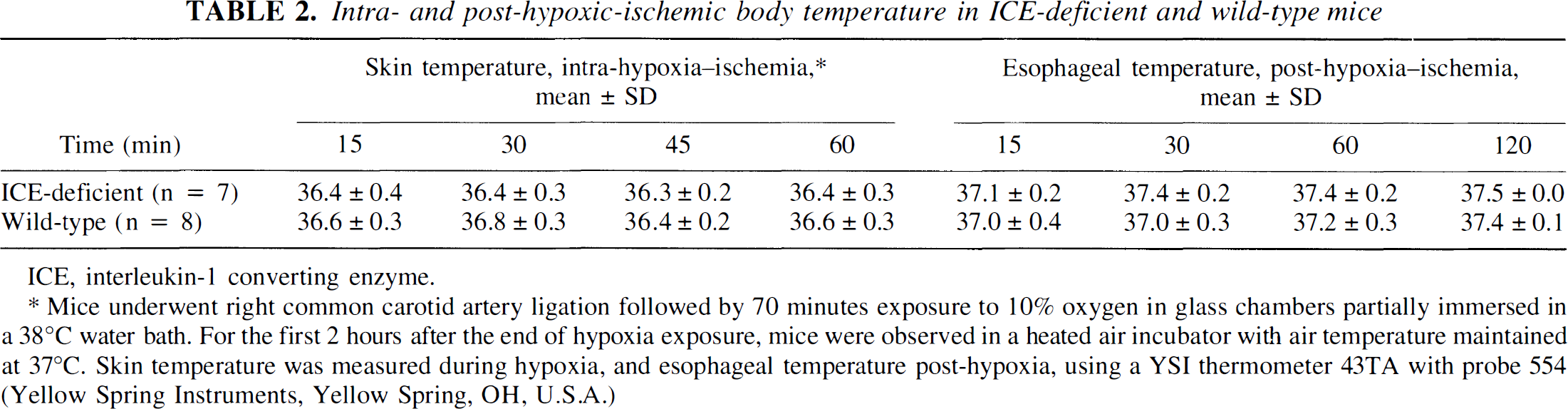
ICE, interleukin-1 converting enzyme.
Mice underwent right common carotid artery ligation followed by 70 minutes exposure to 10% oxygen in glass chambers partially immersed in a 38°C water bath. For the first 2 hours after the end of hypoxia exposure, mice were observed in a heated air incubator with air temperature maintained at 37°C. Skin temperature was measured during hypoxia, and esophageal temperature post-hypoxia, using a YSI thermometer 43TA with probe 554 (Yellow Spring Instruments, Yellow Spring, OH, U.S.A.)
To begin to evaluate possible mechanisms of reduced injury in the ICE-deficient neonatal mice, we measured bilateral cortical blood flow in additional mice from both groups, during 70 minutes of unilateral cerebral HI. Ipsilateral to the carotid ligation there were rapid, abrupt intrahypoxic reductions in cortical blood flow of similar magnitude in both groups (Fig. 4). There was no reduction in contralateral cortical blood flow in either group during the hypoxia exposure period. We also evaluated the neuropathologic outcome in the mice used for CBF measurements 5 days after lesioning. The magnitude of brain tissue loss in both WT and ICE-deficient mice, as evaluated by regional volumes, was less severe than in the preceding experiments; the percent damage ipsilateral to carotid ligation ranged from 12% to 27% in WT mice, and from 8% to 21% in ICE-deficient mice. This difference could reflect a neuroprotective effect of sustained intrahypoxic isoflurane anesthesia, or an independent systematic difference in the experimental conditions (such as a small reduction in mean body temperature). In these animals, to evaluate outcome, regional histopathology in cortex, striatum, hippocampus, and thalamus was evaluated, instead, using a semiquantitative four point injury score, as previously described (Barks et al., 1995). The cumulative injury score (lowest score 0, maximum possible score 12) was compared between groups. Based on this scoring system, ipsilateral damage was again attenuated in the ICE-deficient mice [Mean (±SD) cumulative injury score: 6.6 ± 4.1 WT; 2.2 ± 2.2 ICE-deficient; P < .05, one-tailed t-test].
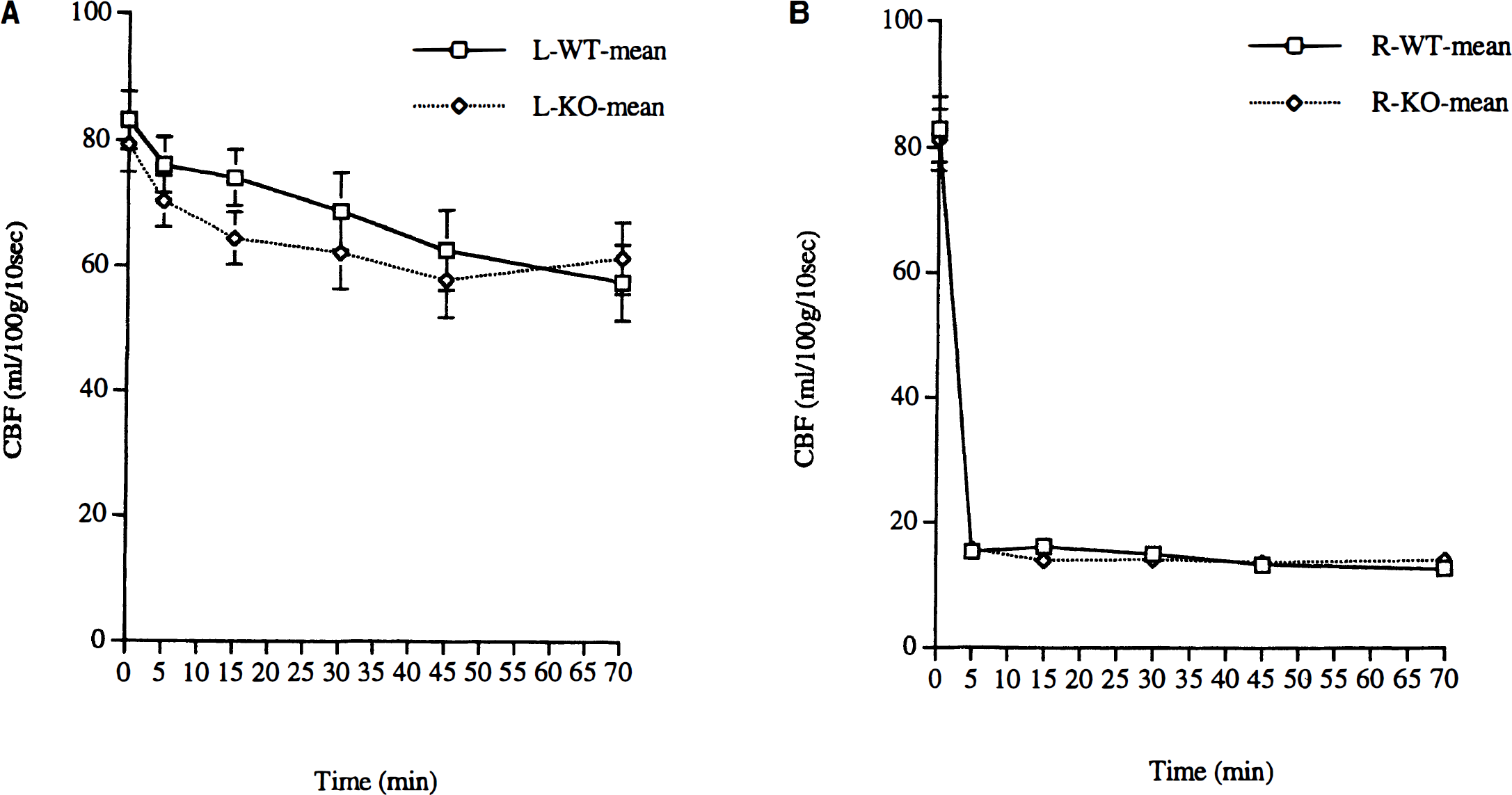
Intra—hypoxic-ischemic cortical CBF in interleukin-1 (ICE)–deficient and Wild-type mice. Right carotid artery ligation was performed in isoflurane anesthetized P9-10 animals. Laser Doppler (see Methods) was then used to measure bilateral cortical blood flow. Measurements were obtained five times during 70 minutes of exposure to 10% oxygen, in isoflurane-anesthetized wild-type (n = 8) and ICE-deficient (n = 9) mice. Mean ipsilateral CBF was reduced to a similar extent in wild-type (WT,
DISCUSSION
ICE was initially identified as the enzyme that cleaves inactive pro-IL-1β to form the biologically active proinflammatory cytokine IL-1β (Dinarello, 1996). Subsequently, ICE was determined to be the prototypic member of a family of cysteine proteases, referred to as caspases. The caspases, including ICE, are implicated as pivotal mediators of apoptotic cell death (Miura et al., 1993). There is recent intriguing evidence that apoptotic mechanisms contribute to neonatal HI-induced brain injury (Hill et al., 1995; De Torres et al., 1997; Cheng et al., 1998); in addition, there is a rapidly expanding body of evidence that apoptosis contributes to ischemic damage in the mature CNS (Du et al., 1996; Chen et al., 1998; Ni et al., 1998). ICE might mediate neuronal death either via pro-inflammatory pathways leading to necrosis, or by initiation of apoptosis, either directly via a caspase cascade, or indirectly via IL-1β. Morphologic evidence suggests that neuronal death in the immature brain may occur along a continuum from apoptosis to necrosis (Portera-Cailliau et al., 1995).
Our results show that in neonatal mice, the absence of ICE confers resistance to HI brain injury. Under conditions that resulted in mild to moderately severe tissue injury in WT controls, the severity of brain damage was substantially attenuated in ICE-deficient mice. The ICE-deficient animals remained vulnerable to severe HI insults. Under conditions that result in mild to moderate tissue injury in WT controls, intrahypoxic reductions in CBF were the same in both groups, as were intra-HI and early post-HI body temperatures. Because some interventions (e.g. hypothermia) may delay rather than prevent injury, it was important to confirm neuroprotection with a longer survival period. The persistence of the protective effect of ICE deficiency up to 3 weeks after neonatal cerebral HI provided direct evidence that the beneficial effects of ICE deficiency were sustained.
Our finding that ICE activity contributes to the evolution of cerebral ischemic injury in neonatal mice is consistent with the hypothesis that IL-1β is a mediator of neonatal HI brain injury. Brain IL-1β activity and mRNA expression are upregulated acutely after unilateral cerebral HI in neonatal rats (Szaflarski et al., 1995; Hagberg et al., 1996). Systemic or intracerebroventricular injection of IL-1 receptor antagonist protein attenuated HI damage in neonatal rats; systemic treatment resulted in up to 100% reduction in damage (Martin et al., 1994). The ICE-deficient mice used in our studies are known to be deficient in systemic IL-1β (and IL-1α) production in vivo, in response to endotoxin (Li et al., 1995). It is unlikely that these mutants are able to produce active IL-1β in the brain during cerebral ischemia; however, it is theoretically possible that these mice could produce mature IL-1β from pro-IL-1β via other proteases (Fantuzzi et al., 1997). CNS IL-1β production was not evaluated in these mice because currently available antibodies do not distinguish mature IL-1β from pro-IL-1β. Although IL-1β is a mediator of the febrile response, we found no evidence of potentially neuroprotective hypothermia in ICE-deficient mice compared to WT controls.
Attenuation of HI injury in ICE-deficient mice could be attributable, at least in part, to interruption of an ischemia-induced, IL-1β-mediated apoptotic cascade. Transgenic mice overexpressing a dominant negative mutant ICE are unable to produce mature IL-1β, are resistant to trophic factor withdrawal-induced neuronal apoptosis, and are resistant to temporary and permanent focal cerebral ischemia (Hara et al., 1997a; Friedlander et al., 1997). Yet, the physiologic role of ICE in the CNS is uncertain. There is no evidence that ICE plays a role in physiologic apoptosis in the developing CNS. In ICE-deficient mice, brains appear normally developed; this finding is in marked contrast with the abnormal brain development of caspase-3–deficient mice (Kuida et al., 1996). It is also possible that ICE proteolytic activity has other deleterious effects in the ischemic brain, but no additional substrates for ICE are currently known.
In addition to promoting apoptosis, I1-1β has many deleterious pro-inflammatory effects that could contribute to necrotic cell death, including recruitment and activation of leukocytes via adhesion molecules or chemotactic factors, and stimulation of free-radical generating enzymes, including cyclo-oxygenase 2, nitric oxide synthase, and phospholipase A2 (Dinarello, 1996). Several lines of evidence indicate that an inflammatory cascade contributes to the pathogenesis of neonatal cerebral hypoxic ischemic damage. Neutrophils and activated macrophages and microglia accumulate in damaged tissue after neonatal cerebral HI (Ivacko et al., 1996; Hudome et al., 1997). A broad range of anti-inflammatory strategies, including neutrophil depletion, platelet activating factor antagonist treatment, IL receptor antagonist protein administration, nitric oxide synthesis inhibition, and free radical scavenger treatment all can attenuate tissue damage after neonatal cerebral HI (Trifiletti, 1992; Palmer et al., 1993; Martin et al., 1994; Liu et al., 1996; Hudome et al., 1997). IL-1β may represent a common stimulus for many components of this inflammatory cascade. These inflammatory mechanisms may be highly relevant clinically, in light of recent evidence that proinflammatory mediators may play a role in the genesis of human perinatal brain damage such as peri ventricular leukomalacia, and in long-term neurologic deficits originating in the perinatal period (Grether et al., 1996; Deguchi et al., 1996; Yoon et al., 1997b).
Support for a direct role of caspases in the pathogenesis of neonatal cerebral ischemic injury is provided by a recent report that intracerebroventricular or systemic administration of a pan-caspase inhibitor attenuated unilateral HI injury in 7-day-old rats; systemic treatment resulted in a 33% to 66% reduction in damage measured 7 days later, depending on the brain region analyzed (Cheng et al., 1998). The percentage injury reduction in ICE-deficient mice 5 days post-HI is of the same order of magnitude as the degree of protection afforded by pan-caspase inhibitor treatment of neonatal rats. The present results are also consistent with previous reports in adult mice. Adult mice of the same ICE-deficient strain are resistant to cerebral ischemic damage induced by permanent middle cerebral artery occlusion, with a 52% mean reduction in lesion volume compared to controls (Schielke et al., 1998). Treatment with several general caspase inhibitors is also neuroprotective in adult rat and mouse models of reversible focal cerebral ischemia (Hara et al., 1997b; Endres et al., 1998). Thus current evidence suggests that caspases contribute to the progression of cerebral ischemia- or ischemia-reperfusion—induced injury in several distinct models in the neonate and adult.
The neuroprotective effect of ICE deficiency did not exhibit regional selectivity in neonatal mice subjected to focal cerebral HI; this suggests that ICE is widely distributed in the neonatal mouse brain. In normal adult gerbils, ICE-like immunoreactivity is localized in scattered hippocampal interneurons. After global forebrain ischemia in gerbils, ICE-like immunoreactivity is localized in microglia in the area of evolving apoptotic neuronal death, but not in the apoptotic neurons (Bhat et al., 1996). Regional or cellular localization of ICE or its mRNA in neonatal or adult rats or mice has not been described.
There are no well established quantifiable indices of white matter injury in the neonatal rodent models. Thus, white matter injury was not systematically evaluated in the present study. In the routine histopathologic specimens, gray and white matter appeared to be affected by HI to a similar degree, and there was attenuation of both gray and white matter damage in ICE-deficient mice. A growing body of literature supports a role for proinflammatory cytokines, including IL-1β, in human perinatal white matter injury (Yoon et al., 1997b; Deguchi et al., 1996). Additionally, it is reported that oligodendroglial death after experimental global forebrain ischemia is apoptotic (Petito et al., 1998). Thus, evaluation of selective white matter injury after neonatal cerebral HI in these ICE-deficient mice may be an important issue for future study.
Maturational stage may influence pathophysiologic mechanisms both of injury and of recovery. Thus, it was not a foregone conclusion that ICE-deficient neonatal mice would be resistant to HI brain injury. Mature IL-1β may have trophic effects that are unique to the developing brain (Brenneman et al., 1992; Strijbos and Rothwell, 1995), and some neurotrophic factors protect the neonatal rodent brain from HI or excitotoxic injury (Nozaki et al., 1993; Holtzman et al., 1996). We did not systematically evaluate potential trophic effects of ICE on the developing brain in the present studies.
There are now several reports of between-strain differences in susceptibility to cerebral ischemia in both neonatal and adult mice. Strains of the 129 lineage, commonly used to generate genetically modified mice, are reportedly resistant to cerebral ischemia, in comparison to many other strains, including C57BL/6 (Yang et al., 1997; Connolly et al., 1996; O'Donnell et al., 1996; Sheldon et al., 1997). Both the ICE-deficient and WT mice use in these studies were derived from the same progenitors, of mixed 129 and C57BL/6 background. Thus, the present findings are unlikely to be due to differences in genetic background between the two strains, but rather are a result of deletion of a functioning ICE gene. Between-strain differences in susceptibility to cerebral ischemia may be attributable to differences in cerebrovascular anatomy (Barone et al., 1993). Yet, equivalent reductions in CBP in WT and ICE-deficient animals showed that the protective effect of ICE deficiency could not be attributed to differences in intra-HI CBP. Although only cortical CBP was measured in these neonatal mice, data from neonatal rats suggest that there are corresponding reductions in intra-HI blood flow in other vulnerable regions (e.g., hippocampus and striatum) (Ringel et al., 1991).
Although brain injury severity was attenuated in ICE-deficient neonatal mice subjected to a moderate cerebral HI insult, ICE-deficient animals remained vulnerable to injury elicited by severe HI insults. One mechanism that could account for these disparate trends would be if apoptotic neuronal death predominates after moderate HI, whereas necrosis predominates after more severe insults (Du et al., 1996), and if ICE deficiency were only protective in a setting where apoptosis predominates. This is unlikely to be the only explanation because even the moderate HI insult commonly elicited lesions with foci of pan-necrosis (Pig. 2). Alternatively, it is conceivable that more severe insults activate multiple necrotic mechanisms that do not depend on IL-1β activity, both in WT and ICE-deficient animals.
In conclusion, our experiments show that ICE activity contributes to the evolution of ischemic brain injury in neonatal mice. This information adds to the growing body of experimental and epidemiologic data suggesting that IL-1β contributes to the pathogenesis of long-lasting neurologic deficits originating in the perinatal period. Whether these deleterious effects of ICE are mediated by IL-1β–induced necrotic mechanisms and/or by pro-apoptotic mechanisms is an important question for future investigations.