
Abstract
We used transgenic mice expressing a dominant negative mutation of interleukin-1β converting enzyme (ICE) (C285G) in a model of transient focal ischemia in order to investigate the role of ICE in ischemic brain damage. Transgenic mutant ICE mice (n = 11) and wild-type littermates (n = 9) were subjected to 3 h of middle cerebral artery occlusion followed by 24 h of reperfusion. Cerebral infarcts and brain swelling were reduced by 44% and 46%, respectively. Neurological deficits were also significantly reduced. Regional CBF, blood pressure, core temperature, and heart rate did not differ between groups when measured for up to 1 h after reperfusion. Increases in immunoreactive IL-1β levels, observed in ischemic wild-type brain at 30 min after reperfusion, were 77% lower in the mutant strain, indicating that proIL-1β cleavage is inhibited in the mutants. DNA fragmentation was reduced in the mutants 6 and 24 h after reperfusion. Hence, endogenous expression of an ICE inhibitor confers resistance to cerebral ischemia and brain swelling. Our results indicate that down-regulation of ICE expression might provide a useful therapeutic target in cerebral ischemia.
Keywords
Apoptosis is a cell suicide program that involves a complex cascade of caspases. The interleukin-1β converting enzyme (ICE) family caspases (Alnemri et al., 1996) are the human homologues of the nematode C. elegans Ced-3 (Ellis et al., 1991; Yuan and Horvitz, 1990). Apoptosis occurs in mammalian cells either physiologically during development (Raff et al. 1993), during degenerative neurological diseases (Thompson, 1995), or after acute tissue insults such as ischemia (Li et al., 1995a; Charriaut-Marlangue et al., 1996) and traumatic injury (Rink et al., 1995). Inhibition of ICE and ICE-like caspases decreases infarct volume in mice (Hara et al., 1997) or rats (Loddick et al., 1996) after intracerebroventricular injection. It has also been reported that in a rat middle cerebral artery (MCA) occlusion model, the administration of the IL-1β receptor antagonist or the IL-1 blocker zinc protoporphyrin, decreased water content and infarct volume in ischemic brain (Relton and Rothwell, 1992; Yamasaki et al., 1995).
Interleukin-1β converting enzyme is one of 10 identified caspase family members that share in common a QACRG consensus sequence at the active site (Alnemri et al., 1996). On activation, ICE is cleaved from a 45 kDa protein to a 20 and 10 kDa fragment. The active ICE enzyme is a tetramer comprised of 2 homodimers (p20–p10) (Wilson et al., 1994; Walker et al., 1994). Interleukin-1β converting enzyme cleaves a 31 kDa proIL-1β protein to generate mature 17.5 kDa IL-1β. IL-1β has been associated with various inflammatory reactions (Dinarello, 1994), with hypoxia-mediated apoptosis in vitro (Friedlander et al., 1996), with global cerebral ischemia in gerbils (Saito et al., 1996), and focal cerebral ischemia in rats in vivo (Liu et al., 1993). Additional substrates of ICE have been recognized including pro-ICE and CPP-32 (Nicholsen et al., 1995; Patel et al., 1996).
Because inflammation and apoptosis have been identified as important components of transient focal ischemia (Johnson et al., 1995), studies were undertaken to examine tissue injury in a transgenic mouse expressing a mutant ICE protein that acts as a dominant negative inhibitor of endogenous ICE (Friedlander et al., 1997). The transgene encodes for a mutant ICE protein in which the amino acid glycine has been substituted for a cysteine at the active cleavage site (position 285). In brief, this transgenic mouse develops normally but expresses a fused mutant ICE-lacZ gene under control of the neuron specific enolase promoter (NSE-M17Z). The gross appearance of its central nervous system and supplying blood vessels are normal. Developmental apoptosis is apparently not inhibited, as evidenced by equal numbers of neurons in the facial motor nucleus compared with wild-type littermates, although mutant dorsal root ganglia neurons are resistant to apoptosis induced by trophic factor withdrawal (Friedlander et al., 1997). In response to lipopolysaccharide challenge, brain IL-1β levels do not increase. We now report that these mutant mice develop smaller infarcts, less neurological deficits, reduced edema, smaller IL-1β levels, and decreased DNA fragmentation after transient MCA occlusion.
MATERIALS AND METHODS
ICE mutant mice
Generation and characterization of the mutant ICEC285G transgenic mouse have been reported elsewhere (Friedlander et al., 1997). Genetic background of the transgenic mice were SV-129 and C57/Black6 strains. Adult mutant mice and their wild-type littermates (male and female, 19–23 g) were initially anesthetized with 1.0% and maintained on 0.4–0.8% halothane in 70% N2O and 30% O2 using a Fluotec 3 vaporizer (Colonial Medical, Amherst, NH, U.S.A.). Regional CBF, blood pressure, heart rate, and core temperature were monitored as described in Hara et al., 1996b. For regional cerebral blood flow measurement, the tip of the laser Doppler probe was placed 2 mm posterior and 6mm lateral from bregma over the MCA perfusion area. Temporalis muscle temperature was also maintained at approximately 36.5°C using a thermostat (BAT-12, Physitemp, Clifton, NJ, U.S.A.).
Ischemia
Filament occlusion of the left MCA was performed as described previously (Hara et al., 1996b). Three hours after ischemia, animals were briefly reanesthetized with halothane, and the filament withdrawn. Twenty-four hours after reperfusion, the forebrains were divided into five coronal (2-mm) sections using a mouse brain matrix (RBM-2000C, Activational Systems, MI, U.S.A.), stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC, Sigma, St. Louis, MO, U.S.A.). The infarcted areas were quantitated by an investigator naive to the groups identity using an image-analysis system (Bioquant IV, R&M Biometrics, Inc., Nashville, TN, U.S.A.) and calculated by summing the volumes of each section determined directly (Huang et al., 1994), or indirectly by the following formula: contralateral hemisphere (mm3)—ipsilateral undamaged volume (mm3) (Swanson et al., 1990). Brain swelling was calculated according to the following formula: (infarct volume + ipsilateral undamaged volume—contralateral volume) × 100/contralateral volume (%).
Neurological deficit
Mice were tested for neurological deficits and scored as described by Bederson et al. (1986) for rats and modified by Hara et al. (1996b) for mice: 0, no observable neurological deficits (normal); 1, failure to extend right forepaw (mild); 2, circling to the contralateral side (moderate); 3, loss of walking or righting reflex (severe). The rater was naive to the treatment group and assessments were made 10 min and 3 h after onset of ischemia, and again at 24 h after reperfusion.
IL-1β assay
To examine for the presence of brain IL-1β levels after MCA occlusion in wild-type and mutant mice, IL-1β immunoreactivity was determined using an enzyme-linked immunosorbent assay kit (Genzyme [Lot B6499F], Cambridge, MA, U.S.A.). Interleukin-1β converting enzyme mutant and wild-type littermates (female, 18–23 g) were subjected to 2 h of MCA occlusion followed by 30 min of reperfusion. Each hemisphere was homogenized for 15 s in phosphate buffered saline (0.1 M, pH 7.4, 4°C) containing 2 mM phenylmethyl-sulfonylfluoride (stock dissolved in dimethylsulfoxide, diluted 1:100 in phosphate buffered saline), 1 μg/ml leupeptin, 1 μg/ml antipain, 1 μg/ml aprotinin, 1 μg/ml pepstatin, 0.05% (weight-to-volume ratio) sodium azide, and 4 mM ethylenediaminetetraacetic acid. The homogenates were centrifuged (30 min at 50,000 × g) and 100 μl of the supernatant was used for each determination. Immunoreactive IL-1β data were expressed as the difference between ischemic and contralateral hemispheres.
DNA fragmentation
For analysis of DNA damage, tissue samples from the ischemic tissue and homologous contralateral hemisphere were obtained 6 h and 24 h after 2 h of transient ischemia. DNA was isolated (Puregene Systems, Minneapolis, MN, U.S.A.), digested with DNAse-free RNAse (Boehringer Mannheim, Indianapolis, MN, U.S.A.) and then extracted with phenolchloroform. The DNA was reprecipitated in ethanol, pelleted, and resuspended. The DNA concentration was quantitated by absorbance at 260 nm. For quantitation of DNA damage, a [32P]ddATP end-labeling method was used (Tilly and Hsueh, 1993; MacManus et al., 1995). Three μg of DNA pooled from three animals per group were used in the labeling procedure. As internal standard 35 ng of a randomly synthesized 100 bp DNA fragment was labeled together with the sample DNA. The DNA was electrophoresed on a 2.0% agarose gel (agarose 3:1, Amresco, Solon, OH, U.S.A.) and detected by autoradiography together with a radioactive standard. The autoradiographs were analyzed by an image analysis system (M4, Imaging Research, St. Catherines, Ontario, Canada). DNA fragmentation was measured as the intensity of the labeled DNA with a size less than than 10 kb.
Statistical analysis
Data are presented as mean ± S.D. Statistical comparisons were made by Student's t-test, two-way analysis of variance (ANOVA) followed by Student's t-tests, or by Mann-Whitney U-test as specified in the results section using the software super ANOVA or StatView 4.5 (Abacus Concepts, Berkeley, CA, U.S.A.). p < 0.05 was considered statistically significant.
RESULTS
Regional CBF decreased to approximately 20% of baseline immediately after MCA occlusion and sustained during 3 h of ischemia. After reperfusion, regional cerebral blood flow immediately increased to 80–100% of baseline. There were no significant blood flow differences between groups at any time point (data not shown), nor were there differences in mean arterial blood pressure, heart rate, rectal and temporalis muscle temperature, blood pH, and blood gases (pO2 and pCO2) detected between groups (Table 1).
Mean blood pressure, physiological parameters (pH, PaCO2 and PaO2), core and temporalis muscle temperature before ischemia and after reperfusion in wild-type and ICE-mutant mice
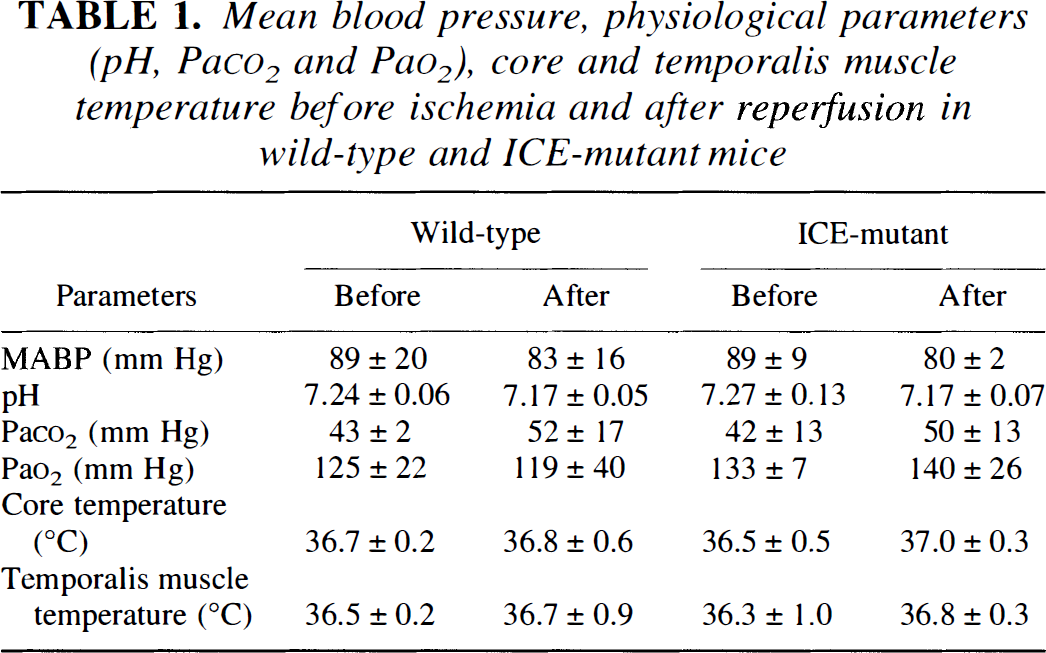
Mean arterial blood pressure (MABP) was monitored using a MacLab/8 data acquisition unit equipped with a transducer amplifier via femoral artery. Arterial blood samples were obtained from the femoral artery. Mean blood pressure was measured before ischemia and 30 min after reperfusion. Thirty-microliter samples were withdrawn twice before ischemia and 30 min after reperfusion. Data are presented as mean ± SD (n = 4 or 5). There are no significant differences between groups (two-way ANOVA).
Infarct volume was significantly smaller in the mutant mice using either a direct or indirect method for analysis after TTC staining (p < 0.01; Student's t-test; Fig. 1A). Infarct areas were smaller in 4 of the 5 coronal sections (4–10 mm) in the mutant group (Fig. 1B; p < 0.05; Student's t-test). Brain swelling was also diminished in the mutant mouse brain after 24 h. Whereas swelling accounted for 9.5 ± 5.4% of the hemispheric volume in the mutant group, it was 17.9 ± 6.8% in the wild-type littermates (p < 0.05; Student's t-test). Neurological deficits were also reduced both after 3 h ischemia and 24 h after reperfusion [2.3 ± 0.5 [wild type, n = 9] versus 1.5 ± 0.9 [mutant, n = 11] after 3 h ischemia, p < 0.05, Mann-Whitney U-test; 1.8 ± 0.3 [wild type, n = 9] versus 0.8 ± 1.0 [mutant, n = 11] 24 h after reperfusion, p < 0.05, Mann-Whitney U-test). Mutant animals showed less turning behavior, posturing and paralysis but more spontaneous motoric activity.
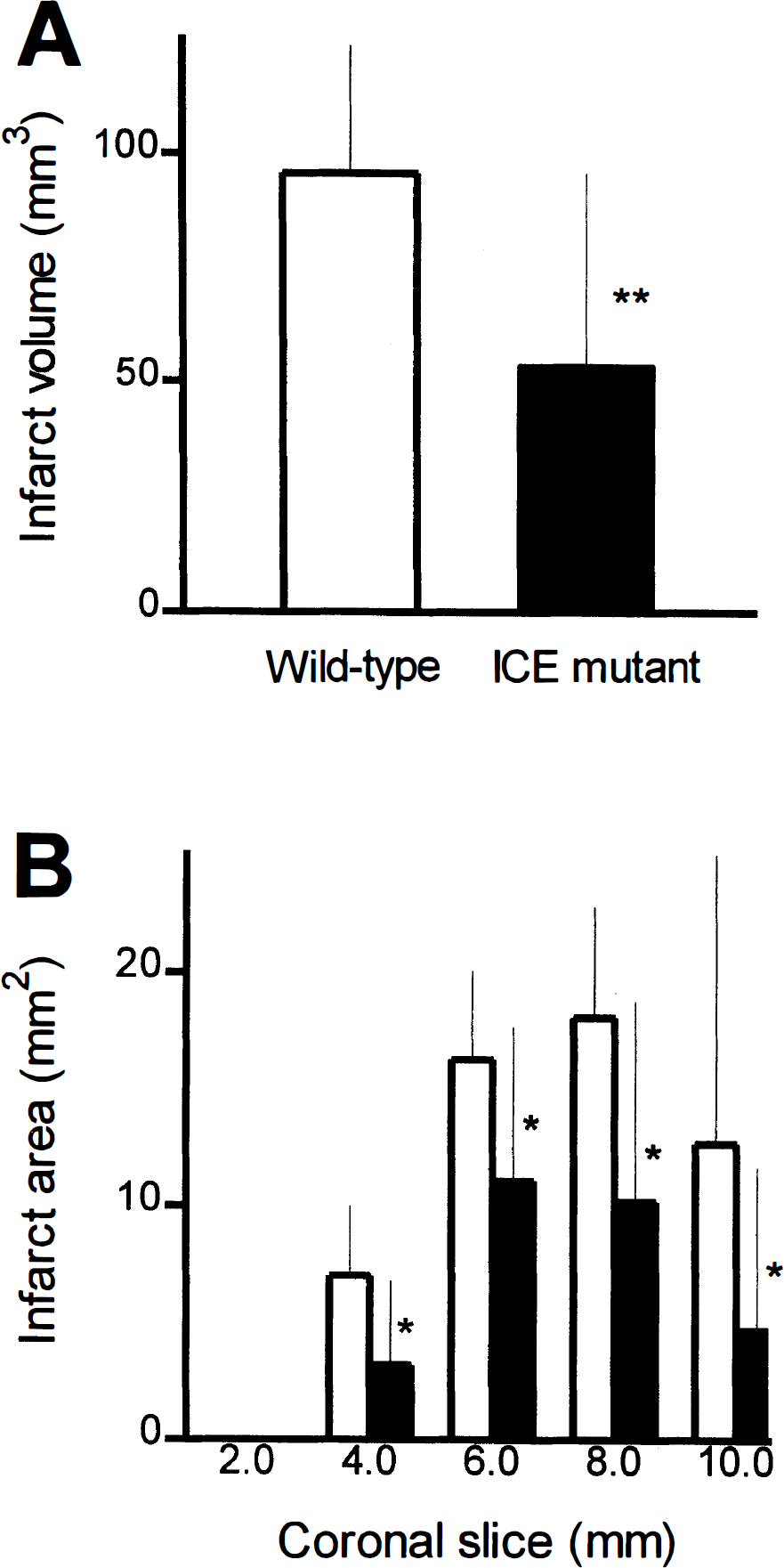
Infarct volume (
Brain IL-1β level differences, which increased after 2 h ischemia and 30 min reperfusion in ischemic tissue in wild-type littermates, were 77% less in mutant mice (88.3 ± 68.0 pg/g brain [wild-type, n = 7] versus 20.6 ± 43.1 pg/g brain [mutant, n = 7], p < 0.05, Student's t-test; absolute concentrations 233 ± 156 and 246 ± 168 pg/g brain in contralateral hemisphere in wild-type littermates and mutant mice, respectively). There were no significant differences between hemispheres in sham animals or in control groups.
Total DNA fragmentation was measured 6 h (earliest observation of laddering in wild-type) and 24 h after 2 h ischemia (Fig. 2A). DNA damage was higher after 24 h than after 6 h. At 6 h ICE mutant mice showed 50% less DNA fragmentation than the wild-type littermates (total DNA damage 2055 ± 43 μCi/ml × b and 1036 ± 429 μCi/ml × b, respectively; n = 3 per group in duplicate; Student's t-test). After 24 h, total DNA damage was 47% lower in the mutant group (total DNA damage 6313 ± 96 μCi/ml × b and 3372 ± 195 μCi/ml × b, respectively; n = 3 per group in duplicate; Student's t-test). DNA laddering was observed at 6 h in only the wild-type littermates but not in the mutant animals whereas at 24 h it was found in all animals.
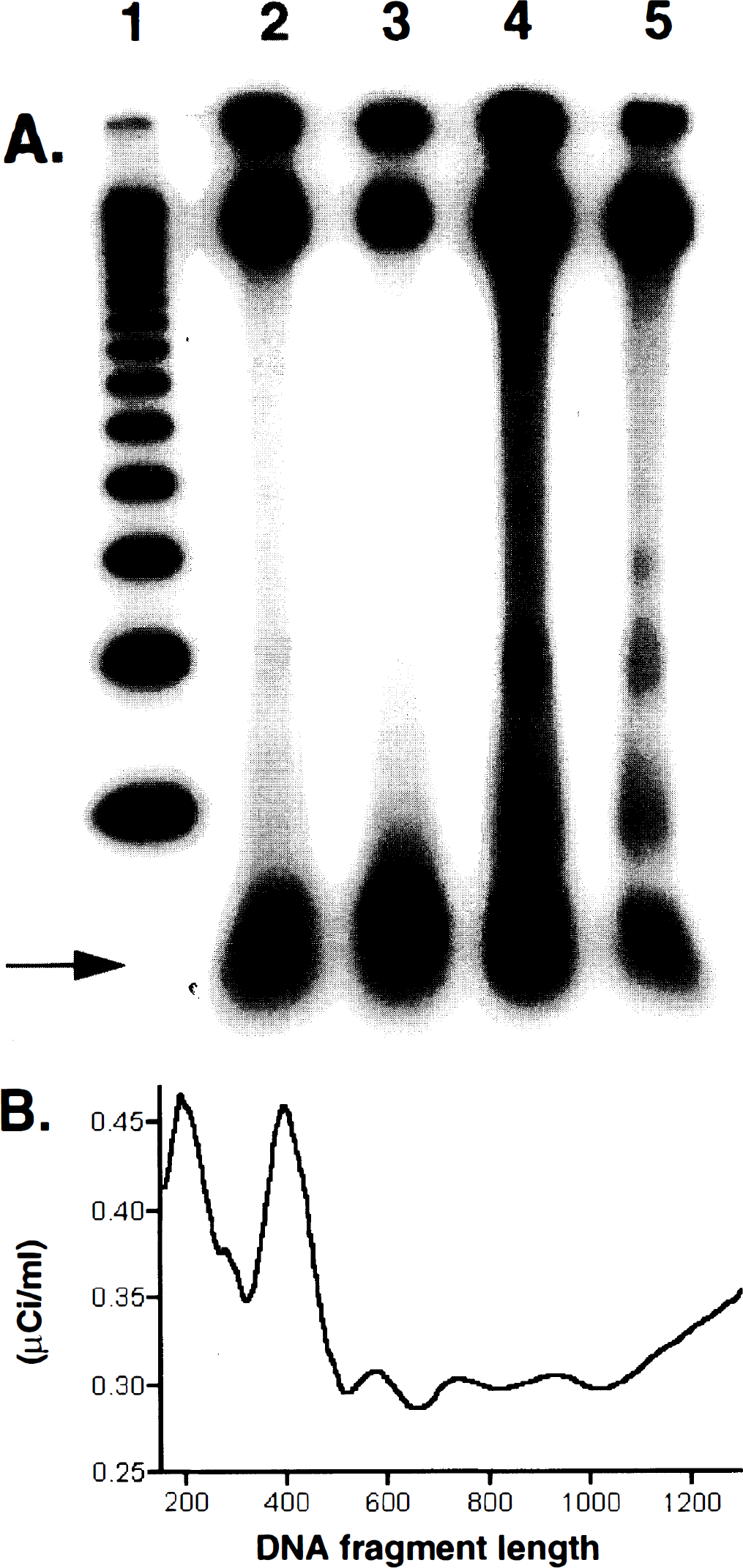
(
DISCUSSION
In this study we report that infarct damage after reversible MCA occlusion was attenuated by 45% in transgenic mice expressing a mutant form of ICE that inhibits the endogenous enzyme(s) (Fig. 1). Both infarct volume and brain swelling were reduced in the mutant mice, thereby suggesting that ICE family members (directly or indirectly) impair neuronal function after stroke, and that blocking ICE activation as well as IL-1β production may be useful to treat cerebral ischemia. Neurological deficits improved as well which may be in part caused by decreased brain swelling. The data seem to be consistent with recent reports showing that neuroprotection follows the administration of competitive irreversible peptide ICE inhibitors (i.e., YVAD.CMK, z-VAD.FMK and z-DEVD.FMK), and this effect sustains for at least 3 days after z-VAD.FMK treatment (Hara et al., 1997). The findings reported here suggest that ischemia can be treated by gene-targeting strategies aimed at downregulating or inhibiting ICE function. Whether such an approach provides therapeutic benefit when gene therapy is initiated after acute ischemia, of course, remains for further study. The strategy may prove more useful in ischemic paradigms wherein cell death is delayed by many hours or days (e.g., global ischemia). Nevertheless, the results suggest that ICE family members inhibitable by the mutant ICEC285G play an important role in cell death induced by ischemic injury.
As noted above, ICE family members have been implicated in both apoptosis and inflammation; both are relevant to cell injury and repair in ischemic brain (Kumar et al., 1995). For example, white blood cells enter ischemic brain hours after transient focal ischemia and probably promote free radical generation, brain swelling, and are of course, hallmarks of tissue inflammation (Kochanek and Hallenbeck, 1992). Markers of apoptosis (e.g., terminal deoxynuclotidyl transferase-mediated dUTP-biotin nick end-labeling staining) already appear after 10 min ischemia followed by 48 h reperfusion (Li et al., 1995b), and suggest that cell death programs are initiated relatively early in ischemia. Oligonucleosomal laddering, identified at 6 h in mouse brain (Fig. 2), provides evidence for the early appearance of apoptotic mechanisms in our model. Although activation of caspase family members may trigger apoptosis (Steller, 1995), the precise steps involved are not completely understood. Other caspase family members such as CPP32 have been implicated (Tewari et al., 1995), and ICE or ICE-like caspases cleave and activate CPP32 (Nicholson et al., 1995). Moreover, activated CPP32 cleaves PARP, lamins, and sterol regulatory element-binding proteins, among other proteins (Wang et al., 1995). Inhibition of CPP32, which does not cleave IL-1β, has also been shown to reduce infarct size (Hara et al., 1997). In addition, mature IL-1β promotes apoptosis in hypoxic cultured cells (Friedlander et al. 1996). Therefore, it seems that both ICE activity and endogenously synthesized mature IL-1β initiate programmed cell death in some situations either by autocrine or paracrine manner. Hence, inflammation and apoptosis seem to share common mechanistic pathways and inhibition of ICE may reduce infarct volume by both mechanisms.
In the mouse MCA occlusion model, DNA laddering, IL-1β levels, and brain swelling indicate that inflammation and apoptosis develop within hours in the wild-type strain; all three plus infarct size were significantly attenuated in transgenic mice expressing the endogenous ICE inhibitor. The observed differences in tissue injury might have been more substantial if ICE inhibition was more complete. Brain IL-1β levels in the mutant mouse may reflect subtotal enzyme inhibition by the mutant protein. Our findings emphasize the need to pursue studies that explore the mechanism of cell death by caspases including interaction among family members, substrates relevant to cell death (in addition to proIL-1β), and factors during ischemia which trigger ICE activation.
In conclusion, our results show that ICE family members play a pivotal role in the pathogenesis of ischemic cell damage and may be targets for future pharmacotherapy of stroke or neurodegenerative diseases.
Footnotes
Acknowledgments:
This work was supported by Massachusetts General Hospital Interdepartmental Stroke Project Grant (NS10828), and an unrestricted award in Neuroscience from Bristol Myers Squibb, New York (M.A.M.); the Deutsche Forschungsgemeinschaft (Fi600/2-1 and En343/1-1; K.F. and M.E., respectively); the National Institute of Neurological Disorders and Stroke, American Heart Association Established Investigatorship Award and Bristol Myers Squibb (J.Y.); and a postdoctoral training fellowship from NIH, and an award from the Joint Section of Cerebrovascular Surgery (Congress of Neurological Surgeons and the American Association of Neurological Surgeons) sponsored by Upjohn, Kalamazoo, MI (R.M.F.).