
Abstract
Increasing evidence suggests that peripheral inflammatory responses to stroke and other brain injuries have an important role in determining neurological outcome. The mediators of this response and the temporal relationships between peripheral and central inflammatory alterations are poorly understood. In this study, we show that experimental stroke in mice induces a peripheral inflammatory response that peaks 4 h after stroke, and precedes the peak in brain inflammation 24 h after stroke. This peripheral response is dominated by the induction of the chemokine CXCL-1 and the proinflammatory cytokine interleukin-6 and could serve as an accessible target for therapy and as a source of biomarkers predictive of prognosis.
Introduction
The local inflammatory response in the brain is well established as a contributor to ischaemic brain damage, and there is increasing evidence that peripheral inflammatory processes and immune alterations are also important (McColl et al, 2007; Offner et al, 2006; Price et al, 2003). Clinically, immune cell mobilisation is accompanied by an acute-phase response (APR), and the upregulation of circulating cytokines and chemokines (Emsley and Tyrrell, 2002). Offner et al (2006) showed an induction of multiple inflammatory mediators in peripheral lymphoid organs as well as in circulation and the brain after experimental stroke. An acute inflammatory challenge in the brain or spinal cord injury induces the synthesis of hepatic inflammatory mediators, notably the chemokine CXCL-1 (CINC-1/KC) (Campbell et al, 2003; Campbell et al, 2005). Circulating levels of the related human chemokine CXCL-8 (interleukin-8 (IL-8)) are increased in stroke patients (Grau et al, 2001), although it is not known whether the liver contributes to this increase. The hepatic chemokine response is believed to be part of the APR and thus may be regulated by similar mechanisms (Campbell et al, 2003). An established trigger of the APR is interleukin-6 (IL-6) (Kopf et al, 1994), which is markedly increased in circulation after clinical and experimental stroke and is a predictor for clinical outcome (McColl et al, 2007; Smith et al, 2004). The temporal and spatial relationship between CXCL-1 and IL-6 after stroke has not been previously investigated and, more generally, the temporal relationships between peripheral and central inflammatory alterations are poorly understood, and therefore form the basis of this study. Our primary objective focused on CXCL-1 and IL-6 and, as a secondary objective, we carried out an exploratory analysis of 14 further inflammatory mediators. In addition to plasma and hepatic responses, we also assess changes in the lung in view of the high concentration of marginating immune cells in the pulmonary vasculature and the role of pulmonary inflammatory events in poststroke complications, such as pneumonia.
In this study, we show a rapid and transient peripheral inflammatory response dominated by the induction of CXCL-1 and IL-6 that precedes brain inflammation after experimental stroke.
Materials and methods
Mice
Experiments were carried out on 10 to 12-week-old (weighing 25 to 30 g) C57BL/6J mice (Harlan-Olac, Bicester, UK) in accordance with the Animals (Scientific Procedures) Act (1986).
Focal Cerebral Ischaemia
Focal ischaemia was induced by transient (60 min) middle cerebral artery occlusion (MCAo), which we routinely observe to cause reproducible striatal and cortical damage 24 h after MCAo (67±17 mm3). Briefly, anaesthesia was induced and maintained by halothane (3.5 and 1.5% in 30% O2 and in 70% N2O, respectively) and core body temperature was maintained throughout the procedure at 37±0.5°C. A 6-0 nylon monofilament (Dermalon, Tyco Healthcare UK Ltd, Gosport, UK), coated with resin (Jet Melt, Radiospares, Beauvais, France)-coated tip (180 μm diameter), was introduced into the external carotid artery and advanced 9 mm along the internal carotid artery to occlude the MCA. After 60 mins, the filament was withdrawn to establish reperfusion for 2, 4, 24, or 72 h. Sham-operated mice underwent the same procedure, except the filament was advanced along the internal carotid artery and then immediately withdrawn. Animals that showed no circling behaviour after occlusion were excluded from the study.
Tissue Harvesting
Plasma was obtained from anticoagulated cardiac blood samples. Mice were perfused transcardially with 0.9% saline, and their liver, lung, cerebral cortex, and striatum were homogenised in buffer (50 mmol/L Tris-HCl pH 7.4, 150 mmol/L NaCl, 5 mmol/L CaCl2, 0.02% NaN3, 1% Triton X) containing protease inhibitors. After centrifugation (17,000 g for 30 mins), supernatants were collected and protein concentrations calculated (BCA assay, Pierce, Thermo-Fisher Scientific, Northumberland, UK) and equilibrated. An additional hepatic sample was immediately placed in RNA later (Sigma, Dorset, UK) at 4°C for 24 h, and then removed and stored at −80°C for quantitative PCR.
Multiplex Protein Assay
The concentrations of CXCL-1, IL-6, and the 14 further inflammatory molecules were quantified in the liver, lung, cortex and striatum (ipsilateral to MCAo), and plasma. Capture antibodies (R&D Systems, Abingdon, UK) were conjugated to LiquiChip Carboxy Beads (Qiagen, Crawley, UK) and adjusted to 2.5 × 106 per mL. Briefly, standards (Duo Set, R&D Systems) or samples were incubated with conjugated beads (1:100) for 2 h. Biotinylated antibody (1:100) (Duo Set, R&D Systems) was added for 1 h. Finally, streptavidin-R-PE (1:1,000; Qiagen) was added for 30 mins. All incubations were carried out in the dark at room temperature and were followed by a wash step. Analyte concentrations were measured using Luminex100 (Luminex Corporation, Austin, TX, USA). At least 100 beads per analyte per well were counted and concentrations calculated using Luminex Data Collector software, Luminex Corporation (Version 1.7). The following were the lower limits of detection (pg/mL): interferon-γ (8.2), IL-1β (9.6), IL-10 (8.2), IL-17 (19.2), IL-2 (8.9), IL-4 (13.7), IL-12p40 (10.3), IL-6 (8.2), tumor necrosis factor-α (28.8), CXCL-1 (15.1), tumor growth factor-β (49.63), CX3CL-1 (180.37), CCL-2 (41.92), CXCL-2 (50.14), CCL-5 (95.41), P-selectin (38.91).
Real-Time PCR
Total RNA was extracted using RNeasy Midi-Kit (Qiagen) and converted into cDNA using Advantage RT-for-PCR Kit (Clontech, Basingstoke, UK) as per the manufacturer's instructions. Real-time PCR for CXCL-1, normalised against β-actin and GAPDH (glyceraldehyde-3-phosphate dehydrogenase), was carried out using the Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems, Warrington, UK). A total of 50 ng cDNA was loaded per reaction with 10 pmol/L primer and 2XSYBR Green PCR Mastermix (Applied Biosystems). Primer sequences were the following:
CXCL-1 forward, CCGAAGTCATAGCCACACTCA;
CXCL-1 reverse, CAAGGGAGCTTCAGGGTCAA;
GAPDH forward, CTCGTCCCGTAGACAAAATGG;
GAPDH reverse, TGACCAGGCGCCCAATA;
β-actin forward, AGATGACCCAGATCATGTTTGA;
β-actin reverse, CACAGCCTGGATGGCTACGT.
Standard curves were generated from the lysates of IL-1β-treated cortical neurones as shown previously (Tsakiri et al, 2008). To ensure continuity and that amplification was not due to genomic DNA contamination, reference samples from neuronal lysates were used between plates and no template controls were performed.
Statistical Analysis
For the primary objective, to investigate the effect of stroke and time on CXCL-1 and IL-6 concentrations, data were log transformed and analysed by two-way ANOVA (analysis of variance) followed by Student's t-test with Bonferroni correction. For our secondary objective, an exploratory analysis of the 14 further mediators, uncorrected P-values obtained from Student's t-tests comparing sham and MCAo groups are reported. For mRNA analysis, Mann–Whitney tests were carried out. All statistics were carried out using JMP software (8th edition, SAS, Marlow, UK).
Results
Plasma CXCL-1 showed an increasing trend at 2 h and was significantly increased at 4 h (P<0.001), before a return to sham levels by 24 h (Figure 1A). CXCL-1 expression was similar in the liver and lung with significant increases at 4 h (P<0.001 and P=0.003, respectively), returning to sham levels by 24 h. In the brain, CXCL-1 increased at 4 h (P=0.019 cortex), with the peak increase occurring at 24 h (P<0.001 cortex, P=0.001 striatum), before returning to sham levels by 72 h. The same pattern of expression was confirmed using ELISA (enzyme-linked immunosorbent assay) (data not shown).
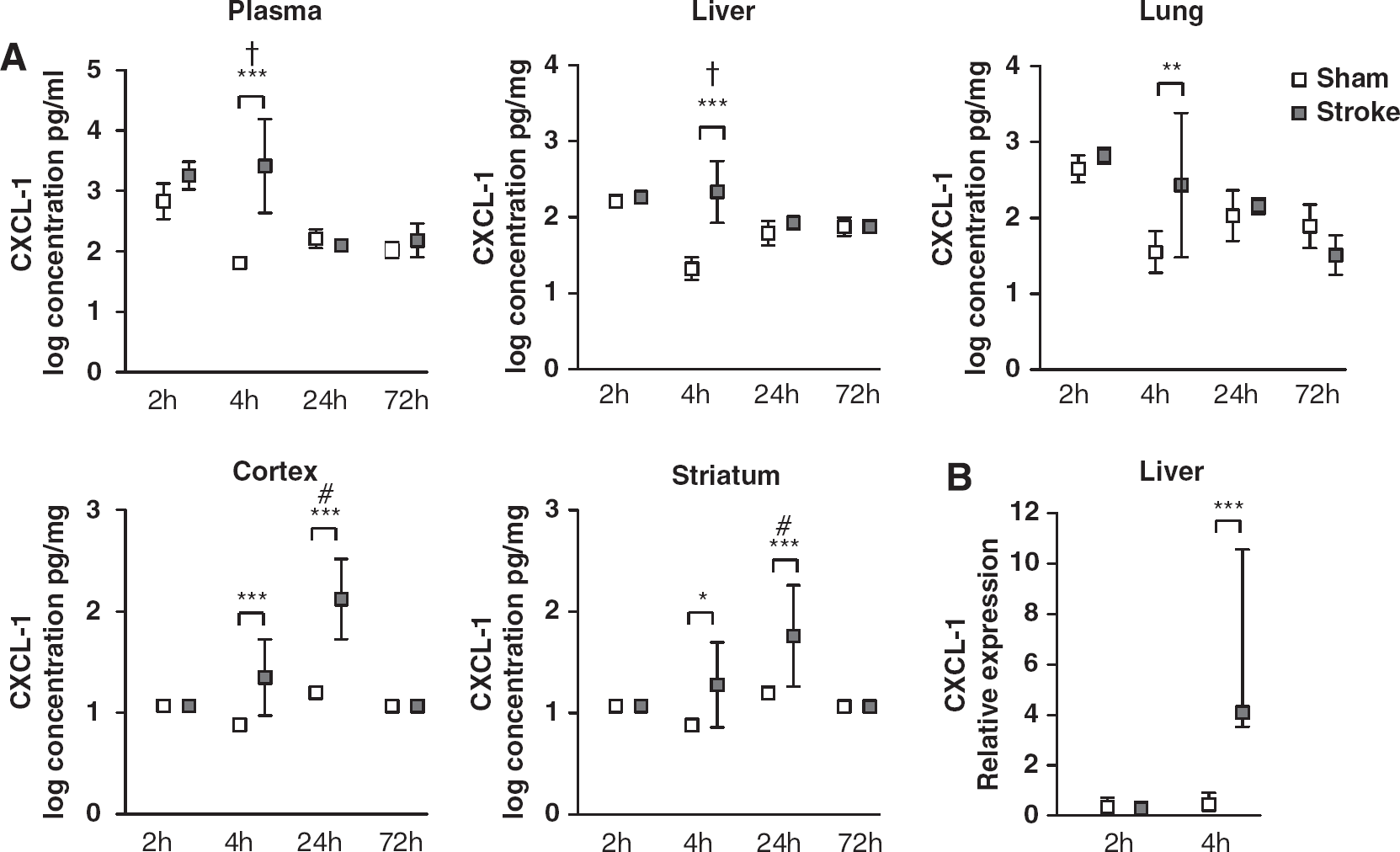
Log transformed CXCL-1 concentrations. (
To verify that the upregulation of hepatic CXCL-1 was not a result of uptake from circulation, CXCL-1 mRNA expression was determined, and found to be significantly increased 4 h after MCAo (P=0.001) (Figure 1B). No increase in expression was observed at 2 h.
Plasma IL-6 peaked rapidly, with an increasing trend at 2 h and a significant increase at 4 h (P<0.001), before returning to sham levels by 24 h (Figure 2). The peripheral IL-6 response was followed by a later induction in the brain, being significantly increased in the cortex and striatum at 24 h (P<0.001 and P=0.003, respectively), before returning to sham levels by 72 h. There was no induction of IL-6 in the liver or lung at any time point (data not shown).
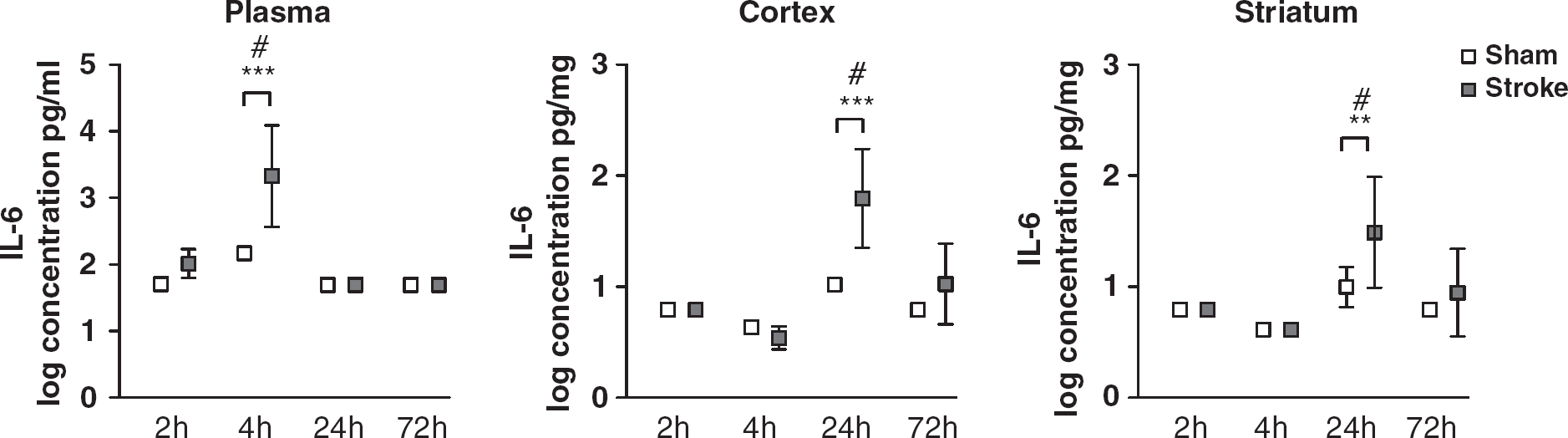
Log transformed IL-6 concentrations. Data are shown as mean with s.d. ∗∗P<0.01, ∗∗∗P<0.001 versus sham two-way ANOVA with post hoc Student's t-tests and Bonferonni corrections, n=5 to 8, #P<0.001 denotes that concentration is significantly different from all other time points.
For our secondary objective, multiplex analysis showed that at each single time point the majority of inflammatory molecules did not show detectable changes in response to experimental stroke (see Supplementary Data). Any changes in other mediators that were observed followed a similar profile to CXCL-1 and IL-6, with early increases in plasma (2 to 4 h), intermediately in peripheral tissues (4 to 24 h) and later in the brain (24 to 72 h).
Discussion
In this study, we show that experimental stroke induces a rapid and transient peripheral inflammatory response, most notably changes in CXCL-1 and IL-6, which precedes corresponding inflammatory changes in the brain. CXCL-1 was significantly upregulated by 4 h in the plasma and peripheral tissues, yet peaked at 24 h in the cortex and striatum, coinciding with declining peripheral levels. These findings are consistent with previous work showing an upregulation in circulating rat CXCL-1 before increasing brain expression (Yamasaki et al, 1995). This spatial and temporal profile of expression could create chemokine gradients that regulate the mobilisation and migration of neutrophils (Call et al, 2001). An increase in circulating neutrophils after experimental stroke has previously been noted as early as 8 h, with a later accumulation in the brain, and a potentiation of this response occurs alongside an increase in CXCL-1 (McColl et al, 2007). Neutrophils contribute to brain damage through multiple mechanisms after stroke, and the temporal and tissue-specific profile of CXCL-1 expression observed in this study supports a key role for this chemokine in neutrophil-mediated brain injury. This is highlighted by recent studies showing neuroprotective effects of antagonists of CXCR2, the receptor for CXCL-1 in mice (Villa et al, 2007). We show for the first time that experimental stroke also induces hepatic and pulmonary CXCL-1 expression with similar temporal profiles to the plasma, indicating that these tissues may contribute to plasma elevation. Interestingly, hepatic CXCL-1 has been shown previously to mediate important effects on neutrophil trafficking to the central nervous system after acute injury (Campbell et al, 2003; Campbell et al, 2005).
Interleukin-6 showed a similar profile of induction to CXCL-1, which suggests spatially and temporally distinct actions in response to experimental stroke that could provide a basis for its pleiotropic effects (Suzuki et al, 2009). The neuroprotective effects of centrally acting IL-6 have been shown previously (Loddick et al, 1998), whereas elevated peripheral levels of IL-6 correlate with poor prognosis in stroke patients (Smith et al, 2004). As an inducer of the hepatic APR, peripherally acting IL-6 could regulate CXCL-1, which is likely to have a detrimental effect on outcome. The earliest increase in IL-6 was detected in the circulation and there was no induction in peripheral tissues such as the liver and lungs, raising the question as to how and where IL-6 is initially induced. Furthermore, these data suggest that IL-6 may act as a general signal to peripheral tissues to trigger the induction of specific inflammatory mediators, including CXCL-1, after experimental stroke. Possible sources of IL-6 include endothelial cells or circulating leukocytes/platelets, although our recent data (unpublished) suggests that the latter is unlikely.
As a secondary exploratory objective of this study that harnessed the breadth of information provided by multiplex analysis, we assessed the profiles of 14 additional inflammatory mediators after experimental stroke. Although some minor changes were observed in these mediators, none were to the extent observed for CXCL-1 and IL-6. However, it is possible that changes could be below the detection limit, although this is unlikely given the high sensitivity of the multiplex technique for the majority of mediators. We also do not discount that mediators not included in our multiplex panel may be altered. Any changes that were observed occurred early in the periphery and late in the brain reinforcing the concept that peripheral inflammatory changes are rapid and precede comparable changes in the brain. Therefore, the acute inflammatory response to experimental stroke appears to be relatively specific, with changes restricted to a small number of mediators, suggesting tight regulatory mechanisms.
Overall, this study shows a rapid and transient peripheral upregulation of specific inflammatory mediators after experimental stroke. In view of the rapidity of these changes, and their potential contribution to stroke outcome, these peripheral inflammatory mediators could serve as accessible targets for therapy and/or biomarkers predictive of prognosis, with studies planned to test these hypotheses.