
Abstract
Study investigates the role of endothelin (ET) receptors in mediating early changes in cerebral blood flow—as measured by laser Doppler flowmetry (CBFLDF)—during experimental pneumococcal meningitis. Meningitis was induced with heat-killed pneumococci and confirmed by a significant increase in CBFLDF (baseline 100%; 225.3 ± 21.8% after 6 hours; mean ± SD), intracranial pressure (ICP), brain water content, and white blood cell count in the CSF. Intravenous administration of the selective endothelin B (ETB) receptor antagonist BQ-788 immediately before pneumococcal challenge (but not 4 hours afterward) significantly attenuated these pathophysiologic alterations (e.g., CBFLDF 6 hours after pneumococcal challenge: 116.7 ± 17.4%). Pretreatment with BQ-123, a selective endothelin A receptor antagonist, had no significant effect on ICP and brain water content, but augmented the increase in CBFLDF and CSF white blood cell count. Since ET is known to trigger the release of nitric oxide (NO) by ETB receptor activation, we examined specific ET–NO interactions in primary rat cerebromicrovascular endothelial cells after stimulation with heat-killed pneumococci. Pneumococci induced a significant increase in both ET and NO concentrations in endothelial cell culture medium. Treatment with phosphoramidon, an inhibitor of the endothelin-converting enzyme, prevented the production of endothelin and markedly reduced NO generation. Our data provide evidence that ET is involved as a mediator in early pneumococcal meningitis in the rat and contributes to the increase in CBFLDF, ICP, brain water content, and CSF pleocytosis, presumably through ETB receptor–mediated NO production.
Keywords
Bacterial meningitis from Streptococcus pneumoniae infection remains an important cause of mortality and morbidity (Quagliarello and Scheld, 1992). The unfavorable clinical outcome often results from cerebrovascular alterations, brain edema, and raised intracranial pressure (ICP) (Durand et al., 1993). In the last several years, animal studies have provided much insight into the pathophysiologic mechanisms of these intracranial complications. The early phase of experimental pneumococcal meningitis is characterized by an increase in cerebral blood flow, ICP, brain water content, and the migration of leukocytes into the subarachnoid space (Pfister et al., 1990; Weber et al., 1996). The molecular mechanisms underlying these changes involve both bacterial components and host factors such as arachidonic acid metabolites, cytokines, reactive oxygen species, and nitric oxide (NO), as well as leukocyte–endothelial interaction (Tuomanen et al., 1989; Saukkonen et al., 1990; Pfister et al., 1990; Koedel et al., 1995).
Several arguments suggest that endothelin (ET) may be involved in the complex inflammatory network of early pneumococcal meningitis. In the cerebral vasculature, intraluminal ET has been found to elicit vasodilation through the induction of NO and prostanoids through an activation of the endothelin B (ETB) receptor (Kobari et al., 1994a,b). In addition, ET can cause an increase in blood–brain barrier permeability (Stanimirovic et al., 1993) and plasma protein extravasation in rat dura mater (Brändli et al., 1996). Besides its role as a regulator of the vascular tone and integrity, recent findings strongly indicate that ET is a mediator of inflammation: bacterial cell wall components, inflammatory cytokines, and neutrophil–endothelial interactions can stimulate ET production (Ehrenreich et al., 1990; Corder et al., 1995). Neutrophil lysosomal proteases can convert the propeptide big-ET into the mature ET. Moreover, ET interacts at the level of biosynthesis and biological actions with a variety of substances involved in the pathophysiologic mechanisms of pneumococcal meningitis such as arachidonic acid metabolites, cytokines, and NO (Rubanyi and Polokoff, 1994). A further sign that ET possibly plays a pathophysiologic role in meningitis is the occurrence of elevated ET levels in CSF samples of patients with bacterial meningitis that were obtained at the time of admission to the hospital (Koedel et al., 1997).
This study investigates the role of ET receptors in mediating pathophysiologic alterations during experimental pneumococcal meningitis.
METHODS
Rat model of pneumococcal meningitis
A well-characterized rat model of bacterial meningitis was used in this study, which was previously described in detail (Pfister et al., 1990; Koedel et al., 1995). The protocols of this study were approved by the Government of Upper Bavaria. Thirty-two male Wistar rats (250 to 300 g) were anesthetized with 100 mg/kg thiopental (Byk Gulden, Konstanz, Germany) before undergoing tracheotomy, and then given artificial ventilation with a small animal ventilator (AP-10; K. Effenberger, Pfaffing, Germany). The MABP was measured through a cannula inserted into the right femoral artery using a pressure transducer (Statham P23, Viggo-Spectramed, Oxnard, CA, U.S.A.). Arterial blood samples were drawn at the beginning and the end of each experiment as well as every 2 hours and were analyzed for arterial Pa
At the end of the experiment, rats were killed by exsanguination, and brain water content, a parameter for brain edema formation, was determined as described previously (Pfister et al., 1990).
Induction of meningitis
When stable baselines of CBFLDF and ICP were achieved for 30 minutes, 100 μL CSF were removed through the cisterna magna catheter. Meningitis was induced by the intracisternal injection of 100 μL 108 colony forming units (cfu)/mL heat-killed pneumococci (HKP). The preparation of pneumococci used in this study was previously described in detail (Koedel et al., 1995). Heat-killed (60°C for 4 hours), unencapsulated pneumococci stem from an isogenic mutant of the encapsulated strain S pneumoniae serotype 3 (no. 17260) (Koedel et al., 1995).
Experimental protocol of in vivo investigations
Rats were divided into the following experimental groups (seven total): group 1, untreated rats injected intracisternally with HKP (n = 8); group 2, rats injected intracisternally with HKP and pretreated with a selective ETB receptor antagonist, BQ-788 (n = 5) (Ishikawa et al., 1994); group 3, rats injected intracisternally with HKP and treated with BQ-788 4 hours after intracisternal challenge (n = 4); group 4, rats injected intracisternally with HKP and pretreated with a selective endothelin A (ETA) receptor antagonist, BQ-123 (n = 7). To determine whether ET receptor antagonists by themselves can influence CBFLDF, ICP, brain water content, or CSF WBC count, we investigated the following control groups: group 5: untreated rats injected intracisternally with phosphate-buffered saline (PBS) (control; n = 3); group 6: control rats pretreated with BQ-788 (n = 3); and group 7: control rats pretreated with BQ-123 (n = 3). Both ET receptor antagonists were given intravenously in a dosage of 0.3 mg/kg as bolus injection, followed by a continuous infusion of 0.3 mg/kg/h, similar to Ishikawa and others (Ishikawa et al., 1994).
In vitro experiments
Recent studies provide evidence that the cerebral microvascular endothelium plays a central role in the pathophysiologic mechanism of pneumococcal meningitis: namely, in the entry of bacteria into the CSF, the synthesis and release of inflammatory mediators, the adhesion and transmigration of leukocytes, and the breakdown of the blood–brain barrier (Quagliarello et al., 1986; Saukkonen et al., 1990; Koedel et al., 1995. Tuomanen, 1996). Cerebral microvascular endothelial cells can synthesize and release ET, and express both ETA and ETB receptors (Yoshimoto et al., 1990; Nilsson et al., 1997). Stimulation of ETB receptors in endothelial cells can induce the release of NO (Hirata et al., 1993). In recent studies, we showed that HKP can induce cerebral microvascular endothelial cells to produce NO, and that NO is a key mediator of early pathophysiologic alterations in pneumococcal meningitis (Koedel et al., 1995). Thus, we tested whether HKP can stimulate cerebral microvascular endothelial cells to release ET, and whether the release of both ET and NO can be modulated either by Nω-nitro-
Preparation of cerebromicrovascular endothelial cells
Endothelial cells from cerebral microvessels were isolated from 3-week-old Wistar rats as previously described (Koedel et al., 1995). Cortical tissue was cleaned of meninges and superficial vessels with fine forceps under a binocular microscope. The hemispheres were incubated with 0.75% collagenase for 1 hour at 37°C. After centrifugation with 25% bovine serum albumin for 20 minutes at 800 × g, the cell suspension was incubated with 0.1% collagenase/dispase (Boehringer Mannheim, Germany) for 30 minutes at 37°C. After centrifugation, the pellet was fractionated with a 33.3% Percoll gradient. The cell layer of the gradient was plated in Dulbecco's modified Eagle's medium (Biochrom, Berlin, Germany) and then supplemented with 20% fetal calf serum (Biochrom) and 25 μg/mL endothelial cell growth factor (Boehringer Mannheim). After 3 to 5 days, the endothelial cells formed a confluent monolayer (approximately 5 × 105 cells/well). All monolayers were characterized as endothelial cells by morphologic criteria and the ability to bind antibody to von Willebrand factor (Boehringer Mannheim). For the experiments, monolayers without any passage were washed with PBS without Ca2+ or Mg2+ and maintained in Dulbecco's modified Eagle's medium with 20% fetal calf serum and 20 μg/mL endothelial cell growth factor without antibiotics and phenol red.
Experimental groups of in vitro experiments
The following three groups were investigated: endothelial cells stimulated with 107 cfu/mL HKP that were 1) untreated (n = 8), 2) treated with
Determination of nitrite and endothelin concentrations in the cell culture medium
The NO production was assessed by measuring nitrite, a stable metabolic product of NO, using the Griess reaction (Koedel et al., 1995). The ET concentrations in cell culture medium were measured by a radioimmunoassay as described previously (Arendt et al., 1993). For determining nitrite and ET, detection limits of 0.2 μmol/L and 1 pmol/L, respectively, were found for the test samples.
Reagents
We used BQ-123, a selective ETA antagonist, and BQ-788, a selective ETB antagonist from Alexis (Grünberg, Germany), as well as the metalloproteinase inhibitor phosphoramidon and the nitric oxide synthase inhibitor
Statistical analysis
For statistical analysis, data on CBFLDF and ICP obtained 3 hours and 6 hours after intracisternal pneumococcal challenge were compared by one-way analysis of variance and Scheffe's test. P values were corrected for repeated measures using the Bonferroni-Holm procedure. One-way analysis of variance and Scheffe's test were applied for statistical analysis of data on brain water content, CSF WBC count, and concentrations of ET and NO/nitrite in cell culture medium. To detect changes over time within the control group (group 5) in the in vivo experiments, data were compared using repeated measures analysis of variance. A P value < 0.05 was considered significant. Data are given as mean ± SD.
RESULTS
In vivo experiments
Mean arterial blood pressure, Pa
Physiologic parameters in different experimental groups
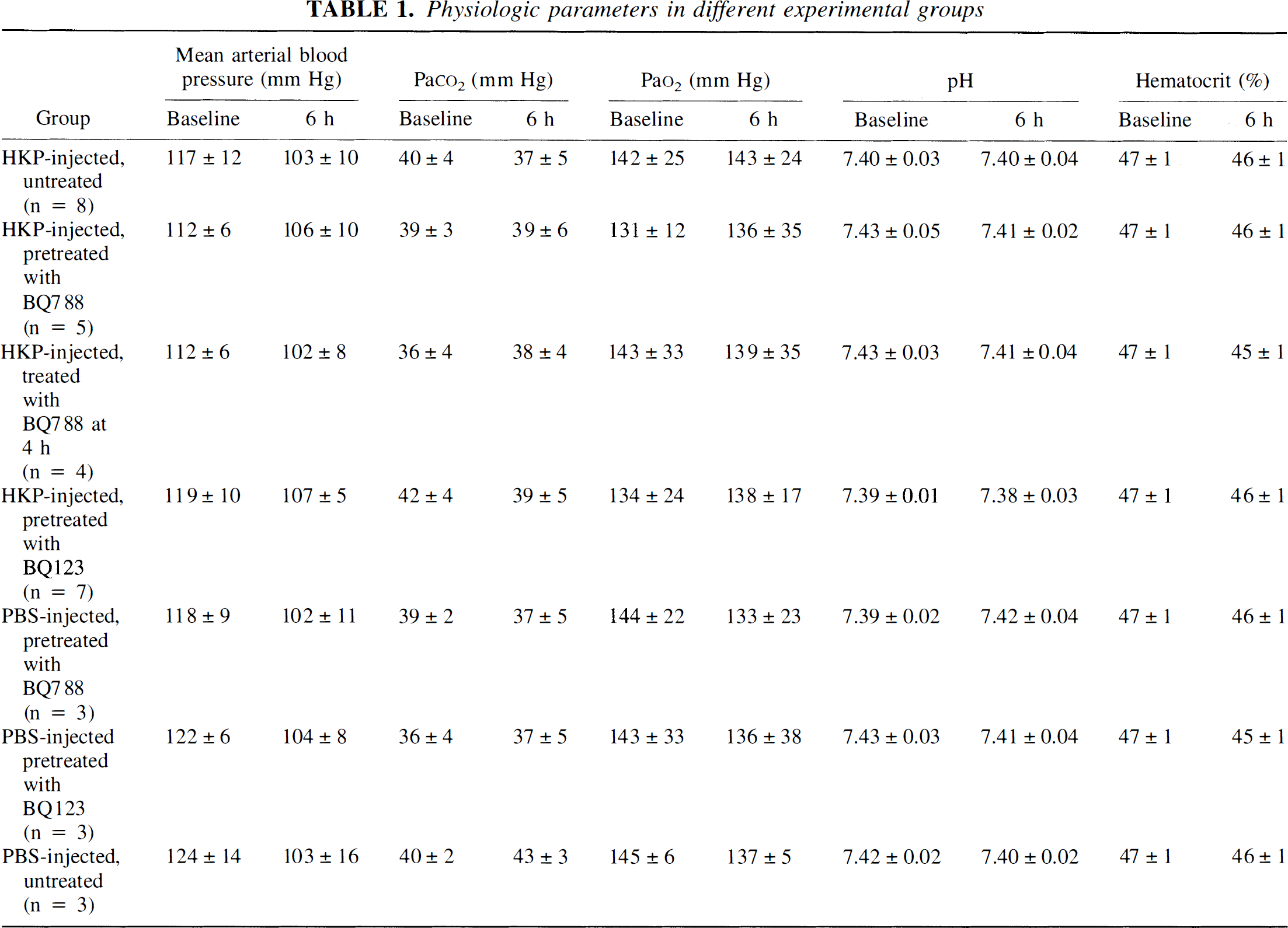
In untreated rats injected intracisternally with PBS (control rats), there was no significant change in CBFLDF or ICP throughout the experiment, and they did not develop CSF pleocytosis. Brain water content measured in control rats was 77.41 ± 0.16%. In rats injected intracisternally with PBS, systemic administration of neither BQ-788 nor BQ-123 led to changes in CBFLDF, ICP, brain water content, or CSF WBC count (P > 0.05, compared with untreated control rats; Table 2).
Effect of endothelin receptor antagonists BQ-788 and BQ-123 in experimental meningitis
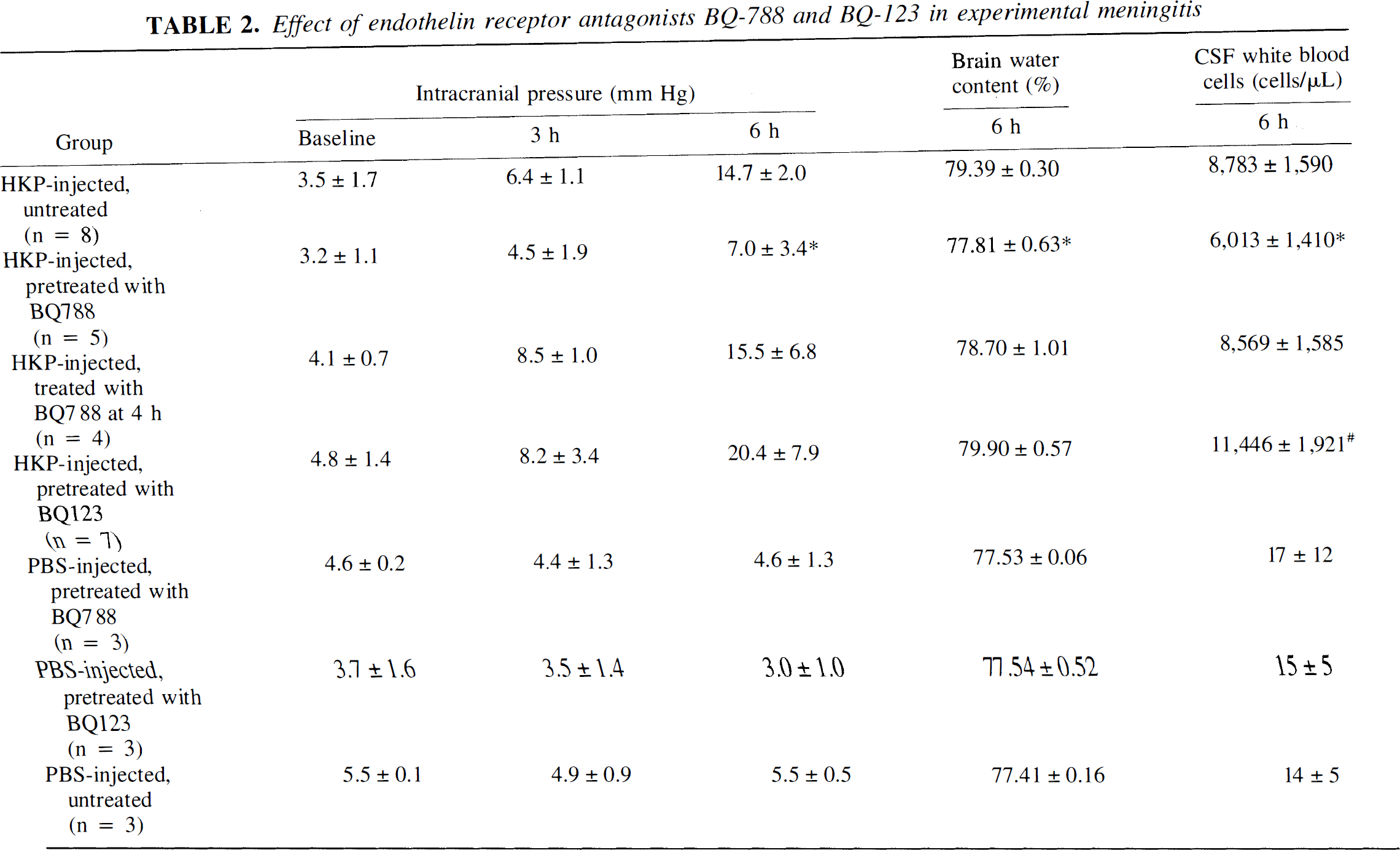
Challenge with HKP induced a significant increase in CBFLDF, ICP, brain water content, and CSF WBC count (Fig. 1; Table 2). Pretreatment with BQ-788 significantly attenuated these pathophysiologic alterations (Fig. 1; Table 2). In contrast, pretreatment with BQ-123 failed to prevent the increase of ICP and brain water content seen after injection with HKP. Moreover, when injecting BQ-123, a significant increase in CBFLDF and CSF pleocytosis was observed (Table 1).
Effect of pretreatment with either BQ-788 or BQ-123 on cerebral blood flow under both physiologic and pathophysiologic conditions. In untreated rats injected intracisternally with PBS (control rats; n = 3), there was no significant change in cerebral blood flow as measured by laser Doppler flowmetry (CBFLDF) throughout the experiment. Intravenous administration of neither BQ-788 (n = 3) nor BQ-123 (n = 3) led to changes in CBFLDF. Challenge with heat-killed pneumococci (n = 8) induced a significant increase in CBFLDF. Pretreatment with BQ-788 (n = 5) significantly attenuated this pathophysiologic alteration (*P < 0.05; compared with both untreated rats injected intracisternally with heat-killed pneumococci and BQ-123–pretreated rats injected intracisternally with heat-killed pneumococci). When BQ-123 was injected (n = 7), a significant increase in CBFLDF was even observed (#P < 0.05; compared with untreated rats injected intracisternally with heat-killed pneumococci).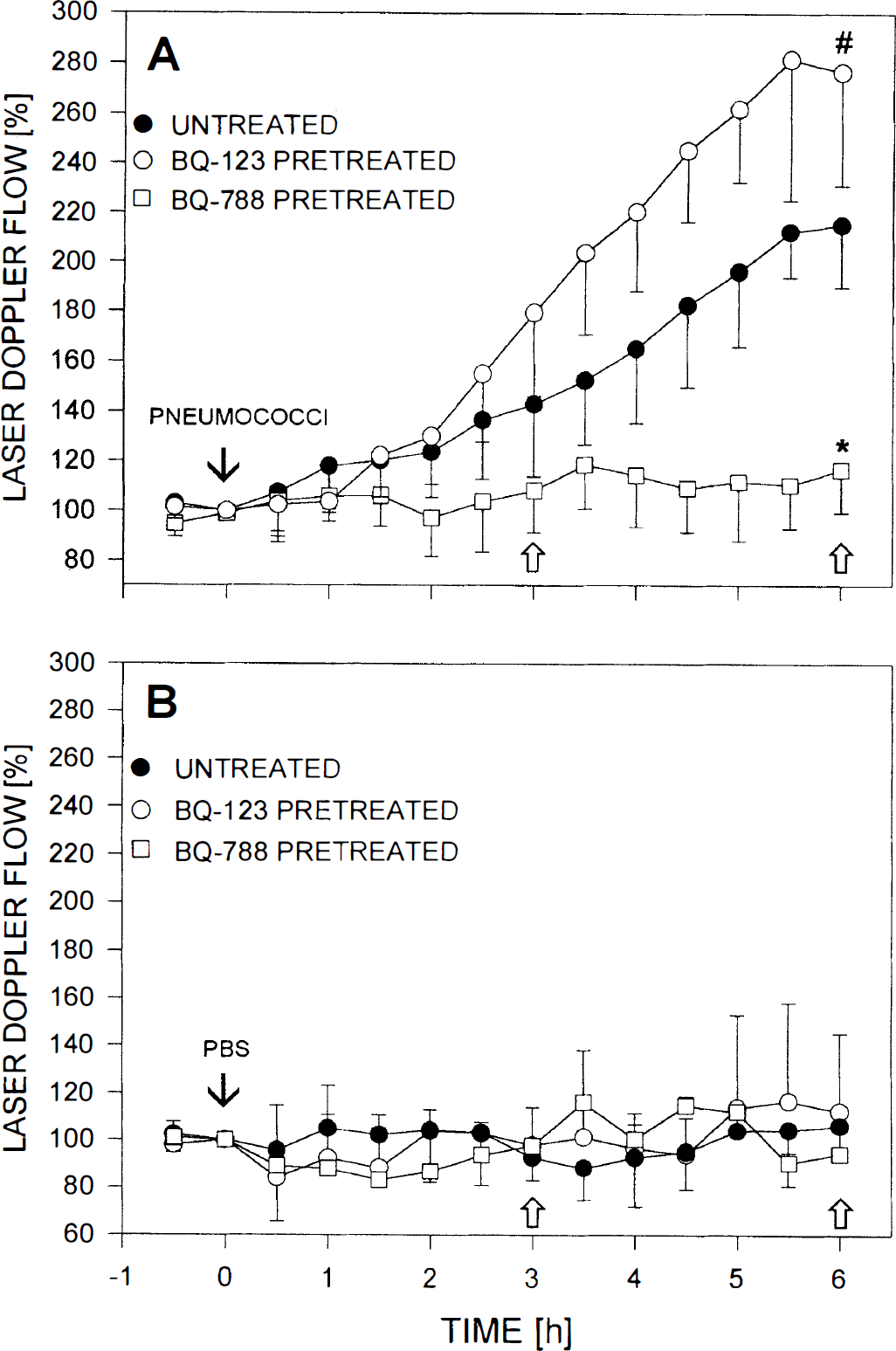
Administration of BQ-788 4 hours after intracisternal challenge did not reverse the increase in CBFLDF (6 hours after pneumococcal challenge: 196.1 ± 25.8%, compared with 225.3 ± 21.8% in untreated rats injected intracisternally with HKP; not significant) and ICP, and had no effect on brain edema formation and CSF pleocytosis (Table 2).
In vitro experiments
Nitrite concentrations in the cell culture supernatant of unstimulated endothelial cells (medium control) were below the detection limit. Stimulation with HKP induced a marked increase in nitrite levels (7.2 ± 1.8 μmol/L; P < 0.05 compared with medium control). Administration of
The ET concentration in cell culture supernatants of unstimulated endothelial cells (negative control) was 5.5 ± 2.4 pmol/L, suggesting a constitutive ET release. This result was supported by previous observations of immunoreactive ET-1 in the supernatants of bovine and rat micro vessel endothelial cells in short-term culture (Durieu-Trautmann et al., 1993; Yoshimoto et al., 1990). Stimulation with thrombin, a known inducer of ET secretion, caused a pronounced elevation of immunoreactive ET-1 in the cell culture medium (49.7 ± 23.6 pmol/L; P < 0.05 compared with negative control). Heat-killed pneumococci induced a significant increase in immunoreactive ET-1 (17.8 ± 2.7 pmol/L; P < 0.05 compared with negative control). No increase in immunoreactive ET-1 concentrations was detected when HKP were incubated in medium in the absence of endothelial cells. Treatment with phosphoramidon but not with
DISCUSSION
The major finding of this study was that the ETB receptor antagonist BQ-788 significantly attenuated the increase in CBFLDF, ICP, brain water content, and CSF WBC count in early pneumococcal meningitis. There are three possible reasons for the failure of BQ-788 to completely inhibit pathophysiologic changes in our study: 1) involvement of other mediators than ET, 2) restricted access of peptidergic antagonists such as BQ-788 to the brain and, thus, to abluminal ETB receptors, or 3) additional ET receptor subtypes, which cannot be blocked by BQ-788.
To date at least two distinct types of ET receptors have been described: ETA and ETB receptors. The ETA receptor has a higher affinity for ET-1 and ET-2 than for ET-3, and the ETB is nonselective for all three ET isopeptides (Sokolovsky, 1995). Pharmacologic data also support the existence of more than these two conventional receptor subtypes. For example, Wren and coworkers (1993) showed that whereas ET-3 can mediate cell proliferation in wounded human umbilical vein endothelial cells, neither ET-1 nor ET-2 can. This response cannot be mediated by the currently defined ETA and ETB receptors. In accordance with Wren and colleagues (1993), a third ET receptor subtype, which preferentially binds ET-3, was suggested to be expressed on bovine endothelial cells (Emori et al., 1990), rabbit vascular smooth muscle cells (Douglas et al., 1995), and in rat cerebellar homogenates (Widdowson and Kirk, 1996). This receptor subtype might be related to the ETC receptor subtype, which was cloned from Xenopus laevis dermal melanophores (Karne et al., 1993). Since binding and functional data pointing to the existence of a third ET receptor subtype are scarce, and a mammalian homologue of this receptor has not yet been identified, it remains unclear whether the failure of BQ-788 to completely inhibit pathophysiologic changes in our study is related to the ETC receptor.
The presence of receptors for ET in rat and human brain has been demonstrated in autoradiographic binding and in situ hybridization studies. Specific ET-binding sites were observed in neurons, astrocytes, microglial cells, vascular smooth muscle cells, and cerebromicrovascular endothelial cells (Rubanyi and Polokoff, 1994; Sokolovsky, 1995). The mammalian brain predominantly contains the ETB receptor subtype (Fernandez-Durango et al., 1994). The ETB receptor is believed to exist mainly on endothelial, neuronal, and glial cells (Harland et al., 1995; Lysko et al., 1995; Nilsson et al., 1997). The ETA receptor is mainly expressed by vascular smooth muscle cells, but it is also abundant on endothelial, neuronal, and glial cells but to a far less degree (Vigne et al., 1990; Lysko et al., 1995; Nilsson et al., 1997). Studies report that ET-1 failed to cross the normal blood–brain barrier (Koseki et al., 1989). Similarly to ET, its peptidergic receptor antagonists, such as BQ-123 and BQ-788, presumably are unable to pass through the blood–brain barrier (Clozel and Watanabe, 1993). In our experiments, we found that BQ-788 had an effect when given by the intravenous route. The effect of BQ-788, as observed in our study, indicates that the agent is able to gain access to ETB receptors mediating pathophysiologic alterations in pneumococcal meningitis. The most probable explanation is that ETB receptors are located in the intravascular compartment, namely on cerebromicrovascular endothelial cells. Since the permeability of the blood–brain barrier is compromised during pneumococcal meningitis (Quagliarello et al., 1986), it is also conceivable that BQ-788 may gain direct access to abluminal ETB receptors on glial and neuronal cells.
Another important finding of this study was that systemic administration of the ETA receptor antagonist BQ-123 significantly augmented the increase in CBFLDF and CSF WBC count. Our data on the efficacy of intravenous BQ-123 contradict most of the available information about the site of BQ-123 action in the brain. For example, Clozel and Watanabe (1993) demonstrated that BQ-123 prevented early cerebral vasospasm that follows subarachnoid hemorrhage after intracisternal but not intravenous injection. However, the observation of the effectiveness of intravenously administered BQ-123 in preventing cerebral postischemic hypoperfusion in gerbils agrees with our findings (Spatz et al., 1996). Moreover, Kobari et al. (1994a) describe a decline in cerebral blood volume after intracarotid administration of a low dose of ET-1; this was prevented with BQ-123 given by the same route. These authors suggest that BQ-123 may act on vasoconstrictive ET receptors located in the cerebrovascular endothelium.
Functional studies have pointed to a variety of biochemical effects elicited by activation of ETA and ETB receptors in the mammalian brain. Originally ET was described to be a highly potent vasoconstrictor (Yanagisawa et al., 1988). Although ET (i.e., ET-1) consistently has potent constrictive effects on isolated cerebral vessels in vitro (Salom et al., 1995), findings on the action of exogenous ET on the cerebral circulation in vivo vary. When applied from the abluminal side, ET-1 induced a pronounced constriction of cerebral arteries and a decrease in cerebral perfusion, which could be prevented by selective ETA receptor antagonists (Rubanyi and Polokoff, 1994; Salom et al., 1995). Low doses of abluminal ET-1, however, occasionally may dilate pial arteries, probably through the induction of prostanoids and NO (Armstead et al., 1989). In addition, topical application of the ETB receptor agonists IRL-1620 and BQ-3020 was shown to mediate dilation of rat basilar artery and feline cerebral resistance arterioles, which were dependent on NO production (Kitazono et al., 1995; Patel et al., 1996).
When ET was given intravascularly, ET was reported to have diverse effects on cerebral vessels: some observed a vasodilatory effect, whereas others reported either no or even a vasoconstrictive effect. Intraarterial infusion of ET-1 and BQ-3020 caused no change in diameters of canine major cerebral arteries or feline cerebral resistance arterioles, respectively (Shigeno et al., 1989; Patel et al., 1996). Even a decrease in cerebral perfusion was demonstrated after direct injection of ET-1 into the cerebral circulation of the goat (Salom et al., 1991). In contrast, an increase in cerebral perfusion was found after intracarotid infusion of high doses of either ET-1 or IRL-1620 in cats (Kobari et al., 1994a, 1994b). Similarly, Willette and others (1990) report increased cortical microvascular perfusion in rats either after intracarotid or intravenous administration of ET-1, and Weitzberg and associates (1993) describe cerebral vasodilation after intravenous infusion of ET-1 in humans. These discrepancies may depend on differences in vascular beds and species investigated in the aforementioned studies.
Since ET is constitutively produced by cerebrovascular endothelial cells, as observed in our and previous studies (Yoshimoto et al., 1990; Durieu-Trautmann et al., 1993), a basal ET-mediated cerebrovascular tone may be present. However, our observation that neither BQ-788 nor BQ-123 modulated CBFLDF when administered in control rats contradicts this hypothesis, but agrees with a variety of recently published studies demonstrating that ET receptor antagonists have no effect on diameters of cerebral vessels or cerebral perfusion under physiologic conditions (Clozel and Watanabe, 1993; Kobari et al., 1994a).
The ET and ET receptor subtypes may play a major role in the pathophysiologic mechanisms of CNS diseases, namely, as a mediator of vasoconstriction and reduced cerebral perfusion. Three reasons support this suggestion: 1) far greater amounts of ET-1 have been detected in the CSF of patients with a variety of neurologic disorders such as cerebral ischemia and subarachnoid hemorrhage than in normal human CSF; 2) the expression of ET receptors was increased in the aforementioned diseases; and 3) inhibitors of ET production and selective ET receptor antagonists showed beneficial effects on the respective disease in animal models of the disease (Rubanyi and Polokoff, 1994; Salom et al., 1995).
In the current study, we report that BQ-788 attenuated the increase in CBFLDF and CSF WBC count, whereas BQ-123 augmented these pathophysiologic alterations. There are several possible explanations for our observation. First, as mentioned earlier, the vasomotor effect of ET probably is composed of relaxant and contractile components that compete with each other. Blockade of ETA receptors, which mainly mediate the vasoconstrictive effects of ET, may thus lead to some potentiation of ET-induced vasodilation and, therefore, to an enhanced increase in cerebral perfusion, as we observed. Second, ETA blockade may be accompanied by a greater access of ET to ETB receptors, thus amplifying ETB receptor–mediated biochemical effects. Through ETB receptor activation, ET was shown to stimulate its own production in rat aortic endothelial cells and in human umbilical endothelial cells (Fujitani et al., 1992; Saito et al., 1995), the synthesis of NO by bovine endothelial cells (Hirata et al., 1993), cytokine production by human umbilical endothelial cells (Stankova et al., 1996), prostacyclin release in guinea pig aortic rings (Matsuda et al., 1993), the expression of adhesion molecules on rat aortic endothelial cells (Hayasaki et al., 1996), and the enhancement of human neutrophil chemotaxis (Elferink and de Koster, 1996). In addition, ET-induced increases of either cerebral perfusion or diameters of cerebral vessels in cats and rats (Willette et al., 1990; Kobari et al., 1994a, 1994b), as well as an increase of the vascular permeability in the rat dura mater (Brändli et al., 1996), have been reported to be mediated by ETB receptor activation. All of these biochemical and pathophysiologic mechanisms have been implicated in the pathophysiologic mechanism of pneumococcal meningitis. Thus, an amplification of these mechanisms may contribute to the observed augmentation of the increase in CBFLDF and WBC counts after BQ-123 administration.
During the last several years, experimental studies have shown that the pathophysiologic mechanisms of pneumococcal meningitis are complex. Animal studies have provided evidence that cytokines (Saukkonen et al., 1990), arachidonic acid metabolites (Pfister et al., 1990), neuropeptides (Weber et al., 1996), reactive oxygen species (Pfister et al., 1990), NO (Koedel et al., 1995), and leukocyte–endothelial interaction (Tuomanen et al., 1989) are involved in the pathophysiologic mechanisms. Several reports indicate that ET may interact with these mediators: ET can stimulate the release of cytokines (Stankova et al., 1996), arachidonic acid metabolites (Matsuda et al., 1993), neuropeptides (Ritz et al., 1992), and NO (Hirata et al., 1993). Also, ET can up-regulate the expression of adhesion molecules including ICAM-1, VCAM-1, and E-selectin (McCarron et al., 1993; Hayasaki et al., 1996) and can induce neutrophil adhesion to endothelial cells (Hayasaki et al., 1996). Furthermore, ET can exert chemoattractant activity for neutrophils (Elferink and de Koster, 1996) and can modulate phagocyte function, such as potentiating fMet-Leu-Phe–activated superoxide production (Ishida et al., 1990).
In this study, cerebromicrovascular endothelial cells were induced to release both NO and ET on stimulation with pneumococci. Inhibition of the endothelin-converting enzyme with phosphoramidon prevented the increase in ET concentrations and markedly reduced NO release. These data help elucidate one possible mode of action of the ETB receptor antagonist: BQ-788 inhibits ET-binding to ETB receptors, thus reducing the generation of NO, which has been identified as a central mediator of pneumococcal meningitis (Koedel et al., 1995). The role of ET in the pathophysiologic mechanism of pneumococcal meningitis was supported by our previously published observation that elevated ET levels can be detected in CSF samples of patients with bacterial meningitis obtained on admission to the hospital (Koedel et al., 1997).
In conclusion, our data indicate that ET is a mediator in early pneumococcal meningitis in the rat and contributes to the increase in CBFLDF, ICP, brain water content, and CSF pleocytosis, presumably by ETB receptor–mediated NO production.