
Abstract
Obstructive sleep apnea (OSA) is associated with cerebrovascular diseases. However, little is known regarding the effects of OSA on the cerebrovascular wall. We tested the hypothesis that OSA augments endothelin-1 (ET-1) constrictions of cerebral arteries. Repeated apneas (30 or 60 per hour) were produced in rats during the sleep cycle (8 hours) by remotely inflating a balloon implanted in the trachea. Four weeks of apneas produced a 23-fold increase in ET-1 sensitivity in isolated and pressurized posterior cerebral arteries (PCAs) compared with PCAs from sham-operated rats (EC50=10−9.2 mol/L versus 10−10.6 mol/L;
Keywords
Introduction
Obstructive sleep apnea (OSA) is a highly prevalent disorder involving collapse of the upper airway during sleep to significantly reduce (hypopnea) or completely block (apnea) the movement of air into and out of the lungs.1, 2, 3 Episodes of OSA generally occur repetitively throughout sleep with each episode producing transient hypoxia and hypercapnia. 1 When the airway is completely blocked, a continued effort to breathe against the closed airway can result in excessive negative pressures in the thoracic cavity. Each episode of OSA continues until the individual arouses and reestablishes a patent airway. 1 The severity of OSA is categorized by the number of OSA events that occur on average during each hour of sleep (apnea hypopnea index or AHI). Mild, moderate, and severe OSA are defined as having 5 to 15, 16 to 30, and >30 events per hour, respectively. 1 Recent estimates suggest that 5% to 25% of the adult western population experience clinically significant OSA.1, 2 Risk factors for OSA include obesity and aging, suggesting that the prevalence of OSA is likely to increase in the future given the current demographic trends. 1
Obstructive sleep apnea alters cerebrovascular responsiveness and is strongly associated with cardiovascular and metabolic diseases that significantly impact the brain.1, 2, 3 Patients experiencing OSA reportedly show decreases in resting cerebral blood flow, impaired autoregulation, and attenuated dilatory responses to hypoxia and hypercapnia. 2 As a result, it is not surprising that OSA is strongly associated with numerous disease states involving the cerebrovascular circulation. Cross-sectional and longitudinal studies have identified OSA as an independent risk factor for stroke, with moderate and severe OSA increasing the risk of stroke by threefold. 2 In addition, OSA augments the damage after a stroke and increases the likelihood of subsequent strokes. 2 Although not as extensively studied as stroke, OSA appears to be associated with silent brain infarcts, white-matter disease, and the onset and severity of dementia. 2
Efforts to elucidate the pathophysiologic mechanisms of OSA in the cerebral circulation are complicated by the fact that OSA patients commonly have comorbidities, including obesity, diabetes, and hypertension.1, 2, 3 For this reason, animal models of OSA are important for studying the effects of OSA in the absence of confounding factors. The most commonly used model, chronic intermittent hypoxia (CIH), recapitulates the hypoxia/reoxygenation component of OSA by exposing experimental animals, generally rodents, to repetitive episodes of hypoxia during the sleep cycle.
Two studies in the literature have addressed mechanisms of cerebrovascular dysfunction using the CIH model of OSA.4, 5 After 14 days of CIH in rats, endothelial mediated dilations in middle cerebral arteries were significantly attenuated. 5 In a second study, CIH in mice attenuated the increase in cerebral perfusion in response to neural stimulation, which was attributed to alterations in the vasoconstricting endothelin-1 (ET-1) system. 4
In an effort to more closely recapitulate the physiologic consequences observed in OSA patients, we have developed a model of OSA that incorporates apneas during the sleep cycle in rats. 6 Apneas were produced by inflating a balloon in the trachea. The severity of OSA in our model was equivalent to moderate OSA in humans. In addition to hypoxic episodes, as occurs in CIH, our model incorporated progressive hypercapnia, increased negative intrathoracic pressures, and arousals. Furthermore, we showed that endothelial dilations involving nitric oxide production were attenuated in the middle cerebral artery after 4 weeks of apneas. 6
Although the ET-1 pathway is associated with cerebrovascular dysfunction during CIH, the responsiveness of cerebral arteries to ET-1 has not been studied in CIH or any other model of OSA. Furthermore, it is not known whether the more moderate hypoxia occurring in our model even affects ET-1 signaling. Therefore, we sought to determine whether constrictions to ET-1 are altered in rat cerebral arteries after OSA, and if so, which of the ET-1 receptors (ETA receptor, ETAR and ETB receptor, ETBR) are involved.
Using isolated pressurized perfused posterior cerebral arteries (PCAs) we show that constrictions to ET-1 were enhanced after two and four weeks of OSA. We report that the increased ET-1 sensitivity was dependent on ETBR, and likely involves transient receptor potential channels (TRPCs) and Rho kinase (ROCK).
Materials and methods
All animal procedures were performed in accordance with the
Endotracheal Obstruction Device Implantation
Long Evans rats, 8 to 9 weeks old, were anesthetized with CCM DEAIII-Rodent Cocktail (Ketamine 37.5 mg/mL, Xylazine 1.9 mg/mL, Acepromazine 0.7 mg/mL; 1 μL/g body weight). An obstruction device or balloon, which was fabricated from silicone tubing, was inserted into the lumen of the trachea caudal to the thyroid. 6 The silicone obstruction device was attached to PE-50 tubing that was tunneled under the skin and exited the back of the neck. The externalized PE-50 tubing was passed through a spring attached to the rat by a harness at one end and a swivel mounted at the top of the cage at the other end (Figure 1A). The swivel allowed free movement of the rat within the cage. The obstruction device did not significantly impede the movement of air when deflated but completely occluded the airway in the trachea when inflated by pressurizing the system. 6 Beginning the day of surgery, the animals were treated for 3 days with an analgesic (5 mg/kg Ketoprofen, Fort Dodge Animal Health, Fort Dodge, IA, USA) and an antibiotic (5 mg/kg Baytril, Bayer HealthCare LLC, Shawnee Mission, KS, USA).
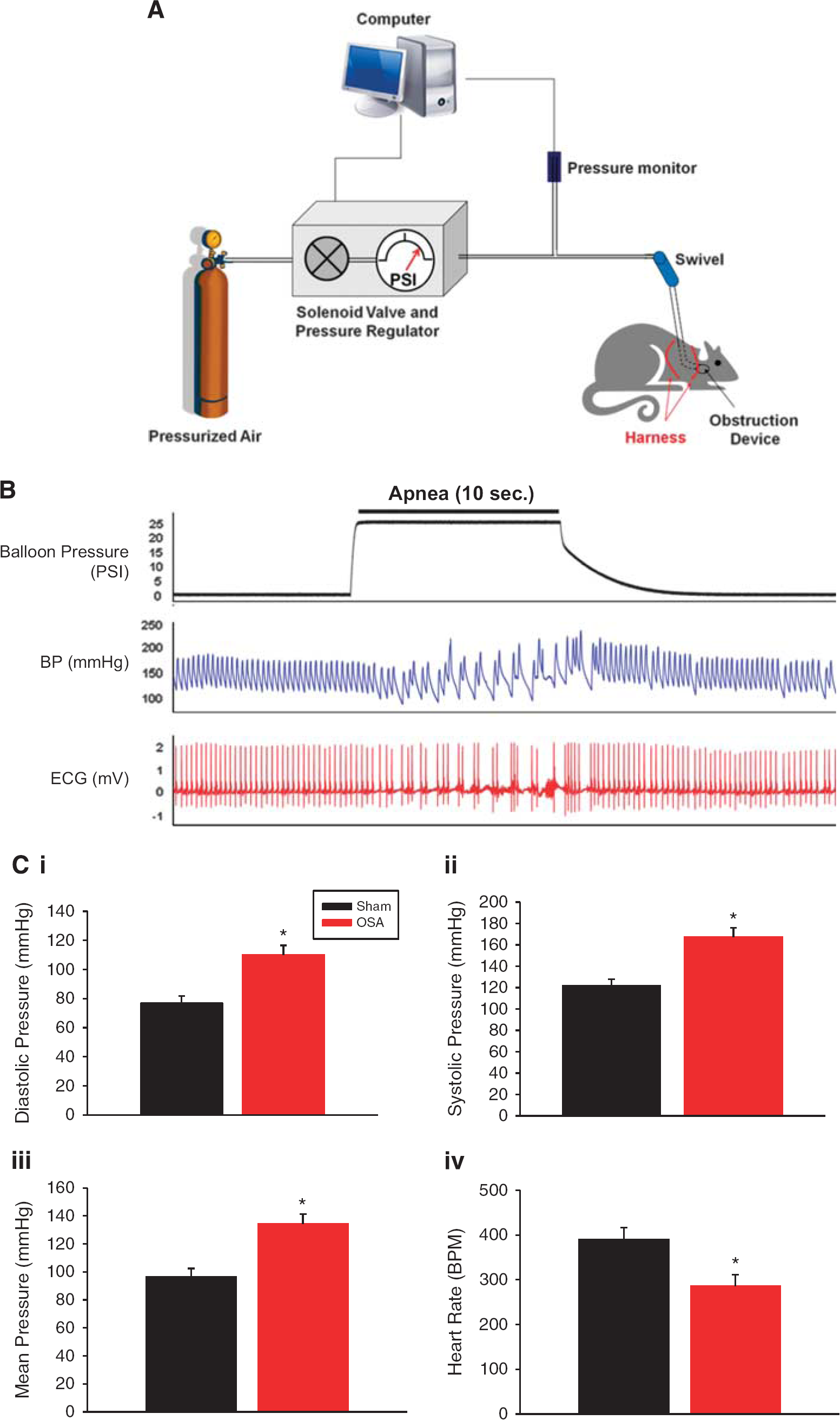
Diagram of tethered rat with implanted obstruction device and system controlling apneas. The system consists of a computer to initiate apneas, a solenoid valve to regulate pressure to inflate the obstruction device, and a pressure transducer (
Computer Controlled Inflation of Obstruction Device
After a 1-week recovery from surgery, rats were subjected to episodes of apnea (10 seconds in duration) for 8 hours a day (0800 to 1600 h) during the sleep cycle. Durations between apneas were varied to prevent the rats from predicting when an apnea would occur. Initial studies (Figures 2, 3, and 4) were performed after 4 weeks of OSA at an average frequency of one apnea every 2 minutes (AHI=30). Subsequent studies (Figures 5 and 6) were performed after 2 weeks of OSA at an average frequency of one apnea every minute (AHI=60). This latter approach was used to increase the throughput of the experiments. Similar changes in cerebral vessel ET-1 and IRL-1620 sensitivity were confirmed in 2- and 4-week groups.
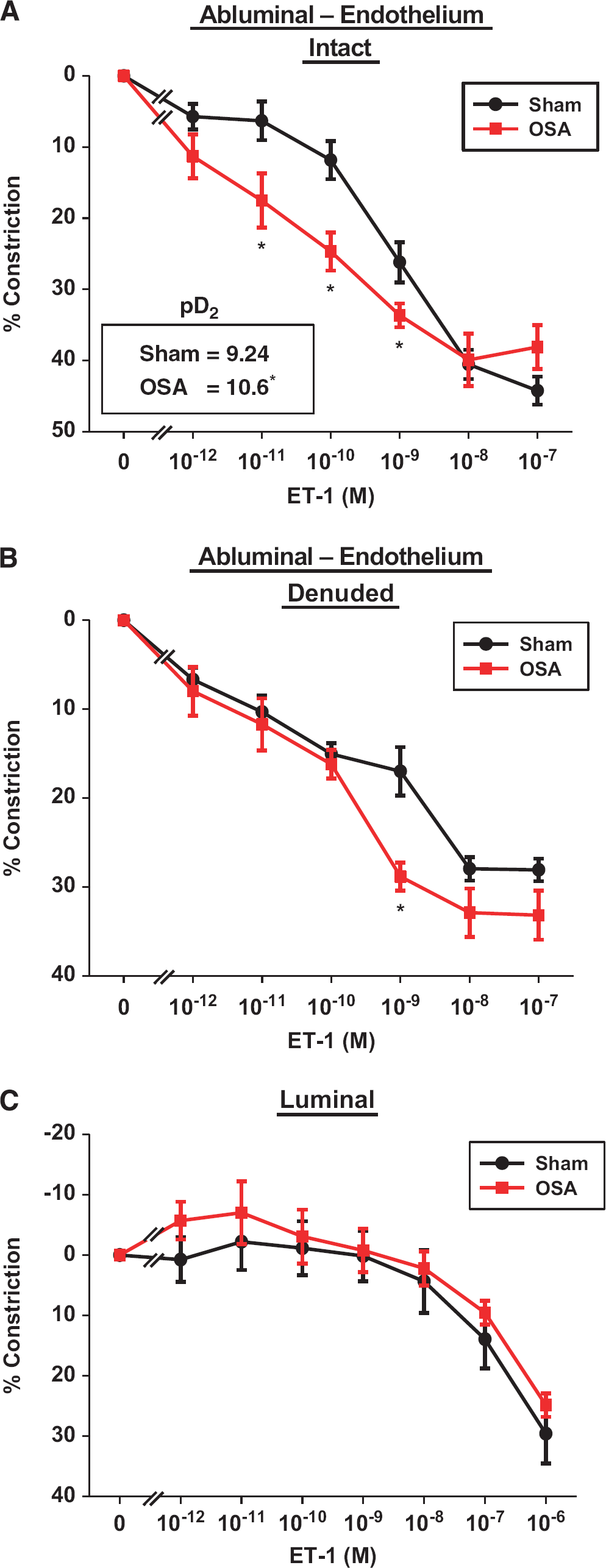
Four weeks of apneas during the sleep cycle augmented posterior cerebral artery (PCA) sensitivity to endothelin-1 (ET-1) applied abluminally in endothelium intact (
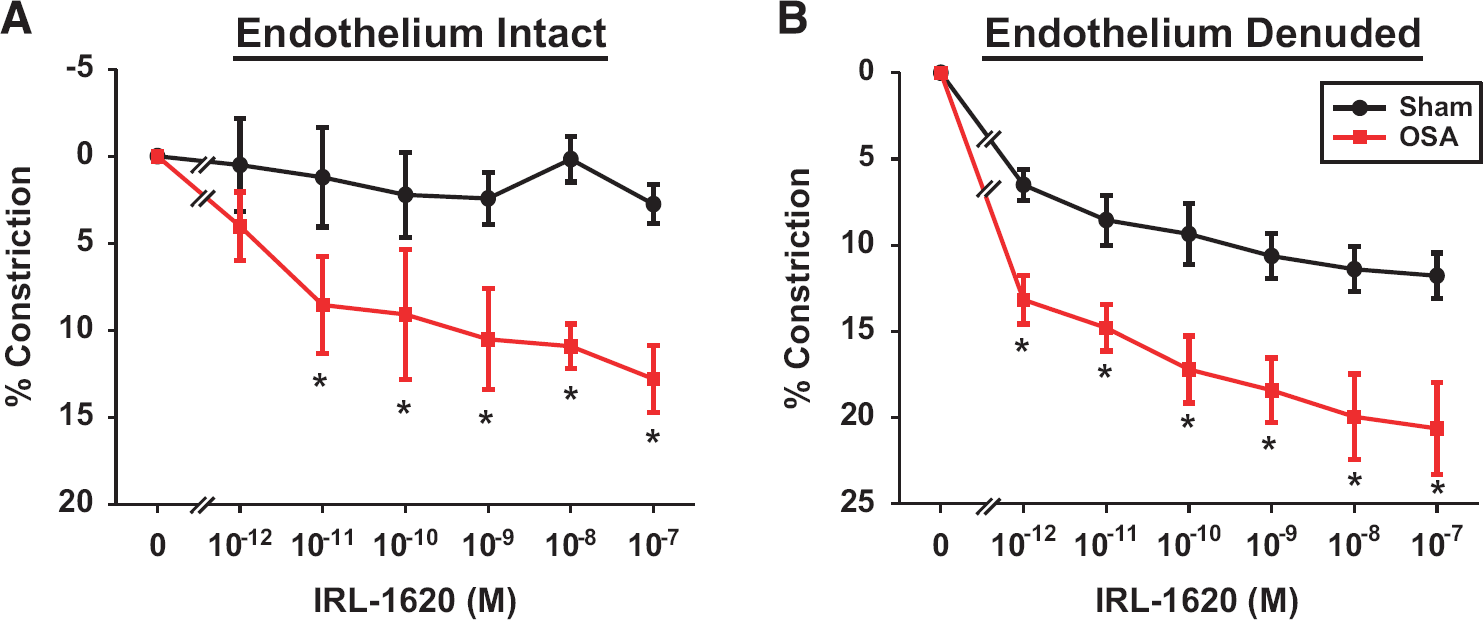
After 4 weeks of apneas during the sleep cycle, the ETB receptor (ETBR) agonist, IRL-1620, induced greater vascular constrictions in obstructive sleep apnea (OSA) posterior cerebral arteries (PCAs) with endothelium intact (
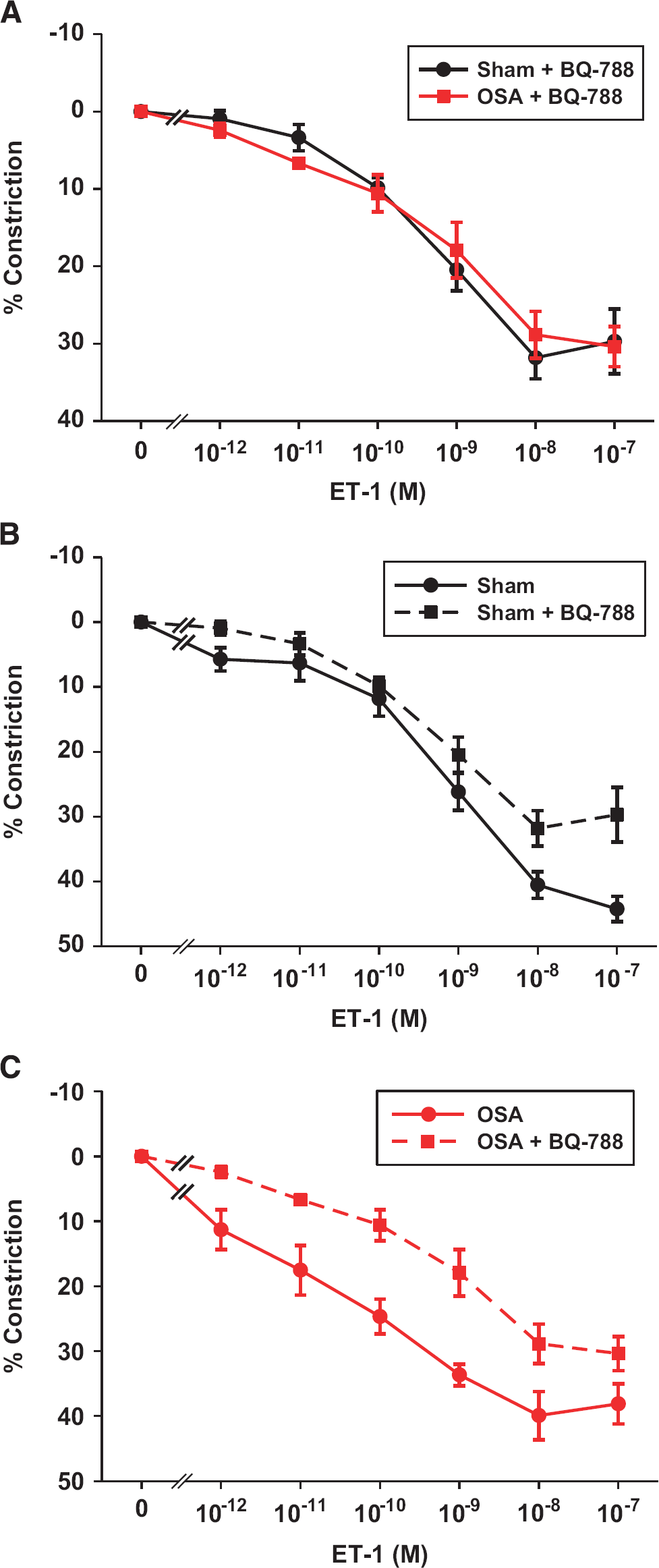
Pretreatment of vessels with the ETB receptor (ETBR) antagonist BQ-788 abolished the increased ET-1 sensitivity of obstructive sleep apnea (OSA) vessels (
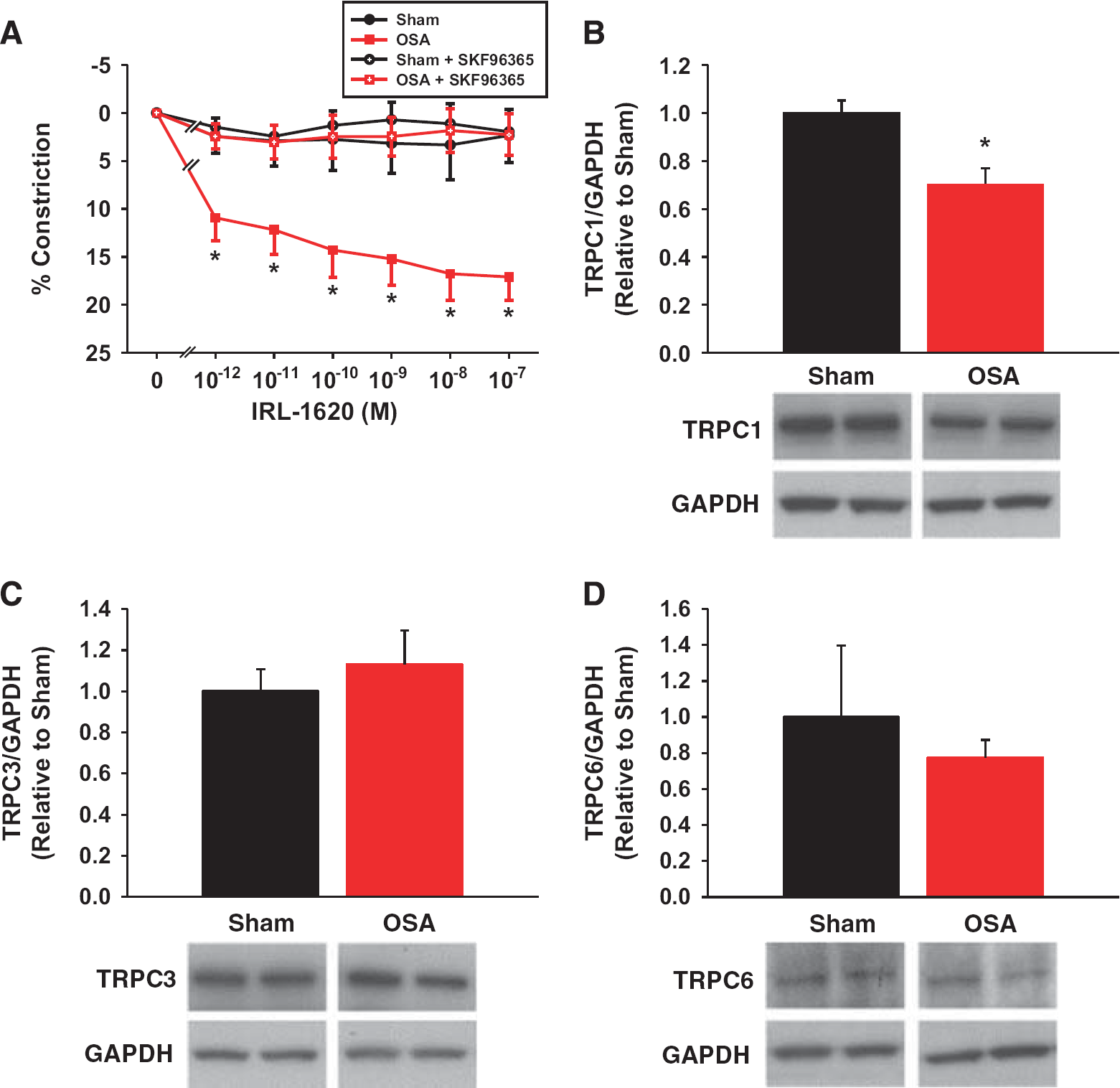
Pretreatment of vessels with the transient receptor potential channel (TRPC) inhibitor SKF96365 abolished the increased IRL-1620 sensitivity of obstructive sleep apnea (OSA) vessels (
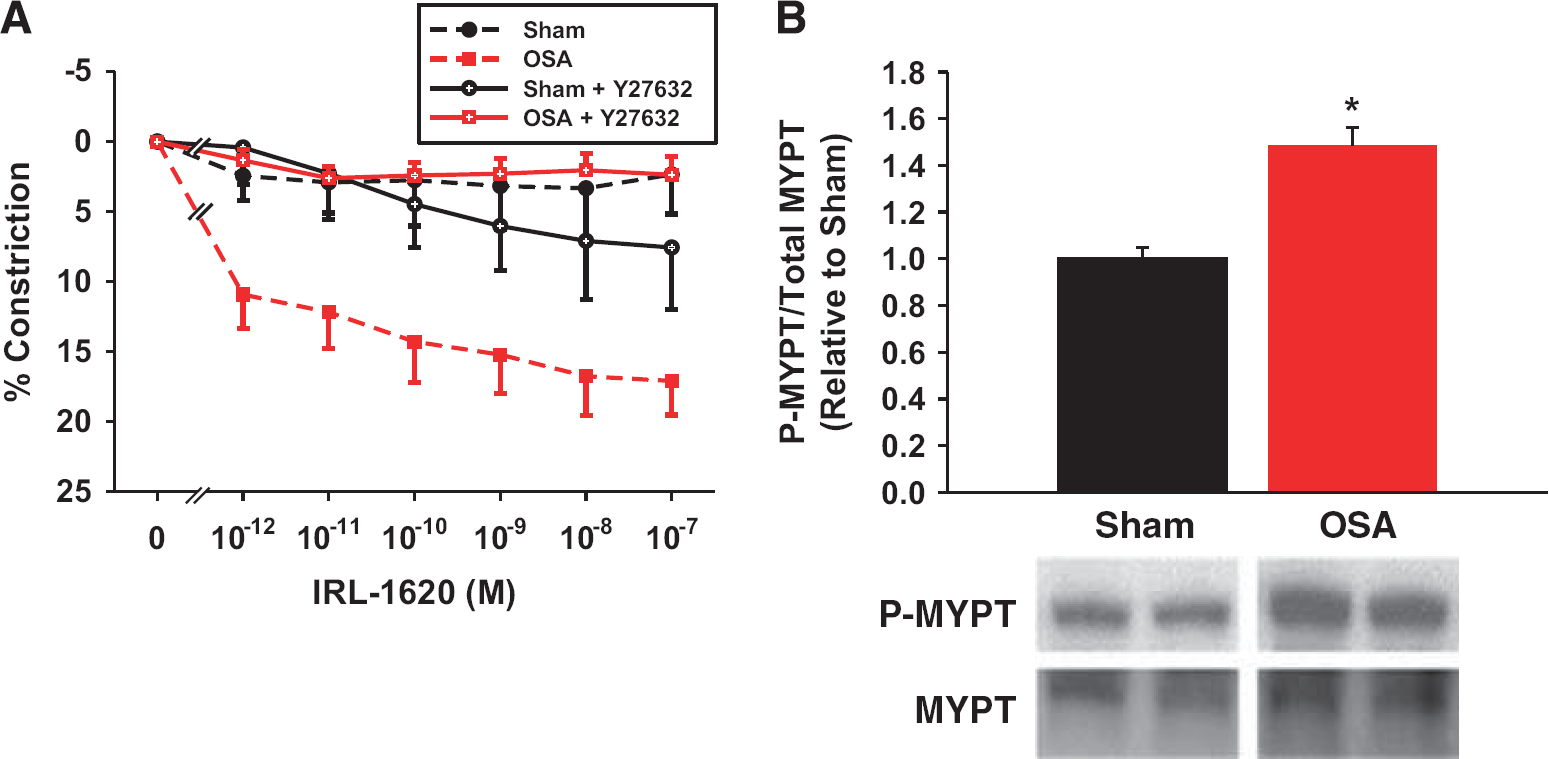
Pretreatment of vessels with the Rho kinase (ROCK) inhibitor Y27632 abolished the increased IRL-1620 sensitivity of obstructive sleep apnea (OSA) vessels (
Apneas were produced by inflating the balloon using compressed air. The timing and duration of the inflations were controlled by an AD Instruments Power Lab stimulator output using LabChart 7 (AD Instuments; Colorado Springs, CO, USA) and a custom-built solenoid valve/ gas regulator. The balloon pressure was monitored at all times using a pressure transducer (Figure 1A). On occasion leaks in the inflation system occurred, which could be readily determined when pressures did not achieve the predetermined set-point. When leaks did occur, the source of the leak was located (swivel or tube connections) and repaired. If the leak was in the balloon, then the animal was removed from the study.
In a subset of rats, blood pressure (BP) and heart rate were continuously monitored by a telemetry system (Millar; Houston, TX, USA) consisting of a solid state pressure sensing probe implanted in the abdominal aorta and biopotential leads attached to the chest wall, respectively.
Sham rats underwent identical surgical procedures and device implantation, but endotracheal obstruction devices were never inflated. In the present study, the experimental rats undergoing periods of apnea are referred to as ‘OSA rats’ and the sham controls are referred to as ‘sham rats’.
Isolated Posterior Cerebral Arteries
After 2 (AHI=60) or 4 (AHI=30) weeks of apneas for the OSA group or the equivalent time for the sham group, rats were anesthetized with isoflurane and decapitated. Each brain was rapidly removed and placed in cold Krebs Buffer. Posterior cerebral arteries were carefully harvested and placed in a vessel chamber that was filled with Krebs buffer consisting of: (mmol/L) 119 NaCl, 4.7 KCl, 1 MgSO4, 1.2 KH2PO4, 0.026 EDTA, 1.6 CaCl2, 5.5 glucose, and 25 NaHCO3. The buffer was gassed with 20% O2, 5% CO2, balanced with N2 to obtain a pH of 7.4 and recirculated at a rate of 9 mL/min. The ends of each PCA were mounted on glass micropipettes and secured with 12-0 nylon sutures. Care was taken to ensure that the mounted vessel segments did not have side branches or leaks. The PCAs were warmed to 37°C, and pressurized to 85 mm Hg by raising buffer-filled reservoirs connected to the glass micropipettes. After 40 minutes of pressurization, luminal flow (150 μL/min) was initiated by adjusting the heights of the inflow and outflow reservoirs. Posterior cerebral arteries were allowed an additional 20 minutes to develop spontaneous tone before the experimental protocol began.
In a subset of experiments, the endothelium was removed by passing air through the lumen of the vessel using a syringe pump. The vessel was exposed to air for 6 minutes at an intraluminal pressure of 85 mm Hg. After the denuding procedure, the lumen was refilled and repressurized with Krebs buffer and allowed 20 minutes to develop steady tone. The absence of dilation to luminally applied ATP (10−5 mol/L) showed the effectiveness of the denuding procedure. Dilation by stimulating inwardly rectifying potassium channel with abluminal KCl (15 mmol/L) showed that the vascular smooth muscle (VSM) was still viable after the denuding process.
Posterior cerebral arteries were magnified x450 and digitally recorded by a CCD camera connected to a DVR. Data were analyzed offline using edge detection software to follow changes in vessel outer wall diameter. Percent constriction was calculated by:
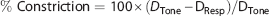
where
Pressurized and perfused PCAs were used to investigate various components of the ET-1 pathway. Dose response curves to ET-1 and the ETBR agonist, IRL-1620, were performed by applying each agonist to the abluminal chamber (10−12 to 10−7 mol/L) and allowing 15 minutes between doses. In one study, ET-1 was administered luminally (Figure 2C) by adding it to the luminal perfusate. Abluminal administration preferentially activates receptors on the VSM, while luminal administration preferentially activates receptors on the endothelium. In some studies, ET-1 or IRL-1620 dose response curves were performed after incubating the pressurized perfused vessel (luminally and abluminally) with the ETBR antagonist, BQ-788 (100 nmol/L; 15 minutes), the TRPC antagonist, SKF96365 (25 μmol/L; 30 minutes), or the ROCK inhibitor, Y27632 (1 μmol/L; 30 minutes).
Immunohistochemistry
Rats were anesthetized with 3% isoflurane and decapitated. Brains were quickly removed and a wedge of cortex containing the PCA was dissected and frozen in 2-methylbutane at a temperature of −80°C. The wedges were coated in optimal cutting temperature compound (Sakura, Tokyo, Japan), cut into 12 μm cross sections of the PCA, mounted on glass slides, and stored at −80°C until processing.
Tissue sections were fixed with either 4% paraformaldehyde (ETBR) or acetone (ETAR) for 10 minutes. After three washes with phosphate-buffered saline, the sections were blocked and permeabilized for 30 minutes at room temperature with 10% serum from the host species of the secondary antibody, 0.5% BSA, and 0.1% Tween-20 in phosphate-buffered saline. Primary antibodies were diluted in the block/permeabilization buffer described above and exposed to sections overnight at 4°C in a humidifying chamber. The ETAR antibody (Alomone Labs, Jerusalem, Isreal) and the ETBR antibody (Enzo Life Sciences, Plymouth Meeting, PA, USA) were diluted to 2.5 and 5 μg/mL, respectively. Immunoflourescence for each antibody was compared with a control group in which the primary antibody was replaced with nonimmune IgG from the primary antibody host species.
Sections were washed and exposed to the secondary antibodies for 30 minutes in the dark. Secondary antibodies used for ETAR and ETBR were Alexa Flour 594-goat anti-rabbit and Alexa Flour 594-donkey anti-sheep (4 μg/mL; Life Technologies, Grand Island, NY, USA), respectively. After a final wash, the sections were treated with Vectashield containing DAPI (Vector Laboratories, Burlingame, CA, USA) and coversliped. Z-stack images were collected with an Olympus IX81 microscope (x60 objective) and then mathematically deconvolved (3i SlideBook 4.2, SlideBook Software, Denver, CO, USA).
Quantitative RT-PCR
Cerebral arteries were quickly harvested from the brain and snap frozen. Frozen arteries were ground with a pestle and total RNA was isolated using the RNeasy Micro Kit (Qiagen, Valencia, CA, USA). Isolated RNA was treated with DNAse I to eliminate potentially contaminating genomic DNA. RNA purity and concentration were assessed using a NanoDrop UV-Vis spectrophotometer (Thermo Scientific, Wilmington, DE, USA). All samples were diluted to 30 ng/μL.
Original TaqMan assays were designed from rat sequences available in GenBank using Primer Express 2.0 (Applied Biosystems, Grand Island, NY, USA). Primer and probe sequences were forward 5′-TGCAGATTGCCTTGAATGAC-3′, reverse 5′-CGAGGACCAGGCAGAATACT-3′, and probe 5′-FAM-CTTAAAGCAGAGACGAGAAGTGGCCAAG-TAMRA-3′ for
Endothelin-1 ELISA
Endothelin-1 levels were assessed in cerebral arteries and plasma using a commercially available ELISA kit (Enzo Life Sciences). Cerebral arteries were first homogenized and purified using C18 columns (Agilent Technologies, Santa Clara, CA, USA).
Western Blotting
Protein expression was measured in cerebral arteries isolated from rats after 2 weeks of sham or OSA (AHI=60), using standard western blotting techniques. Antibodies for transient receptor potential channels 1, 3, and 6 (TRPC1 (100 KDa): T8276, TRPC3 (110 KDa): T5067, TRPC6 (110 KDa): T6442; Sigma-Aldrich, St Louis, MO, USA), phosphoThr 696 myosin binding subunit of myosin phosphatase (P-MYPT1 (145 KDa): ABS45; EMD Millipore, Billerica, MA, USA), total MYPT1 (140 KDa: 2634; Cell Signaling, Beverly, MA, USA), and glyceraldehyde 3-phosphate dehydrogenase (37 KDa: 6C5; EMD Millipore) were used for protein detection. Glyceraldehyde 3-phosphate dehydrogenase was used as a loading control.
Drugs and Reagents
Endothelin-1 was purchased from EMD Chemicals, Inc. (Gibbstown, NJ, USA). BQ-123 was purchased from American Peptide Company (Sunnyvale, CA, USA). IRL-1620 and BQ-788 were purchased from Tocris Bioscience (Ellisville, MO, USA). SKF96365 was purchased from Cayman Chemical (Ann Arbor, MI, USA). Y27632 was purchased from (Sigma-Aldrich). All agonists and antagonists were dissolved in deionized water. All other reagents were purchased from Sigma-Aldrich.
Statistics
Data are expressed as mean±s.e.m. For comparing a single measure in two groups, a Student's
Results
Apnea Acutely, But Not Chronically, Alters Blood Pressure and Heart Rate
Figure 1B shows BP and ECG, as measured using telemetry, immediately before, during and after a 10-second episode of apnea. Similar to humans we observed profound swings in diastolic and systolic BP during 10 second apneas (marked by the increase in balloon pressure required for inflation; Figure 1B). Blood pressure and heart rate normalized within approximately 5 seconds of apnea resolution. Figure 1C shows that diastolic, systolic, and mean pressures, averaged over the 10-second apnea, were each significantly increased when compared with pressures in sham rats. Each 10-second apnea was accompanied by hypercapnia, acidosis, and a 12% oxygen desaturation of hemoglobin.
6
Resting mean arterial blood pressure after 2 weeks of apneas (AHI=60) was 105±2 mm Hg in the Sham group and 101±6 mm Hg in the OSA group (
Obstructive Sleep Apnea Increases Cerebrovascular Sensitivity to Abluminal Endothelin-1
Maximum diameters for rat PCAs were 263±9 μm (
To remove any influence of the endothelium in the constrictor response to abluminally applied ET-1, studies shown in Figure 2A were repeated in PCAs after removal of the endothelium (Figure 2B). Of note, endothelium removal resulted in similar constriction between sham and OSA PCAs (10.3±1% and 9.7±1.5% constriction, respectively). Two-way repeated measures ANOVA revealed a significant interaction between groups (sham/OSA) and ET-1 concentration (
Obstructive Sleep Apnea Does Not Alter Endothelin-1 Levels or Expression of Endothelin-1 Receptors
Supplementary Figure 1A shows that mRNA expression of
Increased Endothelin-1 Sensitivity in Obstructive Sleep Apnea PCAs Involves ETB receptor
Abluminal application of the ETBR agonist, IRL-1620 (K
To remove the contribution of endothelial ETBR on the IRL-1620 response, studies were conducted in endothelium-denuded arteries. Abluminal application of IRL-1620 dose dependently constricted denuded PCAs from sham and OSA rats (Figure 3B). There was a significant effect between sham and OSA groups (
To further assess the possibility that OSA results in increased ETBR sensitivity, we treated isolated PCAs with the ETBR antagonist BQ-788 (100 nmol/L; IC50=0.9 and 280 nmol/L for ETB and ETA receptors, respectively) and reassessed the responsiveness to ET-1. 9 In the presence of ETBR blockade, the constrictor responses to ET-1 were identical in PCAs from OSA and sham rats (Figure 4A). Thus, the increased sensitivity in OSA arteries was abolished with ETBR blockade (compare Figure 4A with Figure 2A). Figures 4B and 4C compare the response of PCAs to ET-1 in the presence (data from Figure 4A) and absence (data from Figure 2A) of ETBR blockade in sham (Figure 4B) and OSA (Figure 4C) rats. Note that the effect of BQ-788 on ET-1 constriction was greater in PCAs from OSA rats (Figure 4C) than in those from sham rats (Figure 4B). This is especially true for the concentrations from 10−11 to 10−9 M ET-1 where there was a more pronounced constriction after OSA (Figure 2A).
Enhanced Vasoconstriction Through ETB Receptor After Obstructive Sleep Apnea Is Dependent on Transient Receptor Potential Channel and Rho Kinase Signaling
Previous studies in cerebral and peripheral vessels have suggested that TRPCs have an important role in Ca2+ influx after ET-1 receptor signaling.10, 11, 12, 13 TRPC isoforms 1, 3, and 6 appear to be the predominant isoforms involved in VSM cell constrictions.11, 14, 15 To examine the role of TRPCs after OSA, we pretreated isolated PCAs with the pan-TRPC antagonist, SKF96365. The TRPC antagonist had no effect on sham vessels, which showed no constriction to IRL-1620, similar to sham vessels without SKF96365 (Figure 5A). In the presence of SKF96365, the constrictor responses to IRL-1620 were significantly attenuated in OSA PCAs (Figure 5A). Thus, the increased sensitivity resulting from apneas was abolished in the absence of TRPC activity. We also examined protein levels of TRPC1, 3, and 6 in cerebral arteries. We found a significant decrease in TRPC1, and no change in TRPC3 or 6, protein levels in cerebral arteries after OSA (Figures 5B to 5D).
Increased ET-1 constrictions after OSA may be secondary to increased Ca2+ sensitivity. Rho kinase has a key role in the regulation of Ca2+ sensitivity by phosphorylating and inhibiting the myosin binding subunit (MYPT1) of myosin phosphatase. In the absence of active myosin phosphatase, myosin light chain remains in the phosphorylated and pro-constrictive state. 16 To investigate the potential role that ROCK may play in the increased ET-1 constrictions after OSA, we pretreated isolated PCAs with the nonspecific ROCK1/2 isoform antagonist Y27632 (1 μmol/L; IC50=140 and 300 nmol/L for ROCK1 and ROCK2, respectively).17, 18 In the presence of Y27632, the constrictor responses to IRL-1620 were abolished in denuded PCAs from OSA rats (Figure 6A). Thus, the increased sensitivity resulting from apneas was abolished in the absence of ROCK1/2 activity. We also examined protein levels of total and phosphorylatedThr696 MYPT1 in cerebral arteries. We found a significant increase in phospho-MYPT1 protein levels in cerebral arteries after OSA (Figure 6B).
Discussion
The goal of this study was to determine whether apneas during the sleep cycle of rats altered constrictor responses to ET-1 in cerebral arteries. Endothelin-1 and its signaling pathways are often upregulated during pathologic states.7, 19, 20, 21, 22 For these studies, we used our recently developed rat model of OSA. 6 We report three major findings from this study. (1) Cerebral arteries from OSA rats are over 20-fold more sensitive to ET-1 when compared with sham rats. (2) The enhanced vasoconstrictor response to ET-1 in OSA rats occurs primarily through enhanced VSM ETBR signaling pathways. (3) The enhanced response to ETBR stimulation involves TRPC and ROCK signaling. It is important to note that these measured changes in ET-1 signaling occurred in OSA rats at a time that preceded any chronic alterations in systemic BP, inflammation, or oxidative state. 6
The first major finding of this study is that OSA increased the sensitivity of PCAs by 23-fold to ET-1 applied to the abluminal surface. Removal of the endothelium led to an enhanced baseline contractile state of PCAs. Nevertheless, PCAs still showed an enhanced constrictor response in OSA rats at higher ET-1 concentrations. In contrast, ET-1 applied through the lumen (preferential exposure to the endothelium) of sham and OSA PCAs exhibited similar ET-1 sensitivity (Figure 2).
The increased contractility of PCAs in the OSA group was primarily dependent on potentiated ETBR signaling in the VSM. This conclusion is based on studies involving both an ETBR agonist and an ETBR antagonist. First, the ETBR antagonist, BQ-788, abolished the difference in constrictions to ET-1 in PCAs from the OSA and sham groups (Figure 4A). Second, the constrictions after administration of the ETBR agonist, IRL-1620, were enhanced in PCAs from OSA rats compared with those from sham rats (Figure 3). Since, this enhanced response to IRL-1620 persisted with or without the presence of endothelium (Figure 3), the location of the ETBR, which was responsible for the enhanced constrictions, must have been the VSM. Interestingly, we did not observe any differences in mRNA levels of
In addition to our OSA model, there is precedence for an enhanced constrictor response to ET-1 in cerebral arteries through upregulation of ETBR on VSM after ischemic stroke and subarachnoid hemorrhage in rats.23, 24 Contrary to our studies with OSA, ETBR expression was upregulated through an MAPK-dependent mechanism.25, 26 Increased ETBR-mediated constrictions have also been shown in rat parenchymal arterioles after cerebral ischemia and reperfusion. 27 Additionally, upregulation of cerebral and peripheral vessel VSM ETBR has been observed in numerous disease models.22, 23, 28, 29, 30, 31, 32
Endothelin-1 signaling is also enhanced after CIH; however, the effects appear to primarily involve ETAR rather than ETBR. Chronic intermittent hypoxia is another model commonly used to evaluate the hypoxic component of sleep apnea. In one study, the enhanced contractile response to ET-1 in mesenteric arteries after 4 weeks of CIH was attributed to increased ETAR, but not ETBR, expression.
33
In another study, Capone
Although the mechanism by which OSA potentiates the ETBR pathway is not yet fully resolved, Figure 7 summarizes our data suggesting a critical role of TRPCs (Figure 5) and ROCK (Figure 6). Transient receptor potential channels are nonselective cation channels that contribute to increased Ca2+ influx and membrane depolarization in VSM. Increased activity of TRPCs after ETBR activation could thus promote an enhanced contractile effect.10, 11, 12 In a complementary manner, ROCK could contribute to increased contractility through its well-documented capacity to increase sensitivity of the VSM contractile machinery. 16 It should be noted that ETBR has been shown to signal through Gq11,12 and 13, which are involved in the activation of both TRPCs and ROCK.7, 34, 35 Given that inhibition of either TRPCs or ROCK alone fully restored vascular contractility to sham levels, we propose that these mechanisms may act in series. Although further investigation is required, ROCK has been shown to activate TRPCs.36, 37 While we have only examined the effects of OSA on ET-1-mediated vasoconstriction, it is likely that other vasoconstriction pathways involving TRPCs and ROCK may show similar increases in sensitivity.
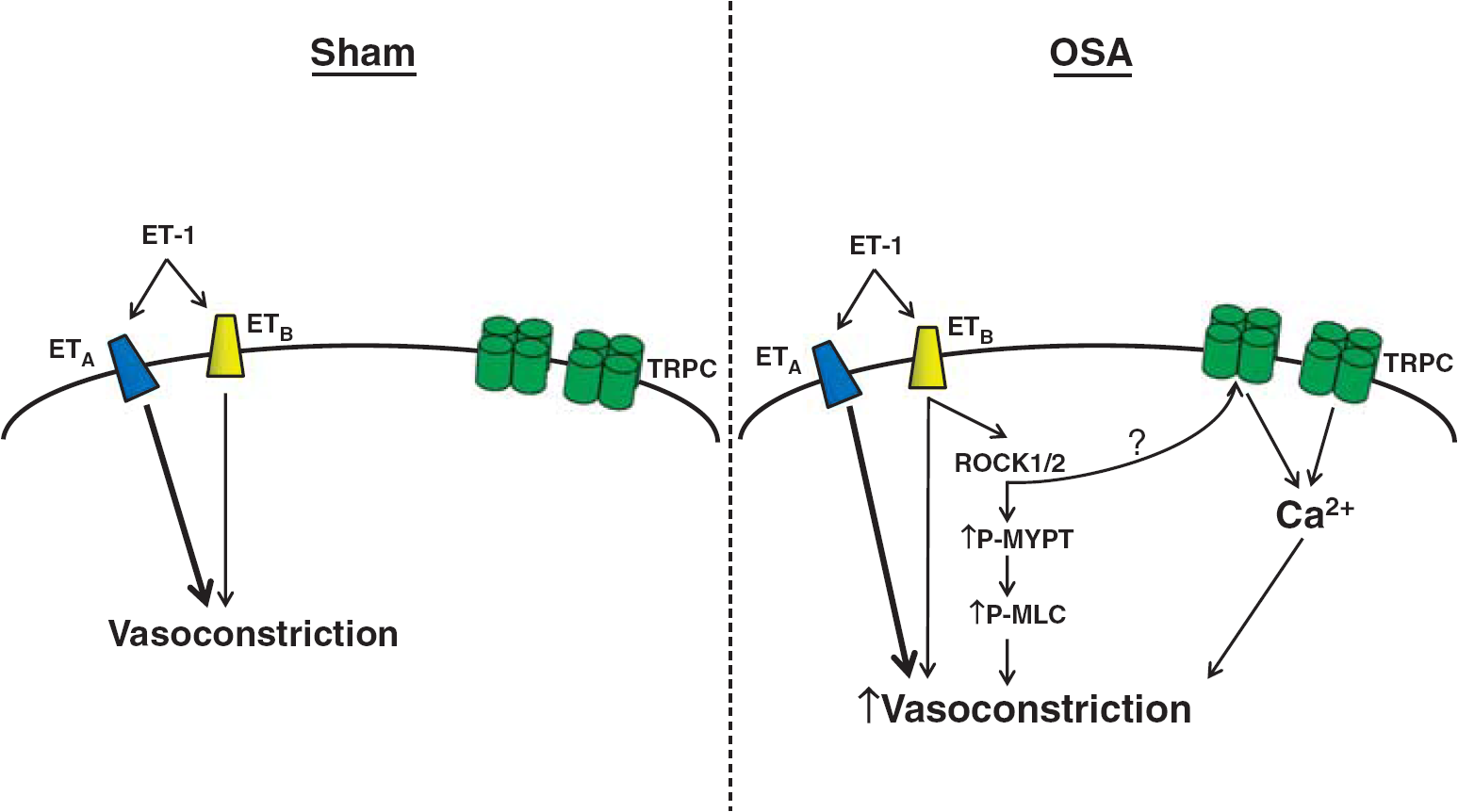
Diagram of potential mechanisms contributing to increased vascular smooth muscle (VSM) ETB receptor (ETBR)-mediated vasoconstriction after obstructive sleep apnea (OSA). ET-1-mediated constriction of sham cerebral arteries primarily involves activation of ETAR. After OSA, ET-1 sensitivity is significantly increased and dependent on VSM ETBR. In our proposed model, increased ETBR-mediated constrictions involve activation of transient receptor potential channels (TRPCs) and Rho kinase (ROCK), leading to increased VSM Ca2+ influx and sensitivity. The ? represents the potential for ROCK activation of TRPCs. ET-1, endothelin-1.
Our data indicate that SKF96365, a blocker of multiple isoforms of TRPC, had no effect on IRL-1620 responsiveness in sham vessels, but completely abolished the increased constrictions in PCAs from OSA rats (Figure 5A), providing functional evidence for TRPCs as a potential mechanism of increased vascular contractility. Since TRPCs are permeable to both Ca2+ and Na+, not only does Ca2+ move into the VSM with activation, but the Na+ influx also depolarizes the VSM to further increase Ca2+ through activation of voltage-operated calcium channels. 38 Expression of TRPCs was either unchanged (TRPC3 and TRPC6) or decreased (TRPC1) in PCAs from OSA rats (Figures 5B to 5D). At first glance, these data appear to be at odds with the idea that TPRCs are involved with the enhanced constrictor effect to ETBR stimulation in PCAs from OSA rats. However, the observation that ETBR-mediated constrictions in sham vessels were the same in the absence or presence of SKF96365 suggests that the role of TRPCs is negligible under normal conditions (Figure 7). However, the ability of SKF96365 to completely block the increased ETBR-mediated constrictions of OSA PCAs suggests that OSA results in VSM TRPC activation, which has a potential role in the ETBR-mediated constrictions (Figure 7).
In addition to Ca2+ influx, increased constriction after OSA could be explained by increased ROCK activity and increased VSM Ca2+ sensitivity. Activation of ROCK phosphorylates and inactivates the myosin binding subunit of myosin phosphatase (MYPT1), suppresses myosin phosphatase mediated dephosphorylation of myosin light chain, and enhances vasoconstriction. 16 After pretreatment of vessels with the ROCK inhibitor, Y27632, the increased constriction of OSA PCAs was abolished (Figure 6A). Furthermore, the ratio of phospho-MYPT1 to total MYPT1 was significantly increased in OSA PCAs (Figure 6B). These data suggest that increased cerebrovascular ET-1-mediated constrictions after OSA involves increased ROCK-mediated Ca2+ sensitization (Figure 7) and are consistent with the previous studies showing that activation of G12/13 protein by ETBR enhances Ca2+ sensitivity through ROCK. 7
In conclusion, we show that OSA significantly alters the constrictor response of cerebral arteries to ET-1. We observe increased ET-1 sensitivity in the absence of elevated BP or upregulation of key components of the ET-1/ET-1 receptor system. The elevated ET-1 sensitivity exhibited by PCAs after OSA is ETBR dependent, and accordingly abrogated by an ETBR antagonist. Figure 7 illustrates how ETBR-mediated hypercontractility of OSA PCAs involves TRPCs and increased Ca2+ sensitivity through activation of ROCK. Taken together, these findings illustrate OSA induced cerebrovascular dysfunction, which may contribute to the increased incidence of adverse cerebrovascular outcomes in OSA patients, and suggest that ETBR antagonists may offer a therapeutic advantage.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.