
Abstract
The role of tumor necrosis factor-alpha (TNF-α) in brain injury is controversial. We studied the effect of anti–TNF-α antibody in a rat model of reversible middle cerebral artery occlusion. During focal ischemia and early reperfusion, TNF-α was rapidly and transiently released into circulation. Pretreatment with intravenous anti–TNF-α antibody reduced cortical (71%, P < 0.015) and subcortical (58%, P < 0.007) injury, enhanced the cerebral blood flow during reperfusion, and improved the neurologic outcome. This further supports the contention that TNF-α is a deleterious cytokine in stroke, whereas circulating antibody against TNF-α may protect brain from reperfusion injury.
The role of tumor necrosis factor-alpha (TNF-α) in brain injury is controversial. It has been shown that TNF-α under in vitro conditions may protect neurons against metabolic–excitotoxic insults and cytotoxic effects of amyloid-β peptide and iron by promoting maintenance of intracellular calcium homeostasis and by suppressing the accumulation of reactive oxygen species (Cheng et al., 1994; Barger et al., 1995). The “neuroprotective” theory has been supported by findings that damage to neurons caused by focal cerebral ischemia and epileptic seizures was exacerbated in TNFR-KO mice, which are genetically deficient in TNF receptors (Bruce et al., 1996). An increase in reactive oxygen species in response to excitotoxic stimuli and reduced levels of manganese superoxide dismutase in TNFR-KO mice led to the concept that TNF-α may protect neurons by stimulating antioxidant pathways (Bruce et al., 1996).
On the other hand, a vast body of evidence indicates that TNF-α is a major deleterious cytokine in stroke and traumatic brain injury. It is known that TNF-α is expressed in ischemic brain (Liu et al., 1994), and immunocytochemical studies confirm the presence of TNF-α antigen in brain as early as 30 minutes after occlusion of the middle cerebral artery (MCA) in the same regions where TNF-α mRNA-positive cells were detected (Buttini et al., 1996). Early increases in TNF-α levels after transient cerebral ischemia in gerbil brain also have been demonstrated (Saito et al., 1996). It has been reported that intracerebroventricular administration of TNF-α increases the volume of brain injury after MCA occlusion in a dose-dependent fashion (Barone et al., 1997). Furthermore, topical application of the type I soluble TNF-α (TNFbp) receptor (Nawashiro et al., 1997) or anti–TNF-α antibody (Ab) (Meistrell et al., 1997) resulted in amelioration of brain infarction in a model of permanent MCA occlusion, whereas intravenous pretreatment with TNFbp reduced brain injury after permanent MCA occlusion in spontaneously hypertensive rats (Dawson et al., 1996). In a closed head injury, systemic administration of agents that block TNF-α synthesis (e.g., pentoxifylline) or TNFbp was associated with cerebral protection (Shohami et al., 1996). Studies in transgenic mice demonstrate that overexpression of TNF-α causes abnormalities in CNS function, and the direct action of TNF-α in the pathogenesis of the disease was confirmed by peripheral administration of TNF-α Ab (Probert et al., 1995).
To further clarify whether TNF-α is a cerebroprotective or deleterious cytokine in brain, a rat model of reperfusion brain injury after MCA occlusion was used (Kittaka et al., 1996; Wang et al., 1997). This reperfusion model is characterized by significant formation of reactive oxygen species in the brain (Chan, 1996; Siesjo, 1992). The TNF-α plasma activity and therapeutic effects of systemic administration of anti–TNF-α Ab were studied in relation to brain injury and CBF changes.
MATERIALS AND METHODS
The studies were performed using Sprague-Dawley rats weighing 260 to 300 g obtained from Charles River Breeders (Hollister, CA, U.S.A.). The animals were housed under diurnal light conditions with unlimited access to food and water. All procedures were done in accordance with the Animal Care Guidelines at the University of Southern California and approved by the National Institutes of Health.
Plasma levels of tumor necrosis factor–alpha
Plasma TNF-α levels were quantitated using the commercially available rat specific enzyme-linked immunoassay kit (Genzyme Cambridge, MA, U.S.A.) in a group of seven animals subjected to 1-hour MCA occlusion. Blood samples (300 μL) were drawn from an indwelling PE-50 catheter placed in the right femoral vein at 30 and 60 minutes after initiation of MCA occlusion, then 30, 60, and 120 minutes and 24 hours after the initiation of reperfusion. The whole blood was centrifuged at 10,000 rpm for 10 minutes at 4°C, and plasma removed and stored at −20°C until thawed for TNF-α level determination.
Focal ischemia studies
Rats were deprived of food 12 hours before surgery. Reversible MCA occlusion was performed using an intraluminal thread technique (Zea Longa et al., 1989) as modified in our laboratory (Kittaka, et al., 1996) in control rats (n = 9) and rats treated with anti–TNF-α Ab (n = 6). The procedure involved initial anesthesia with metofane and maintenance with 50 mg/kg of intraperitoneal pentobarbital. Atropine methyl nitrate (0.18 mg/kg intraperitoneally) was given as premedication to prevent airway obstruction by mucus formation. The animals were allowed to breath spontaneously. Rectal temperature was maintained at 37°C by a thermostatically regulated heating pad. A PE-50 catheter was introduced into the right femoral artery for continuous monitoring of the MABP, as well as for repeated sampling of blood for serial measurements of Pa
Briefly, the surgical technique was as follows: under the operating microscope, the right carotid complex was exposed through a ventral midline incision, and the external carotid artery and its branches ligated. Temporary ligatures were placed around the common carotid artery and internal carotid artery to prevent bleeding. Through a transverse incision in the artery, a 3-0 monofilament nylon suture with rounded tip was introduced into the external carotid artery lumen, and gently advanced 17.5 mm into the internal carotid artery to occlude the MCA at its origin from the Circle of Willis. After 1 hour of occlusion, the thread was pulled out, the external carotid artery permanently tied at the level of bifurcation, and the common carotid artery and internal carotid artery sutures removed to allow reperfusion.
In experimental animals, the effects of polyclonal rabbit anti-mouse TNF-α neutralizing Ab (0.36 mg/kg) (Genzyme, Cambridge, MA, U.S.A.) dissolved in nonpyrogenic sterile saline were studied in rats after intravenous injection through a previously placed indwelling femoral PE-50 catheter. Antibody was given 1 hour before occlusion of the MCA. Control animals had a similarly placed indwelling catheter and received only vehicle. This anti-mouse TNF-α neutralizing Ab binds well to rat TNF-α (Teti, et al., 1993).
Blood flow and head temperature measurements
Cortical CBF was monitored in a separate study before and during occlusion and for 1 hour of reperfusion by a laser–Doppler flowmetry tissue perfusion monitor (Transonic BLF21, Ithacka, NY, U.S.A.) with the probe (0.8-mm diameter) positioned at 0.1 mm above the dura over the cortical surface. In the hemisphere ipsilateral to the MCA occlusion, coordinates were as follows: point A, 1 mm posterior to the bregma and 5.4 mm lateral to the midline; point B, 1 mm posterior to the bregma and 2.1 mm lateral to the midline; point D, 1 mm anterior to the bregma and 3.4 mm lateral to the midline; and point C in the contralateral hemisphere, 1 mm posterior to the bregma and 5.4 mm from the midline. At each point, a small burr hole was drilled in the skull, and the bone carefully removed to prevent damage to the cortex. Steady-state baseline values were recorded before occlusion, and CBF during occlusion and reperfusion expressed as a percentage of the baseline values.
Head temperature was monitored with a 36-gauge thermocouple temperature probe in the temporalis muscle connected to a digital thermometer/thermoregulator model 9000 (Omega, Stanford, CT, U.S.A.).
Motor neurologic behavioral score
Neurologic examination was performed after 24 hours of reperfusion, just before killing. Neurologic outcome was scored on a six-point scale (Kittaka, et al., 1996): 0, no neurologic deficit (normal); 1 (failure to extend left forepaw fully), mild focal neurologic deficit; 2 (circling to the left), moderate focal neurologic deficit; 3 (falling to the left), severe focal deficit; 4, rats did not walk spontaneously and had a depressed level of consciousness; 5, stroke-related death.
Measurement of volume of injury
The area of injury was delineated by incubation of unfixed 2-mm coronal brain slices in 2% triphenyltetrazolium chloride in 0.173 mol/L sucrose and 50 mmol/L K+ phosphate buffer (pH 7.4) for 20 minutes at 37°C, and then stored in 10% formalin. Serial coronal sections were displayed on a digitizing video screen using an imaging system from Jandel Scientific (San Rafael, CA, U.S.A.), and the areas of nonstaining tissue were determined in each section. The volume of injury was calculated by summing affected areas from each coronal section and multiplying by the thickness of each section. The volumes of the control and lesioned hemispheres were calculated, and the amount of injury was expressed as a percentage of lesioned hemisphere and in absolute terms (cubic millimeters). The total injury volume of gray matter corrected for edema and the edema volume was calculated by subtracting the volume of the normal gray matter in the control hemisphere from the volume of gray matter in the lesioned hemisphere (Swanson et al., 1990). The edema volume was calculated by subtracting the volume of the normal gray matter in the control hemisphere from the volume of gray matter in the lesioned hemisphere (Swanson et al., 1990). Measurements of the volume of injury were done separately for the pallium and striatum. A coronal section from each brain was obtained at the level of the optic chiasm. Representations of the injury areas of individual sections from each animal in control group and the group treated with anti–TNF-α were superimposed to gain an impression of the topography and incidence of infarction. The boundary of infarction was redrawn for each rat on the corresponding coronal section, and the following areas delineated: the infarct area where all rats were affected 100%, the infarct area where 50% or more of rats were affected, and the infarct area where less than 50% of rats were affected.
Statistical analysis
Levels of TNF-α, physiologic variables, and infarction and edema volumes were compared between groups using a Student's t test. A P value of less than 0.05 was considered statistically significant.
RESULTS
Plasma activity of tumor necrosis factor–alpha
The MCA occlusion caused rapid, significant, and transient increase in plasma TNF-α levels in comparison with sham-operated animals (Fig. 1), beginning as early as 30 minutes after the onset of occlusion (71% of rats), with all animals showing increased levels by 1 hour. During reperfusion, high levels of TNF-α were maintained up to 2 hours into reperfusion, then normalized by 24 hours. Peak levels occurred at the onset of reperfusion for 57% of rats and 1 hour into reperfusion for the remaining 43%. The TNF-α levels in the MCA group varied widely between animals (range 78 to 12,878 pg/mL) and are therefore expressed as logarithmic values to demonstrate the consistent trend found, despite individual differences in magnitude. Sham-operated animals demonstrated no TNF-α activity, and no activity was seen before initiation of MCA occlusion. Despite wide variations of TNF-α levels in the MCA group, there was for each studied time point a statistically significant difference in comparison with sham-operated animals (Fig. 1).
Plasma tumor necrosis factor–alpha (TNF-α) levels in rats subjected to middle cerebral artery (MCA) occlusion and reperfusion (•, solid line) are compared with sham-operated rats (▪, interrupted line) by Student's t test; *P < 0.01. In the MCA group, TNF-α levels varied widely between animals and are therefore expressed as logarithmic values to demonstrate the consistent trend found, despite individual differences in magnitude. Time period (I) denotes zero level of TNF-α before occlusion before and after neck dissection, and before filament insertion.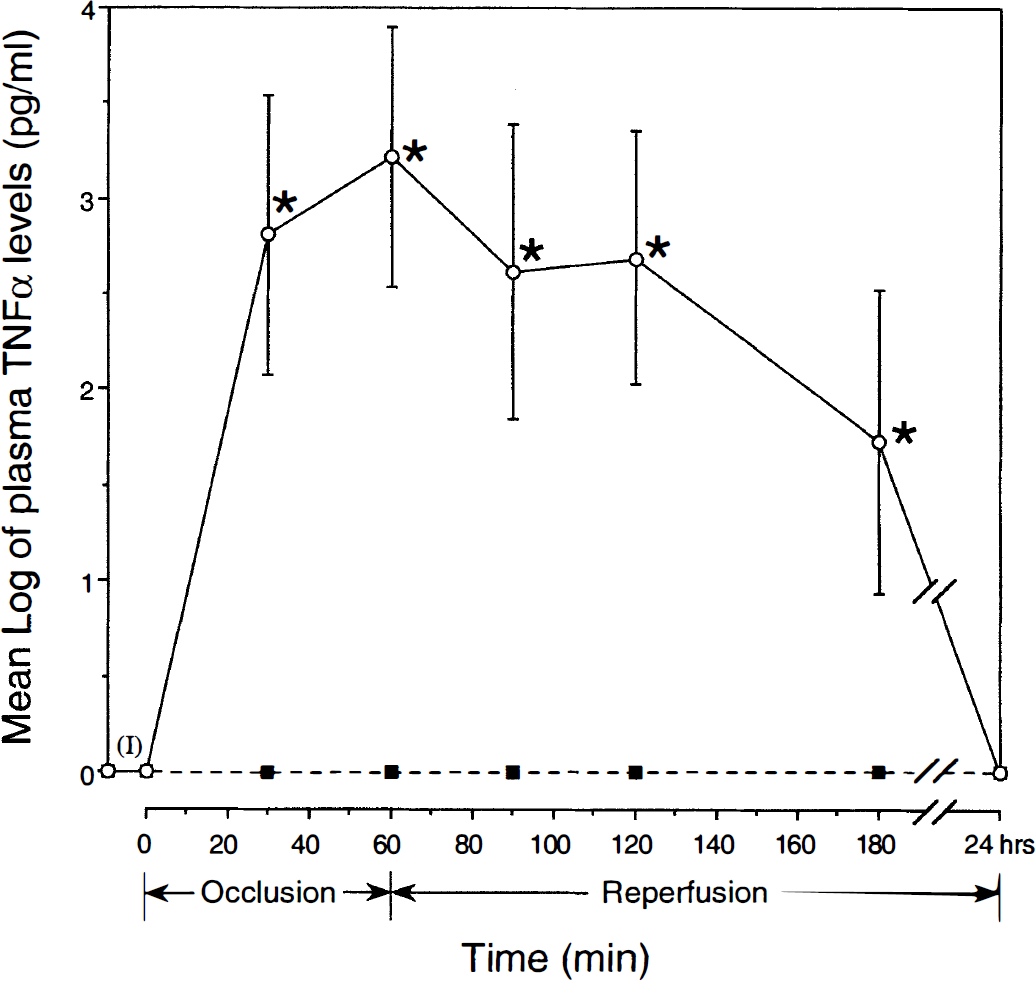
Physiologic variables
Physiologic variables
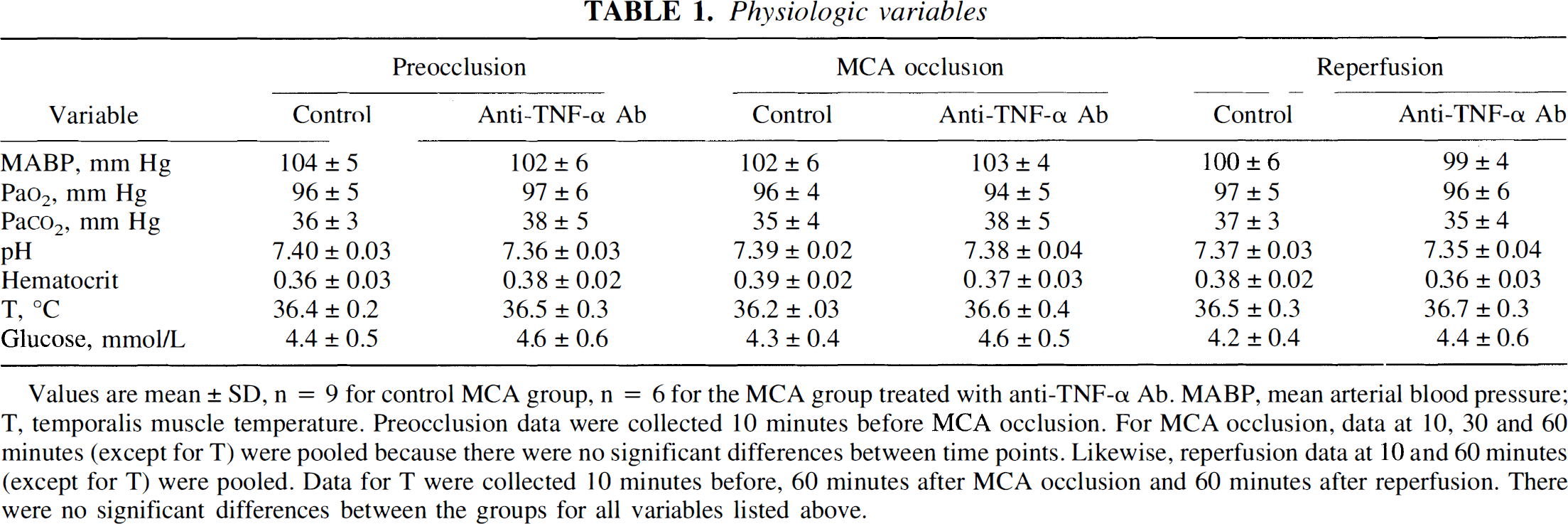
Values are mean ± SD, n = 9 for control MCA group, n = 6 for the MCA group treated with anti-TNF-α Ab. MABP, mean arterial blood pressure; T, temporalis muscle temperature. Preocclusion data were collected 10 minutes before MCA occlusion. For MCA occlusion, data at 10, 30 and 60 minutes (except for T) were pooled because there were no significant differences between time points. Likewise, reperfusion data at 10 and 60 minutes (except for T) were pooled. Data for T were collected 10 minutes before, 60 minutes after MCA occlusion and 60 minutes after reperfusion. There were no significant differences between the groups for all variables listed above.
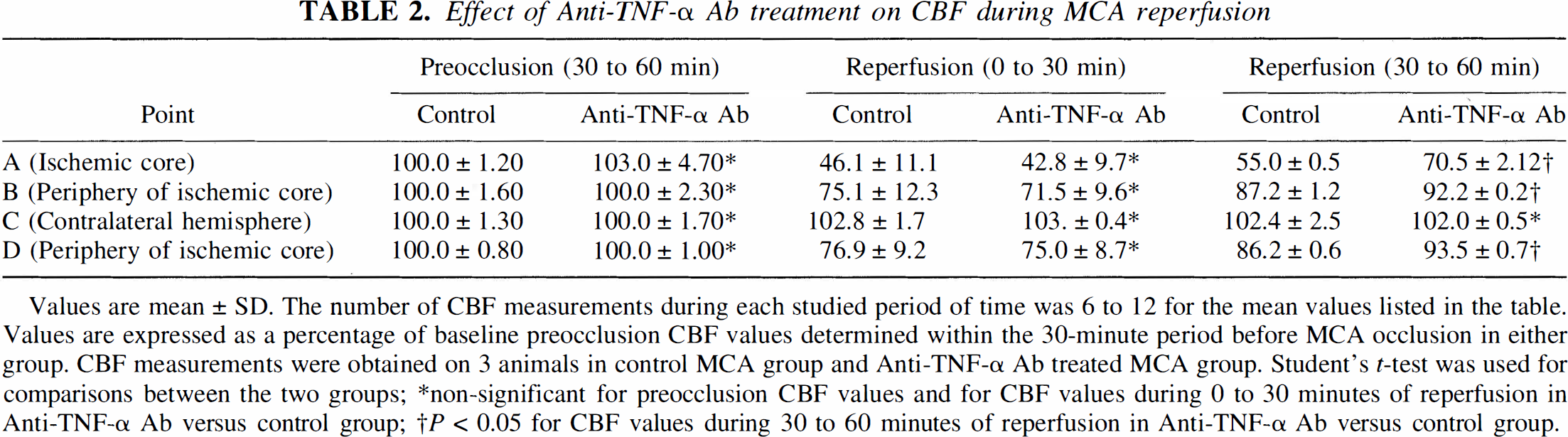
No significant differences in baseline tissue perfusion units were observed between control rats and those treated with anti–TNF-α Ab, indicating similar preocclusion CBF values. Baseline CBF readings also were taken before TNF Ab administration, and there was no significant difference with preocclusion values, indicating that the Ab did not influence the CBF under basal conditions. During MCA occlusion, the reductions in CBF in the ischemic core (point A) were comparable among the groups: 15% to 16% (P < 0.001, control MCA group) and 12% to 14% (P < 0.001, MCA group treated with anti–TNF-α Ab) of baseline values (Fig. 2). In the periphery of the ischemic core (points B and D), the respective reductions in CBF were 35% to 37% (P < 0.001, control MCA group) and 32% to 35% (P < 0.001, MCA group treated with anti–TNF-α Ab) of baseline values. During the first 30 minutes of reperfusion, the trend toward CBF restoration was comparable between the two groups: in the core, the average values were 46% versus 43% in control and MCA group treated with anti–TNF-α Ab, whereas in the periphery of the ischemic core the values ranged from 75% (point B) to 77% (point D) in the control group versus 72% (point B) to 75% (point D) in the MCA group treated with anti–TNF-α Ab (Fig. 1; Table 2). However, during 30 to 60 minutes of reperfusion, the recovery of CBF was significantly improved in the MCA group treated with anti–TNF-α Ab; in the core, the average values were 71% (MCA group treated with anti–TNF-α Ab) versus 55% (control group) (Fig. 1; Table 2), representing a 30% increase in flow. Relatively modest but significant effects of Ab on CBF during reperfusion were observed in both studied points in the periphery of ischemic core. The lack of more pronounced effect of Ab in these regions could be explained by significant spontaneous recovery of CBF even without Ab intervention (Fig. 2; Table 2).
Changes in cerebral blood flow during MCA occlusion and reperfusion in control rats Effect of Anti-TNF-α Ab treatment on CBF during MCA reperfusion Values are mean ± SD. The number of CBF measurements during each studied period of time was 6 to 12 for the mean values listed in the table. Values are expressed as a percentage of baseline preocclusion CBF values determined within the 30-minute period before MCA occlusion in either group. CBF measurements were obtained on 3 animals in control MCA group and Anti-TNF-α Ab treated MCA group. Student's t-test was used for comparisons between the two groups non-significant for preocclusion CBF values and for CBF values during 0 to 30 minutes of reperfusion in Anti-TNF-α Ab versus control group P < 0.05 for CBF values during 30 to 60 minutes of reperfusion in Anti-TNF-α Ab versus control group.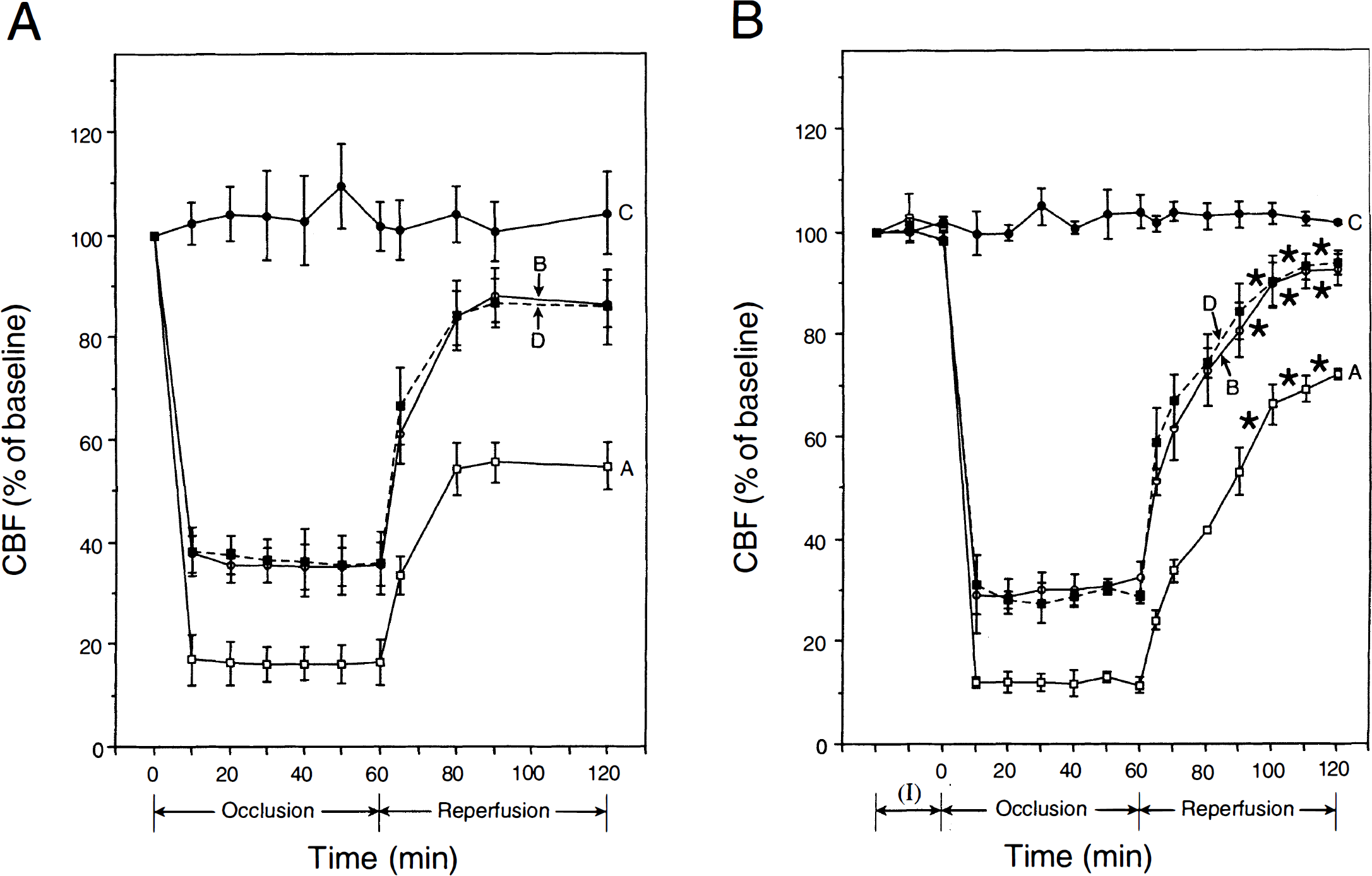
Motor neurologic behavioral score
After 24 hours of reperfusion, the control group had a mean neurologic score of 0.56 ± 0.17, which was reduced by nearly twofold (0.33 ± 0.21) by treatment with anti–TNF-α Ab. The Kruskal-Wallis test, however, did not reach the level of significance. Possible reason for this is that neither of the animals in either group had developed a score greater than 1. Thus, with this method, the scores were limited to only a narrow range of either 0 or 1 (and no values in between), which significantly lowered the sensitivity of the neurologic scale for statistical comparisons and precluded discrete neurologic changes between the groups to be detected.
Neuropathology
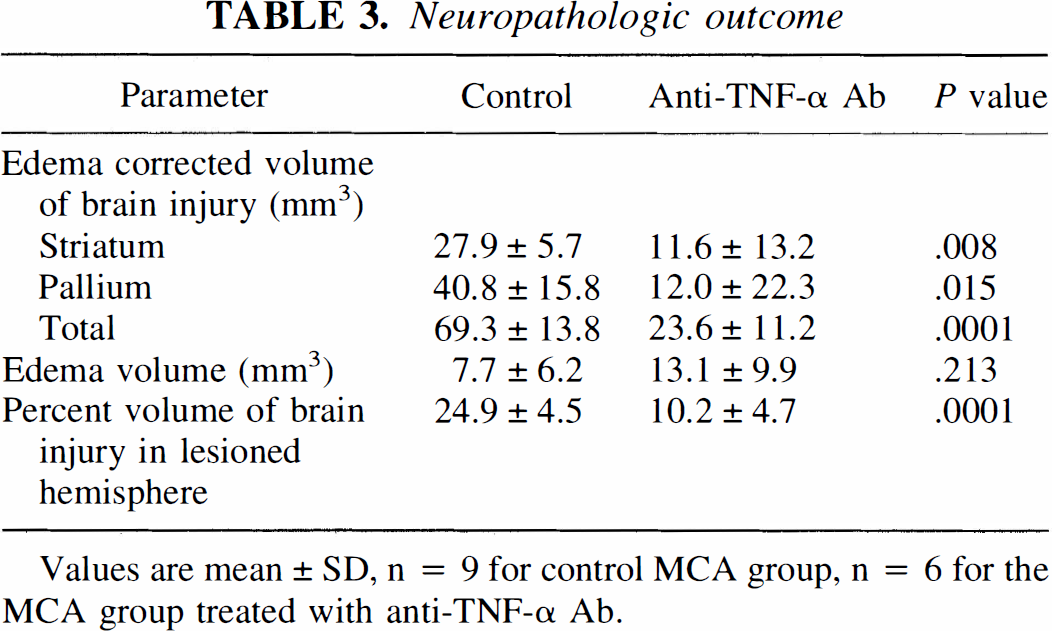
Rats treated with anti–TNF-α Ab had a significant reduction in the volume of brain injury compared with control rats of 71% (P < 0.015) in the cortical region, and 58% (P < 0.007) in the subcortical region (Table 3). Edema volume was not significantly different between the groups. The total volume of injury of gray matter (corrected for edema) was significantly decreased by 66% in the Ab-treated group relative to control rats (P < 0.001). The percent volume of the lesioned hemisphere involved in the Ab-treated group was 59% smaller than the control group (P < 0.0001). Total area of brain injury determined at the level of the optic chiasm (anterior coronal block) and expressed as a percentage of the coronal sectional area is depicted in Fig. 3. Figure 3 illustrates that 100% of control and Ab-treated rats had injury in the lateral striatum, 50% or more exhibited changes in the medial striatum and lateral cortex, and less than 50% showed changes in the dorsolateral cortex with a significant reduction of area in each region for the Ab-treated animals.
Incidence and topography of the infarction at the level of the optic chiasm during reversible MCA occlusion in control rats Neuropathologic outcome Values are mean ± SD, n = 9 for control MCA group, n = 6 for the MCA group treated with anti-TNF-α Ab.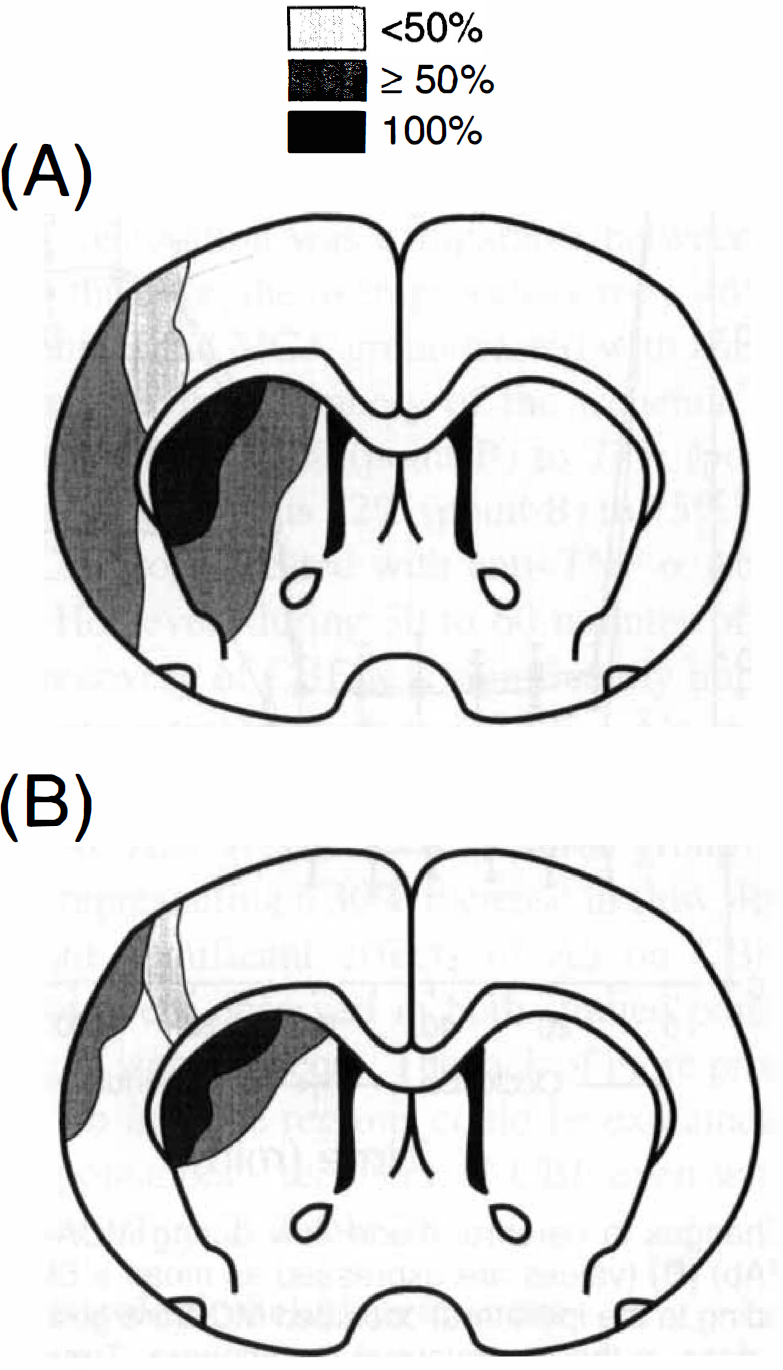
DISCUSSION
The current study demonstrates that TNF-α is rapidly and transiently released into circulation during focal ischemia and early reperfusion, and that circulating anti–TNF-α Ab protects the brain from reperfusion injury. Rapid TNF-α synthesis (within 30 minutes) has been shown in brain after permanent MCA occlusion in normal and spontaneously hypertensive rats (Liu et al., 1994; Buttini et al., 1996) and in gerbils after transient global cerebral ischemia (Saito et al., 1996). Increased plasma TNF-α activity has been shown 4 hours after permanent MCA occlusion in spontaneously hypertensive rats (Dawson et al., 1996). It has been suggested that TNF-α could exacerbate CNS injury by several mechanisms (Feurstein et al., 1994). Our study supports the contention that TNF-α is a deleterious cytokine in stroke and plays a major role in the pathophysiologic mechanism of cerebral ischemia.
Recent studies demonstrated that inhibition of TNF-α by topical, intraperitoneal, or intravenous administration of TNFbp resulted in 10% to 26% reduction of total brain injury after permanent MCA occlusion in mice (Nawashiro et al., 1997), whereas better results were obtained in spontaneously hypertensive rats (Dawson et al., 1996). The degree of neuroprotection with circulating anti–TNF-α Ab in the current model was 58% for the striatum, 71% in the cortex, and 66% for the total volume of injury in gray matter, which was much higher than the protection seen in the studies with TNFbp. The reduction in brain injury that we have shown also is more substantial than that seen after other neuroprotective agents including the noncompetitive N-methyl-
Other studies have demonstrated that systemic administration of anti–TNF-α Ab resulted in significant neuroprotection in transgenic mice overexpressing TNF-α (Probert et al., 1995), whereas a combined systemic and intraventricular administration of anti–TNF-α prevented albumin leakage into the CSF in a meningitis model in rats (Kim et al., 1992). An international, multicenter, placebo-controlled trial has demonstrated that intravenous administration of monoclonal anti–TNF-α Ab is a safe and feasible therapeutic modality in the treatment of patients with sepsis (Cohen and Carlet, 1996). Thus, it is possible that the inhibition of TNF-α by intravenous anti–TNF-α Ab also may be effective in the treatment of stroke patients by ameliorating the consequences of several pathophysiologic processes triggered by TNF-α (Feurstein et al., 1994).
It is not known why in the current study the anti–TNF-α Ab treatment completely spared the ventral portion of the cortex. In the current MCA model (Kittaka et al., 1996; Wang et al., 1997; Kittaka et al., 1997), however, the infarct was located in all animals in the lateral striatum, and in more than 50% of animals in the lateral cortex, whereas only in less than 50% of animals the infarction extended to the ventromedial and dorsolateral cortex. The spatial localization of infarct in the animals treated with anti–TNF-α Ab possibly reflects regional-dependent neuroprotection that could be related to differential regional levels of TNF-α production by brain in response to focal ischemic insult and reperfusion. This hypothesis currently is under study in our laboratory.
Associated with the decreased volume of brain injury after TNF-α inhibition in the current study was a significant increase in the restoration of CBF during reperfusion and in particular within the ischemic core. It has been shown that TNF-α affects the release of the vasoactive agents endothelin-1 and nitric oxide from cerebral endothelial and smooth muscle cells (Estrada et al., 1995) and causes dose-dependent pial vasoconstriction and blood–brain barrier breakdown (Megyeri et al., 1992). This cytokine promotes thrombus formation and procoagulant transformation of the vascular endothelium (Nawroth and Stern, 1986; Pober, 1988), and promotes leukocyte recruitment, infiltration, and adhesion to cerebral capillaries, resulting in increased accumulation and thrombosis (Feurstein et al., 1994). Direct cytotoxic effects on glial and cerebral endothelium and decreased CBF after administration of TNF-α also have been shown (Feurstein et al., 1994; Tureen, 1995; Estrada et al., 1995). Thus, although TNF-α may not be directly neurotoxic in vitro (Barger at al., 1995; Barone et al., 1997), the inhibition of TNF-α by intravenous anti–TNF-α Ab may ameliorate the consequences of several pathophysiologic vascular mechanisms active in cerebral ischemia. Further study of the specific effects of TNF-α are likely to elucidate the precise mechanisms involved.
Finally, it is intriguing whether anti–TNF-α Ab or TNFbp could be used successfully in post-stroke or post–head trauma treatments to block effects of TNF-α. Although Current and previous studies have clearly established TNF-α as a major deleterious cytokine in stroke, they still have not given enough focus to possible therapeutic advantages of TNF-α blockade. One study suggests that administering TNFbp within 30 minutes before and after MCA occlusion was similarly effective in reducing focal ischemic injury, but in most studies TNFbp was given either immediately after closed head injury (Shohami et al., 1996) or MCA occlusion (Nawashiro et al., 1997), or TNFbp and TNF-α Ab were administered using protocols that involves repetitive intracerebroventricular injections before and after MCA occlusion (Barone et al., 1997). Future studies should evaluate dose-related effects of TNF-α blockade at different time intervals after an ischemic insult or head injury to determine the potential of this approach for post-stroke and post–head trauma treatments.