
Abstract
The temporal evolution of cerebral infarction was examined in rats subjected to transient occlusion of both common carotid arteries and the right middle cerebral artery. After severe (90-min) ischemia, substantial right-sided cortical infarction was evident within 6 h and fully developed after 1 day. After mild (30-min) ischemia, no cortical infarction was present after 1 day. However, infarction developed after 3 days; by 2 weeks, infarction volume was as large as that induced by 90-min ischemia. These data suggest that infarction after mild focal ischemia can develop in a surprisingly delayed fashion. Some evidence of neuronal apoptosis was present after severe ischemia, but only to a limited degree. However, 3 days after mild ischemia, neurons bordering the maturing infarction exhibited prominent TUNEL staining, and DNA prepared from the periinfarct area of ischemic cortex showed internucleosomal fragmentation. Furthermore, pretreatment with 1 mg/kg cycloheximide markedly reduced infarction volume 2 weeks after mild ischemia. These data raise the possibility that apoptosis, dependent on active protein synthesis, contributes to the delayed infarction observed in rats subjected to mild transient focal cerebral ischemia.
Although initial evidence of neuronal degeneration after transient global cerebral ischemia in rodents can appear in a delayed fashion up to 2–3 days following the ischemic insult (Kirino, 1982; Pulsinelli et al., 1982), morphological evidence of cell damage in animal models of focal ischemia is thought to present more rapidly (Garcia and Kamijyo, 1974; Little et al., 1974; Garcia et al., 1993). After a 2-h transient focal ischemic insult in rats, the boundaries of the ischemic lesion were visible by 24 h, with development of infarction by 46 h (Chen et al., 1993; Zhang et al., 1994). We have reported that lesion volume, measured by triphenyl tetrazolium chloride (TTC) staining or histology after hematoxylin–eosin staining 6 h after 90-min transient occlusion of the middle cerebral artery (MCA) in our rat model of focal ischemia, is comparable to that measured 1 week later (Lin et al., 1993). Even in permanent ischemia models, in which ischemia duration increases with (is equal to) survival time, volume of the ischemic lesion as defined after 6–12 h of ischemia is maximal, with no further increase during the next 162 h (Garcia and Kamijyo, 1974; Garcia et al., 1993; Zhang et al., 1994). Kirino et al. (1988) looked specifically for evidence of “slow neuronal damage” at intervals up to 6 months after permanent occlusion of the middle cerebral artery in rats, and concluded that evolution of injury was complete by 12–24 h. Fujie et al. (1990) did observe late progressive shrinkage of the thalamus during 3 months after permanent middle cerebral occlusion in rats, but noted that this shrinkage was also triggered by cortical aspiration and hence likely a late secondary change (perhaps reflecting retrograde axonal degeneration and consequent loss of trophic support) rather than delayed evolution of ischemic death. Thus, in experimental studies of focal brain ischemia it has been common practice to assess lesion extent 2–24 h after ischemia onset (Tamura et al., 1981; Kaplan et al., 1991; Xue et al., 1992; He et al., 1993; Maier et al., 1993; Karibe et al., 1994).
However, previous examinations of lesion evolution after focal ischemic insults have used relatively severe insults, either relatively prolonged durations of transient ischemia, or permanent ischemia. The original goal of the present study was to determine whether a 24–48-h endpoint was indeed adequate to assess the extent of brain lesion after mild transient focal ischemia.
MATERIALS AND METHODS
Stroke model
Long-Evans male rats (body weight: 300–350 g) (Charles Rivers, Wilmington, DE, U.S.A.) were used in this study. Housing and anesthesia concurred with guidelines established by the institutional Animal Studies Committee, and were in accordance with the PHS Guide for the Care and Use of Laboratory Animals, USDA Regulations, and the AVMA Panel on Euthanasia guidelines. Rats were allowed free access to water and rat chow (Wayne, Chicago, IL, U.S.A.) until surgery. Methods for focal cerebral ischemia-reperfusion have been detailed in previous publications (He et al., 1993; Lin et al., 1993).
Briefly, animals were anesthetized with isoflurane (induction, 2.5%; maintenance, 1.5% in a mixture of 30% O2 and 70% N2 at a flow rate of 650 ml/min) through a face mask. The right femoral artery was cannulated for monitoring arterial blood pressure and heart rate and for obtaining blood samples for arterial blood gas. Arterial blood gases were determined before, during, and after ischemia and remained stable at pH 7.4 ± 0.1, Pao2 > 80 mm Hg, and Paco2 37 ± 7 mm Hg (measured with a blood gas analyzer, model 238, Ciba Corning, Medfield, MA, U.S.A.). Physiological parameters including arterial blood pressure and pulse rate were monitored using Digi-Med Blood Pressure Analyzer (Micro-Med, Inc., Louisville, KY, U.S.A.). The rectal temperature was recorded and maintained at 37.0 ± 0.5°C via an electronic temperature controller (Versa-Therm 2156, Cole-Parmer, Chicago, IL, U.S.A.) linked to a heating lamp (Yip et al., 1991) and homeothermic blanket control unit (Harvard Apparatus, South Natick, MA, U.S.A.). The right MCA was exposed: a 2-cm vertical skin incision midway between the right eye and ear and after splitting the temporalis muscle; a 2-mm burr hole was drilled at the junction of the zygomatic arch and the squamous bone (Chen et al., 1986). The right MCA was ligated with a 10–0 suture under an operating microscope. Complete interruption of blood flow at the MCA occlusion site was confirmed under the microscope. Both common carotid arteries (CC As) that had been previously isolated and freed of soft tissues and nerves were then occluded using nontraumatic aneurysm clips. Temporary occlusion of the right MCA and both CCAs resulted in focal ischemia (reduction of blood flow by 88–92%) (An et al., 1993) confined to the cerebral cortex in the right MCA territory. At the end of the ischemic period (30 or 90 min), the right MCA ligature and both CCA clips were released. Restoration of blood flow in the previously occluded arteries after release of the arterial ligature and aneurysm clips was confirmed directly under the operating microscope. Free access to food and water was allowed after recovery from anesthesia. All of the rats were kept in air-ventilated cages with temperature maintained at 24 ± 0.5°C until the end of the experiment. In the group of rats treated with cycloheximide, the drug was dissolved in normal saline and given i.p. 15 min before onset of ischemia.
Morphometric analysis of infarct volume
Infarct volumes were measured by morphometric analysis of infarct areas that were defined by TTC (Sigma, St. Louis, MO, U.S.A.). The “indirect” morphometric analysis of infarct volume has been detailed elsewhere (Swanson et al., 1990; Lin et al., 1993). Animals were killed with an overdose of pentobarbital (100 mg/kg) followed by intracardiac perfusion with 200 ml of 0.9% NaCl 1, 3, or 14 days after 30 or 90 min of ischemia. Brains were cooled in ice-cold saline for 5 min, then sliced into 2-mm coronal sections using a rat brain matrice (Harvard Bioscience, South Natick, MA, U.S.A.). The brain slices were incubated in phosphate-buffered saline (pH 7.4) containing 2% TTC at 37°C for 20 min and then stored in 10% neutral-buffered formalin. A cross-sectional area of the TTC-unstained region for each brain slice was determined using an image analyzer (DUMAS, Drexel University, Philadelphia, PA, U.S.A.), and the indirect method proposed by Swanson et al. (1990) (subtraction of residual right hemisphere cortical volume from the cortical volume of the intact left hemisphere). Hemisphere cortical volume was calculated by summation of infarct volumes measured in component brain slices. Infarct volumes defined by TTC stain in our model correlate well with those defined by histological examination after hematoxylin and eosin staining (Lin et al., 1993).
TUNEL staining
Under anesthesia, animals underwent perfusion fixation with 200 ml of normal saline followed by 10% formalin at 24 and 72 h after 30-min ischemia. Paraffin-embedded brain sections (7 μm) were deparaffinized in two changes of xylene for 5 min each, then washed sequentially in 100, 95, and 70% ethanol. Nuclei of tissue sections were stripped of proteins by incubation with 20 μg/ml proteinase K (Sigma, St. Louis, MO, U.S.A.) for 5 min. Slices were washed in distilled water. We adapted a modified TdT-mediated dUTP-biotin nick-end labeling (TUNEL) method (Gavrieli et al., 1992) by using an ApopTag in situ apoptosis detection kit (Oncor, Gaithers-burg, MD, U.S.A.). The brain slices were immersed in the equilibration buffer for 10 min. The terminal deoxynucleotidyl transferase (TdT) and dUTP-digoxigenin were added to the sections and the slices were incubated in a humidified chamber at 37°C for 1 h. The reaction was stopped by incubating slices with the stop/wash buffer at 37°C for 30 min. After washing in phosphate-buffered saline, the sections were incubated with the antidigoxigenin–peroxidase solution for 30 min. The slices were colorized with DAB/H2O2 solution (0.05% diaminobenzidine tetrachloride and 0.02% H2O2 in 50 mM Tris-HCl buffer), and then counterstained with methyl green. For controls, brain slices were treated similarly but in the absence of TdT enzyme, digoxigenin-dUTP, or anti-digoxigenin antibody, respectively.
Analysis of DNA fragmentation
DNA was harvested from the periinfarct area and the uninjured contralateral cortex of the rat brain, 72 h after mild ischemia, or 24–48 h after severe ischemia. The Puregene DNA isolation kit (Gentra Systems, Minneapolis, MN, U.S.A.) was used to extract the DNA and purify the samples. Briefly, fresh cortical tissue was homogenized and treated with proteinase K (200 μ/ml). Protein was precipitated with M NaCl (final concentration), and DNA was subsequently precipitated with isopropanol, washed with ethanol, and rehydrated in Tris-EDTA. DNA (5 n-g/lane) was electrophoresed in the presence of RNAse A on a 1.5% agarose gel (NuSieve: SeaKem 3:1; FMC Products, Rockland, ME, U.S.A.) containing ethidium bromide (0.3 μ/ml) at 50 V for 5 h. The DNA was visualized with ultraviolet photography.
Statistical analysis
To compare difference among groups, we used one-way analysis of variance followed by a post-hoc Tukey test. Comparison between the two groups (mild versus severe ischemia) was by Student's t test. A p value <0.05 was considered significant.
RESULTS
Arterial blood gases and mean arterial pressure obtained both during and after ischemia were within the normal range in all experimental groups (see Materials and Methods). Focal cerebral ischemia for 90 min followed by reperfusion (severe transient ischemia) resulted in a well-delineated lesion in the cerebral cortex of the right MCA territory as early as 6 h after ischemia. Lesion volume showed little change when measured 1, 3, or 14 days later (Figs. 1 and 2).
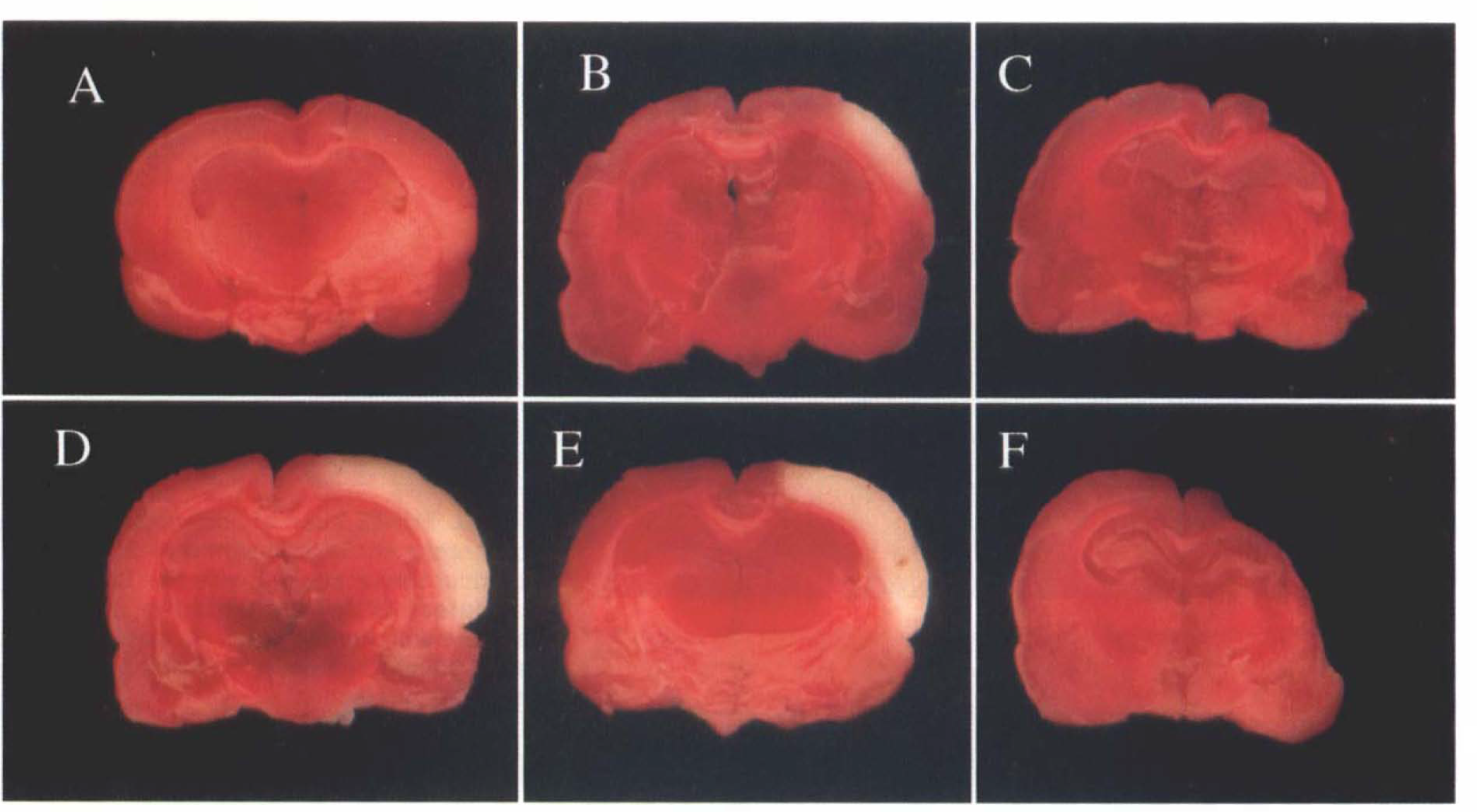
Evolution of infarction after mild and severe focal cerebral ischemia.
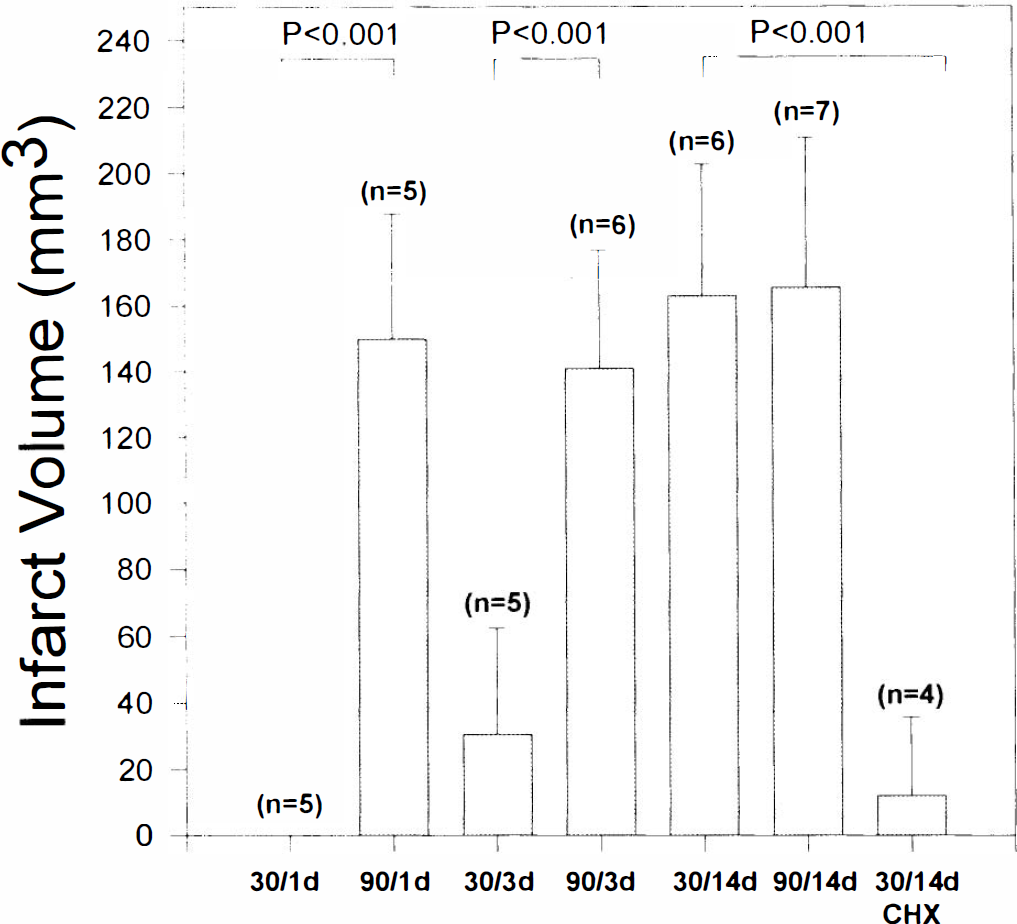
Quantification of infarct evolution. TTC-defined infarct volume after focal cerebral ischemia. Bars are labeled with ischemia duration (30 or 90 min) and maturation time (1, 3, or 14 days). Number of animals is indicated above each bar. CHX denotes 1 mg/kg cycloheximide pretreatment.
One day after 30 min transient ischemia (mild transient ischemia), there was no detectable lesion by TTC stain (Figs. 1 and 2) or histological examination (Fig. 3A). However, by 3 days a small TTC-unstained infarction developed (Figs. 1 and 2). By 2 weeks later, infarct volume had progressed markedly, such that lesion volume was indistinguishable from that produced by severe transient ischemia (Fig. 2). Control experiments showed no lesion at 2 weeks in the sham-operated group (n = 3, data not shown). Visual inspection of the right MCA trunk and branches 1, 3, or 14 days after the ischemic insult did not suggest any gross occlusion or abnormality.
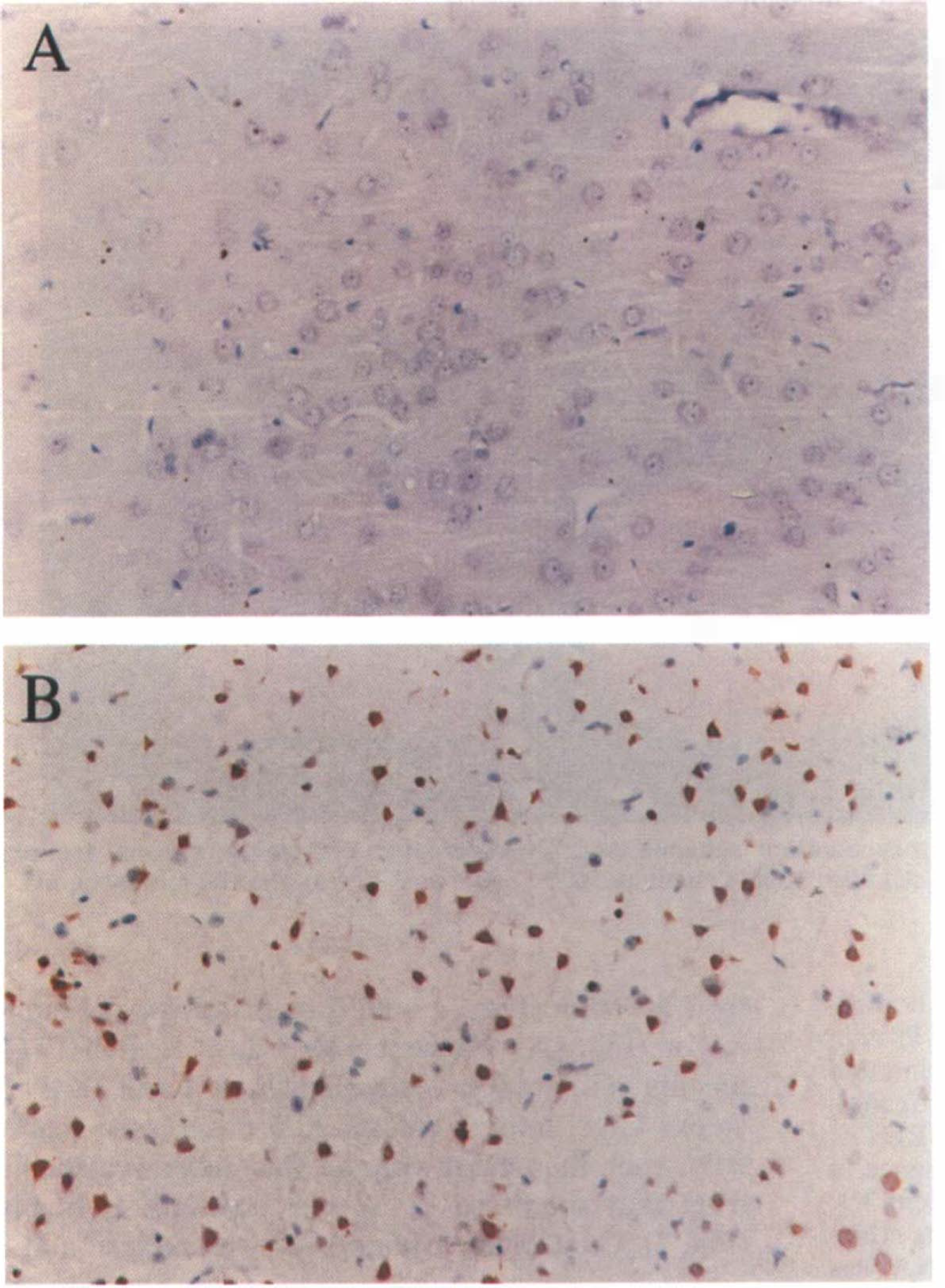
TUNEL stain of cortical tissue. Photomicrographs show representative 20× sections of cortical tissue stained with the TUNEL method to detect DNA fragmentation, and counterstained with methyl green.
Growing evidence has suggested that apoptosis may contribute to the delayed loss of CA1 hippocampal neurons after global ischemic insults, and to cortical neuronal death after focal ischemic insults (see Discussion section). To examine the possibility that apoptosis might be a factor in the unexpected very delayed lesion progression noted above, three additional studies were performed.
First, modified TUNEL staining (Gavrieli et al., 1992) was performed to look for evidence of nuclear DNA breakdown, a common feature of apoptosis (Bursch et al., 1990; Arends et al., 1990; McConkey, 1991; Compton, 1992; Gavrieli et al., 1992; Wijsman et al., 1993). The cortex of sham-operated animals showed no TUNEL staining (data not shown). One day after mild transient ischemia, TUNEL staining was likewise absent in the affected right cerebral cortex (Fig. 3). However, by 3 days substantial cortical neuronal TUNEL staining had appeared, localized to the injured hemisphere (approximately in the core plus penumbral area; Fig. 3). Some of the TUNEL-positive neurons showed typical ischemic changes whereas others appeared grossly intact.
Second, DNA was extracted from the approximate penumbral region area and examined for degradation on an agarose gel. Three days after mild transient ischemia, this DNA showed evidence of internucleosomal “ladder” breakdown, whereas no breakdown was seen in DNA similarly extracted from sham-operated animals (Fig. 4). DNA extracted from the penumbral region 1 or 2 days after severe transient ischemia showed nonspecific DNA breakdown without laddering (Fig. 4).

DNA fragmentation in the periinfarct area after severe and mild ischemia. Control (C) and postischemia (24 and 48 h after severe ischemia and 72 h after mild ischemia) DNA electrophoresis reveals the appearance of nucleosome-size bands in the 72-h sample, indicating the presence of fragmented DNA in the periinfarct area after mild ischemia.
Third, the protein synthesis inhibitor, cycloheximide, was examined for neuroprotective effects. Cycloheximide administered at 1 mg/kg i.p. 15 min before ischemia markedly reduced the progressive appearance of infarction after mild transient ischemia, as assessed 14 days later (Fig. 2).
DISCUSSION
The major finding of the present study is that brain infarction after mild transient focal ischemia can develop in a surprisingly delayed fashion, >3 days after the original insult. In the present model, the magnitude of this delayed infarction component was considerable, indeed much larger than the small infarct originally apparent at the 3-day mark. Present observations thus may have important implications for choosing appropriate endpoints in experimental studies of mild transient ischemia. The widely applied convention of determining brain injury or infarct volume 2–24 h postinsult would have grossly underestimated damage in the current paradigm.
Delayed loss of vulnerable hippocampal CA1 neurons is known to occur after transient global ischemia (Kirino, 1982; Pulsinelli et al., 1982). Despite recovery of blood flow, energy metabolism, and electrical activity, CA1 neurons, which initially appear to have survived the insult, go on to die days later; in the setting of hypothermic neuroprotection, this death can occur more than a week postinsult (Dietrich et al., 1991). The present study suggests that the principle of delayed brain cell death may occur broadly in the settings of both global and focal ischemia.
Further study in different animal models of focal ischemia is warranted to elucidate the extent to which delayed infarction occurs in other settings. Does it occur in humans? Our first reaction to this question was negative, since there is little evidence of delayed deterioration in neurological function after mild cortical stroke in humans, except for that caused by peak edema formation. However, on reflection, this clinical experience may not represent countervening evidence. The possibility cannot be currently excluded that the neurological deficit after mild ischemia is maximal—corresponding to initial infarction as well as brain tissue at risk for subsequent very delayed infarction. Serial brain imaging studies could help address the question, but to our knowledge available studies do not constitute a compelling rebuttal of the hypothesis that very delayed infarction can occur in humans.
Further study will also be needed to define the mechanisms responsible for delayed infarction. Visual examination of the right MCA arterial tree at various times after the ischemic insult did not suggest the appearance of late vascular pathology; indeed, the observed protective effect of cycloheximide itself argues against the possibility that the observed progression of infarct volume was due to late progressive arterial vasculopathy. However, quantitative measurement of regional blood flow over time will be needed to exclude absolutely such a vascular cause.
We present here initial support for the possibility that much of the observed late cell death may be due to apoptosis. Two major forms of cell death, necrosis and apoptosis, have been distinguished morphologically (Kerr et al., 1972; Wyllie et al., 1980; Gerschenson, 1992), although absolute criteria for distinction have not been delineated (Farber, 1994). Necrosis is characterized by prominent acute cell body swelling with subsequent cell lysis. Apoptosis is characterized by compaction of the cell body and internucleosomal DNA cleavage; in many cases, new protein synthesis appears to be required (Johnson and Deckwerth, 1993). We did not verify here that our cycloheximide regimen indeed inhibited brain protein synthesis, but a smaller dose (0.6 mg/kg) s.c. did inhibit forebrain protein synthesis in 7-day-old rat pups (Pavlik and Teisinger, 1980).
Although classically hypoxic–ischemic neuronal death has been considered to occur by necrosis, recently clues have emerged suggesting that apoptosis may also contribute to neuronal loss after both global and focal ischemia. Protein synthesis inhibitors can reduce the delayed death of hippocampal CA1 neurons after global ischemia in vivo (Goto et al., 1990; Shigeno et al., 1990), and the loss of CA1 field excitatory synaptic potentials in the hippocampal slice after an anoxic insult (Papas et al., 1992). Furthermore, other studies of global ischemia in vivo have shown that hippocampal CA1 is the site of internucleosomal DNA fragmentation (Heron et al., 1993; Macmanus et al., 1993; Okamoto et al., 1993; Roberts-Lewis et al., 1993; Kihara et al., 1994; Nitatori et al., 1995). Findings of cycloheximide protection (Linnik et al., 1993) and internucleosomal DNA cleavage (Linnik et al., 1993; Tominaga et al., 1993; Macmanus et al., 1994; Charriaut-Marlangue et al., 1995; Li et al., 1995) have also arisen in recent studies of focal brain ischemia. In our cortical cell cultures, oxygen-glucose deprivation normally induces excitotoxic necrosis; however, if this acute excitotoxic death is blocked by NMDA and AMPA/kainate antagonists, then an apoptosis component is unmasked (Gwag et al., 1995).
A key difference between delayed neuronal death after transient global ischemia, and the delayed evolution of infarction described here, is the widespread involvement of nonneuronal cell types in the latter setting. The protective effect of cycloheximide against the extent of focal ischemic infarction observed by Linnik et al. (1993) previously and here suggests that apoptosis may also play an important role in the delayed death of these nonneuronal brain cells.
Regardless of underlying mechanism, one practical implication of our study is to suggest additional caution in the selection of a temporal endpoint for measurement of focal ischemic injury volume in animal models, especially after mild insults. Moreover, present results support the intriguing possibility that surprisingly delayed therapeutic interventions—perhaps specifically directed against apoptosis death—may prove to be valuable in reducing the brain tissue loss ultimately resulting from mild focal ischemic insults.