
Abstract
Inflammation is a hallmark of stroke pathology. The cytokines, tumor necrosis factor (TNF), interleukin (IL)-1, and IL-6, modulate tissue injury in experimental stroke and are therefore potential targets in future stroke therapy. The effect of these cytokines on infarct evolution depends on their availability in the ischemic penumbra in the early phase after stroke onset, corresponding to the therapeutic window (<4.5 hours), which is similar in human and experimental stroke. This review summarizes a large body of literature on the spatiotemporal and cellular production of TNF, IL-1, and IL-6, focusing on the early phase in experimental and human stroke. We also review studies of cytokines in blood and cerebrospinal fluid in stroke. Tumor necrosis factor and IL-1 are upregulated early in peri-infarct microglia. Newer literature suggests that IL-6 is produced by microglia, in addition to neurons. Tumor necrosis factor- and IL-1-producing macrophages infiltrate the infarct and peri-infarct with a delay. This information is discussed in the context of suggestions that neuronal sensitivity to ischemia may be modulated by cytokines. The fact that TNF and IL-1, and suppossedly also IL-6, are produced by microglia within the therapeutic window place these cells centrally in potential future stroke therapy.
Keywords
Introduction
Tumor necrosis factor (TNF), interleukin (IL)-1, and IL-6 are potent, inflammatory cytokines, and potential targets in future stroke therapy. All three cytokines are able to modulate the size of the ischemic damage in experimental stroke (focal cerebral ischemia) in rodents, and their levels increase in cerebrospinal fluid (CSF) and blood after ischemic stroke in humans (Allan et al, 2005; Simi et al, 2007; McCoy and Tansey, 2008; Whiteley et al, 2009). There is evidence that TNF and IL-1 in the stroke-lesioned rodent brain are produced by resident microglia, intrathecal macrophages, and infiltrating, monocyte-derived macrophages (Buttini et al, 1994; Davies et al, 1999; Lambertsen et al, 2005; Clausen et al, 2008), whereas IL-6 in addition is produced by neurons (Suzuki et al, 1999, 2009). Although TNF, IL-1, and IL-6 are also among the best investigated cytokines in CSF and blood in human stroke patients, we still know very little about their availability and mechanism of action in human brain in the early phase, up till 4 to 6 hours, after induction of experimental stroke, corresponding to the therapeutic window in human stroke (Ginsberg, 2008). Before considering if and how to target these cytokines in patients with stroke, a minimal requirement therefore is to know the availability, including the cellular production, of these cytokines in the ischemic territory in the early phase after stroke onset in rodents and ideally also in the human brain.
The finding that TNF, IL-1, and IL-6 increase many-fold (up to 40- to 60-fold) in the brain within the first 24 hours after experimental stroke (Hill et al, 1999; Lambertsen et al, 2005; Clausen et al, 2005, 2008) has contributed to the view that their effects on infarct evolution critically depend on a pronounced, many-fold increase in cytokine levels in the ischemic territory. However, as will be discussed in this review, the time profile for the evolution of the infarct does not match the increases in cytokine messenger RNA (mRNA) and protein productions, if these many-fold increases in cytokine levels should determine the evolution of the infarct. In the case of permanent (ischemic) strokes, the infarct has accordingly almost attained its final volume several hours before the cytokine production reaches its maximum. However, TNF, IL-1, and IL-6 are extremely potent molecules and along with their pro-forms they are present in low pg/mg protein levels also in normal brain parenchyma in both humans and rodents (Breder et al, 1993; Gahring et al, 1996; Hillhouse et al, 1998; Gregersen et al, 2000; Vitkovic et al, 2000; Lambertsen et al, 2002, 2005; Clausen et al, 2005). Further, evidence is accumulating that glutamate, which is released into the synaptic cleft in the first minutes after stroke onset (Dirnagl et al, 1999), can increase the sensitivity of primarily neurons to cytokines (Cardenas et al, 2002; Pradillo et al, 2005; Gardoni et al, 2011). Knowing the cellular production of TNF, IL-1, and IL-6 in normal brain, and knowing the time profile and the cellular sources of these cytokines within, and the possibilities of their transport into the ischemic territory, in the early phase after stroke onset is crucial to understand the mechanisms by which these cytokines interact with the potentially salvageable neurons in the ischemic ‘penumbra’. The purpose of this review is to clarify the association between the in situ production and total availability of TNF, IL-1, and IL-6 and the evolution of the infarct in experimental stroke in the rodent, and to clarify the association between TNF, IL-1, and IL-6 in CSF and blood, and stroke severity, infarct size, and clinical outcome in human stroke. Based on the evidence that TNF, IL-1, and in part also IL-6 are microglial derived, at the time of and in the early phase after stroke, we finally discuss the role of microglial-derived cytokines in determining the fate of the potentially salvageable neurons in the ischemic ‘penumbra’ and point to future research questions/hypotheses.
Infarct Evolution and Therapeutic Window in Experimental and Human Stroke
The ‘penumbra’ is originally defined as the tissue immediately surrounding the infarct core in which there is reduced blood flow, impaired neuronal functionality, but preserved structural integrity, but which can be recruited into the developing infarct (Astrup et al, 1981), if therapeutic strategies that counteract, inhibit or block deleterius events fail (Ginsberg, 2008). According to the original definition, which is based on studies in the rat, the penumbra is a highly dynamic region, which temporarily may expand, but nevertheless regresses in conjunction with the maturation of the infarct (Figures 1A and 1B). As the penumbra can, in principle, only be visualized by in vivo imaging techniques, although its exact identification in brain sections using histologic techniques are impossible, we will in this review only use the term ‘penumbra’, when a ‘context-related’ definition of the penumbra is provided. Otherwise, we will use the broader term ‘peri-infarct’, which in addition to the penumbra comprises the tissue further away from the developing infarct (Sommer, 2010), tissue which is affected by the stroke, but unlikely to die and to be recruited into the infarct. The ‘ischemic territory’ consists of both the infarct and peri-infarct.
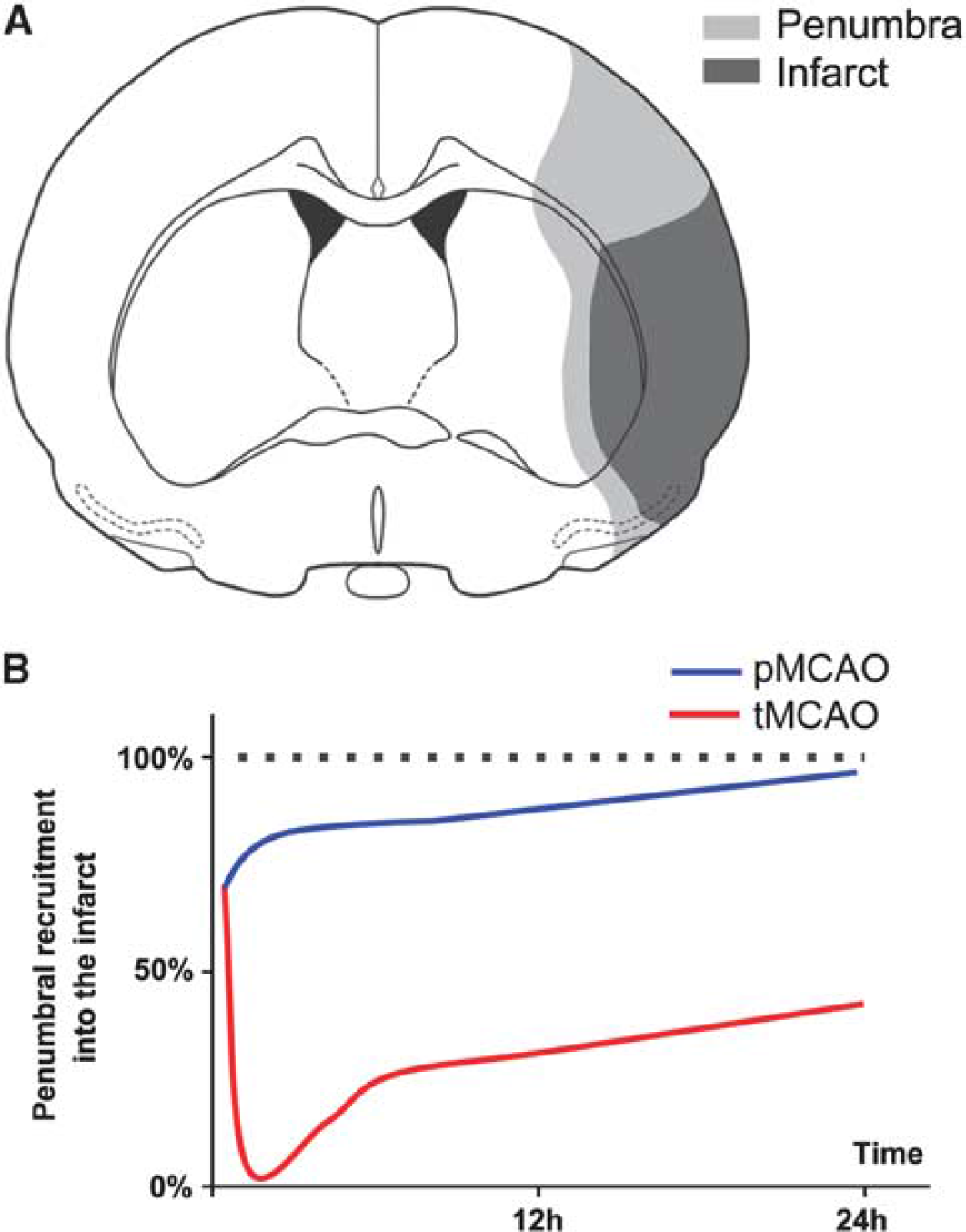
Dynamics of the recruitment of the penumbra into the infarct in experimental stroke. (
Infarct Evolution in Experimental Stroke
The most commonly used stroke models are the models of permanent and transient middle cerebral artery occlusion (MCAO) in the rodent, which mirror thrombo-embolic occlusion of the middle cerebral artery in humans. Transient MCAO (tMCAO) mirrors occlusion of the middle cerebral artery followed by reperfusion, which can be either spontaneous or assisted, as when reperfusion is achieved after successful recanalization therapy in patients. Both stroke models are highly invasive, and may or may not involve craniectomy, which should be taken into consideration when evaluating cytokine responses to the stroke. The largest difference between models is, however, the difference between permanent MCAO (pMCAO) and tMCAO (Hossmann, 2012). Although reperfusion attenuates infarction if instituted rapidly after arterial occlusion, it provides fast access for leukocytes into the infarct core and penumbra, which raises questions about the potential additional contribution of leukocyte-derived cytokines in modulating infarct evolution.
Several groups have worked on bridging the gap between the penumbra as originally defined by the use of in vivo imaging techniques and flow studies, and molecular definitions of the penumbra in tissue sections (Hossmann, 1996, 2012; Back et al, 2004; Sharp et al, 2011; Hoehn et al, 2001; Weinstein et al, 2004; Ginsberg, 2008). There is consensus that what corresponds best to the penumbra at the molecular level is (1) areas of suppressed protein synthesis but preserved ATP content, and (2) areas of heat shock protein 70 induction in neurons (Hossmann, 1996; del Zoppo et al, 2011). By recording the area of reduced protein synthesis and the area of reduced ATP levels in parallel sections from the same mice, the Hossmann group, in a series of elegant experiments, showed that the dynamics of the penumbra—defined as the area of suppressed cortical protein synthesis but preserved ATP—was different after pMCAO and tMCAO (Hata et al, 2000a, 2000b; Hossmann, 2012). After pMCAO, this area rapidly regressed from close to 30% of the final infarct volume after 1 hour of ischemia to ∼18% at 6 hours, and to 5% after 24 hours of ischemia, when the edge of the area of reduced protein synthesis and ATP depletion exactly matched the borders of the histologically defined infarct (Figures 1A and 1B; Hata et al, 2000a). In comparison, after 60 minutes of tMCAO, the penumbra initially increased, as a result of the reperfusion, whereafter it was gradually recruited into the infarct, to eventually regress to a few percent of the final infarct volume 3 days after tMCAO (Figure 1B; Hata et al, 2000b).
Taken together, this suggests that the penumbra is smaller and recruited more rapidly into the infarct after pMCAO than after tMCAO (Figure 1B; Hata et al, 2000a, 2000b). This is in line with other studies in rodents, including high-resolution magnetic resonance imaging-based studies, showing that the recruitment of the potentially salvageable penumbral tissue into the infarct happens most rapidly in the early phase after stroke (Hoehn et al, 2001; Weinstein et al, 2004; Liu et al, 2010). In a recent opinion paper (Hossmann, 2012), it was emphasized that pMCAO and tMCAO have different pathophysiologies, where the condition induced by pMCAO reflects the situation in patients with stroke, who do not qualify for recanalization therapy, and who constitute the majority of stroke patients. The tMCAO model allows us to study stroke pathophysiology under the best possible conditions for successful therapeutic intervention, corresponding to patients with stroke that either show spontaneous reperfusion or are successfully treated with recanalization treatment within 1 hour after stroke onset, which is a small minority of patients (Hossmann, 2012).
Interestingly, it has emerged that also the injury within the infarct core develops in a more dynamic way than the imaging studies originally suggested. This has lead to the concept of the infarct core as consisting of ‘multiple mini-cores’ associated with multiple ‘mini-penumbras’, which over time coalesce to form a solid infarct (del Zoppo et al, 2011), a pattern which is also depicted in stainings for neuronal heat shock protein 70 production and microglial activation (Lehrmann et al, 1997; Lambertsen et al, 2005; Hughes et al, 2010).
Infarct Evolution and Therapeutic Window in Human Stroke
As outlined above, the concept of the penumbra being tissue at risk that is potentially salvageable and therefore the target in stroke therapy has been developed in rodents under controlled conditions and invasive methods. The transfer of this concept, however, to the clinical situation with human stroke patients using modern imaging techniques has proven to be extremely challenging (Heiss and Sobesky, 2008). Positron emission tomography using ligands (e.g., 11C-flumazenil) that allow the discrimination between tissue with and without morphologic destruction or measurements of oxygen supply and blood flow are still the golden standards to identify potentially salvageable brain tissue (Heiss and Sobesky, 2008). Positron emission tomography studies have severe limitations due to invasion and radiation exposure for the patients and require such complex logistics that this technique is available only in few specialized centers. The magnetic resonance imaging-based method that determines the ‘mismatch’ between perfusion-weighted and diffusion-weighted images (PWI–DWI mismatch) is the only widely used possibility to determine penumbral tissue in stroke patients (Back et al, 2004; Rivers et al, 2006; Heiss and Sobesky, 2008; Liu et al, 2010). Using this method, it was shown in 14 patients who underwent serial imaging analysis from ∼7 hours to until 42 days after stroke that also in humans the lesion size increases rapidly (<70 hours) to its maximal size (Schwamm et al, 1998). Thus, similar to the situation in mice and rats, the recruitment of penumbral tissue into the infarct is a rapid process also in humans, which thereby underscores the significance of the ‘time is brain’ concept (Ginsberg, 2008; Lo, 2008). At the moment, the only approved pharmacological therapy for ischemic stroke is the recanalization of the occluded artery by intravenous (i.v.) administration of recombinant tissue plasminogen activator, allowing reperfusion and thereby access of blood-borne leukocytes and cytokines into the ischemic territory. Because of the limited time window (<4 to 6 hours after stroke) and several severe risk factors, such as hemorrhage, this therapy is only applied in <10% of all stroke patients (Huang et al, 2006), thereby emphasizing the need for additional neuroprotective strategies. Furthermore, it appears that early inflammatory events may be required for recovery at later time points (Lo, 2008), so inhibiting or blocking cytokine responses might interfere with tissue recovery.
Infarct Modifying Effect of Early Stroke Cytokines
TNF is probably the most extensively studied cytokine in experimental stroke. Tumor necrosis factor can both exacerbate and counteract infarct evolution in rodents (Figure 2 and Table 1A). Tumor necrosis factor exists in a transmembrane and a soluble form, which both signal through surface membrane receptors TNF-RI (TNF-p55R) and TNF-RII (TNF-p75R; Kriegler et al, 1988). In experimental stroke, administration of neutralizing antibodies to TNF or of TNF-binding protein (TNF-bp, soluble (s) TNF-RI, sTNF-p55R) has protective effects (Figure 2 and Table 1A). Although the results of intervention studies thereby indicate a pathologic role of TNF, studies of TNF-deficient mice suggest that TNF is neuroprotective (Bruce et al, 1996; Gary et al, 1998; Taoufik et al, 2007; Lambertsen et al, 2009), an effect which appears to be mediated via TNF-RI (Figure 2 and Table 1A). Importantly, these observations corroborate the idea that the evolution of the infarct and, thus, the survival of ischemic neurons is modulated by the TNF, which is already present in the neural tissue at stroke onset.
Studies on the neuroprotective or neurotoxic effect of TNF in experimental stroke
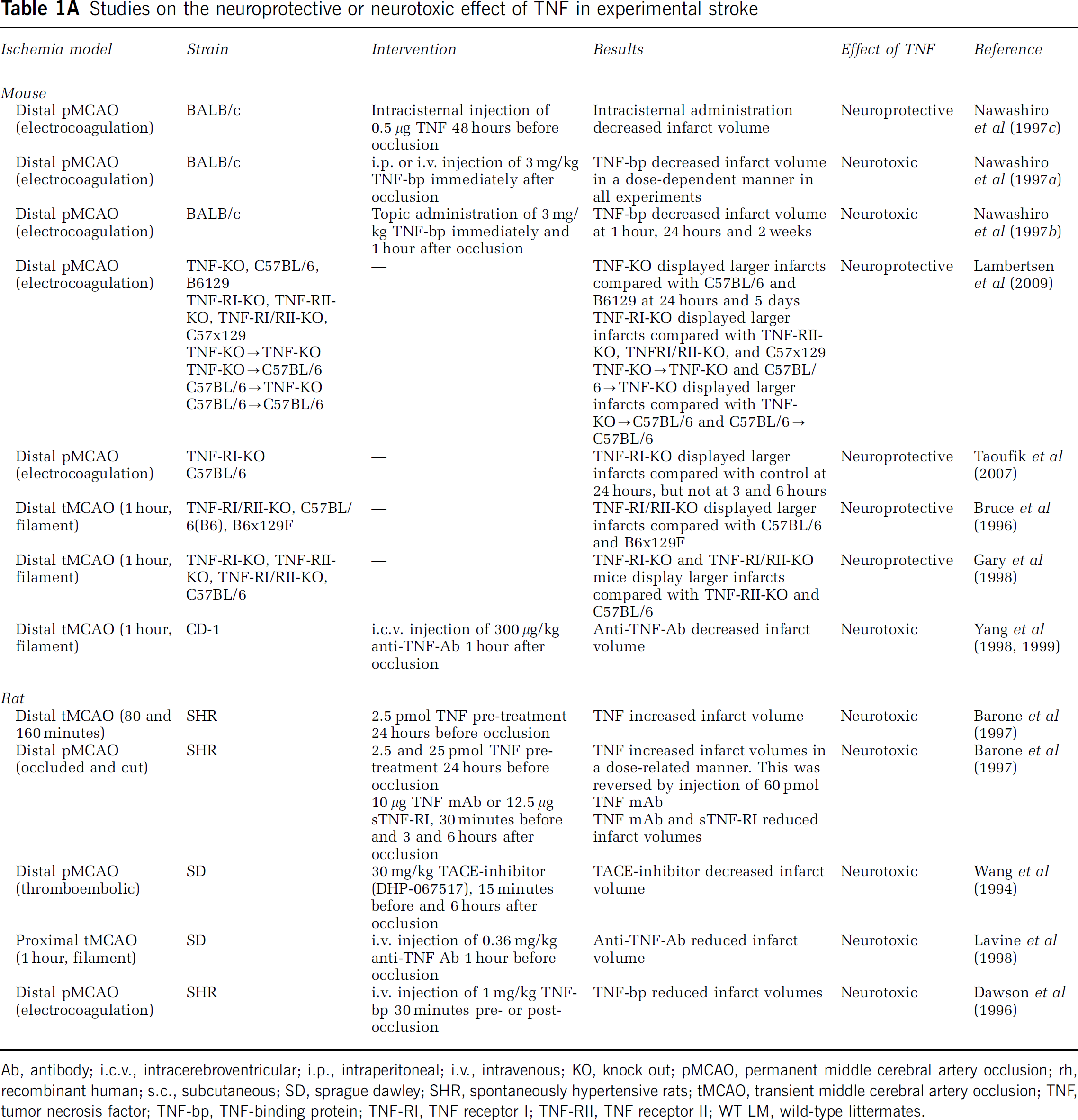
Ab, antibody; i.c.v., intracerebroventricular; i.p., intraperitoneal; i.v., intravenous; KO, knock out; pMCAO, permanent middle cerebral artery occlusion; rh, recombinant human; s.c., subcutaneous; SD, sprague dawley; SHR, spontaneously hypertensive rats; tMCAO, transient middle cerebral artery occlusion; TNF, tumor necrosis factor; TNF-bp, TNF-binding protein; TNF-RI, TNF receptor I; TNF-RII, TNF receptor II; WT LM, wild-type littermates.
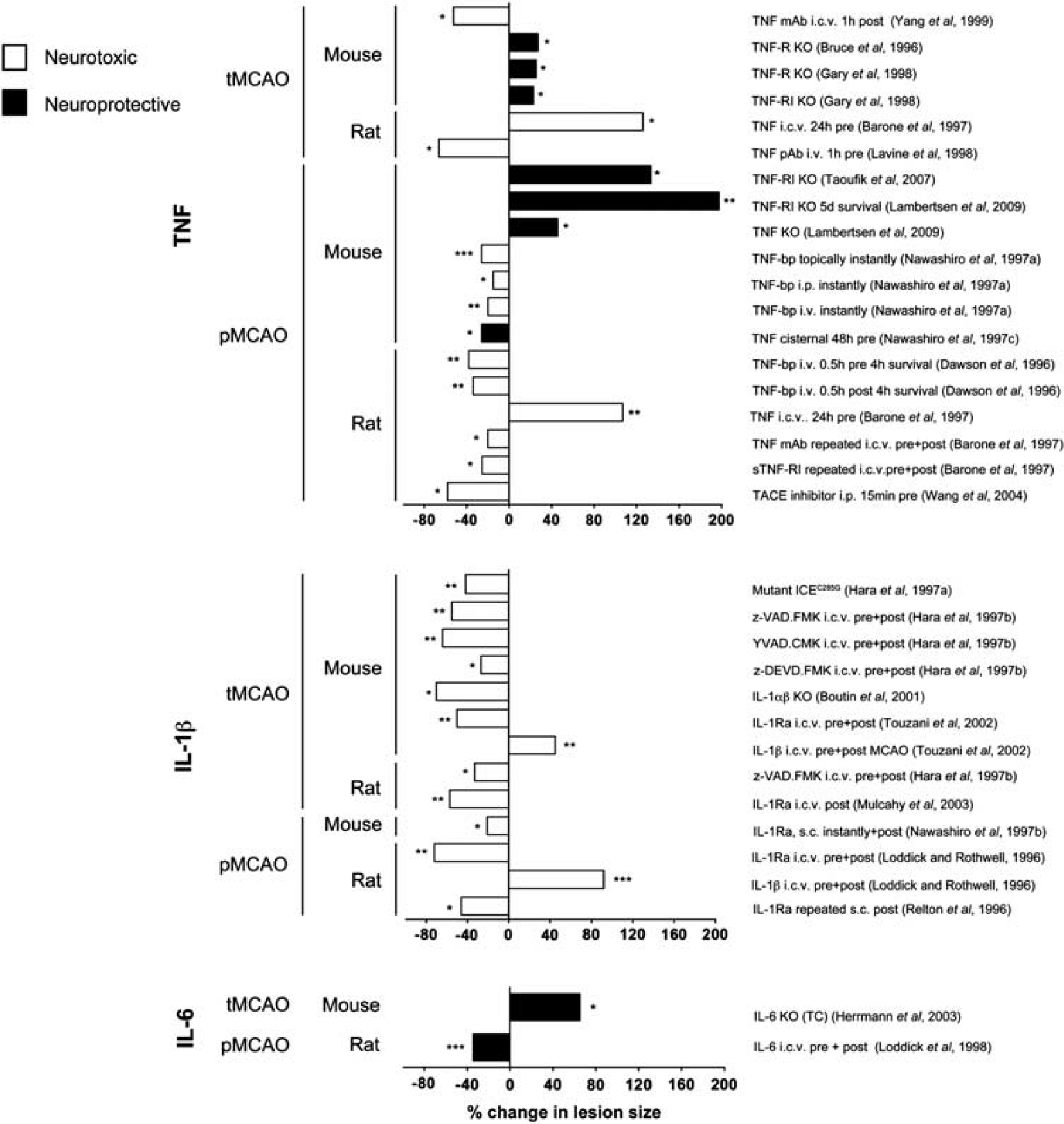
Effect of therapeutic or genetic manipulation of tumor necrosis factor (TNF), interleukin (IL)-1β, or IL-6 bioavailability on infarct size in experimental stroke. Graphic presentation of studies showing a neuroprotective (black) or neurotoxic (white) effect of therapeutic intervention or genetic manipulation of TNF, IL-1β, or IL-6 or their receptors in rodents after transient MCAO (tMCAO) or permanent MCAO (pMCAO). Data are presented as the percent difference in total infarct volume between the experimental and the control group, giving rise to an increase (bars directed to the right) or a decrease (bars directed to the left) in lesion size. Unless stated data were obtained from studies using 24-hour post-surgery survival. Further details on doses and experimental settings are given in Tables 1A, 1B, 1C. bp, binding protein; i.c.v., intracerebroventricular; i.p., intraperitoneal; i.v., intravenous; KO, knock out; mAb, monoclonal antibody; pAb, polyclonal antibody. ∗P<0.05, ∗∗P<0.01, ∗∗∗P<0.001.
Studies on the neuroprotective or neurotoxic effect of IL-1β in experimental stroke
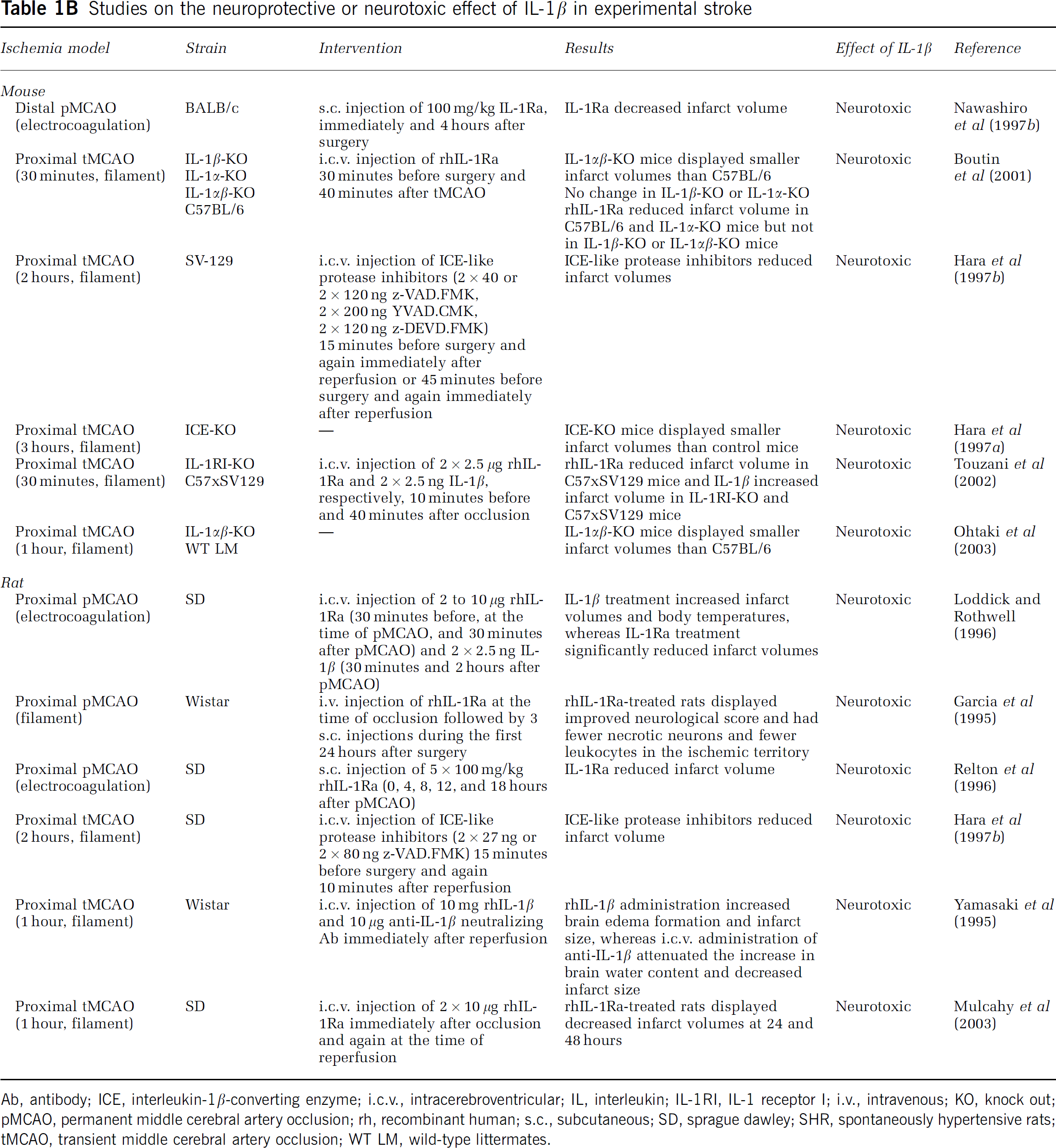
Ab, antibody; ICE, interleukin-1β-converting enzyme; i.c.v., intracerebroventricular; IL, interleukin; IL-1RI, IL-1 receptor I; i.v., intravenous; KO, knock out; pMCAO, permanent middle cerebral artery occlusion; rh, recombinant human; s.c., subcutaneous; SD, sprague dawley; SHR, spontaneously hypertensive rats; tMCAO, transient middle cerebral artery occlusion; WT LM, wild-type littermates.
Studies of neurotoxic and neuroprotective effects of IL-6 in experimental stroke
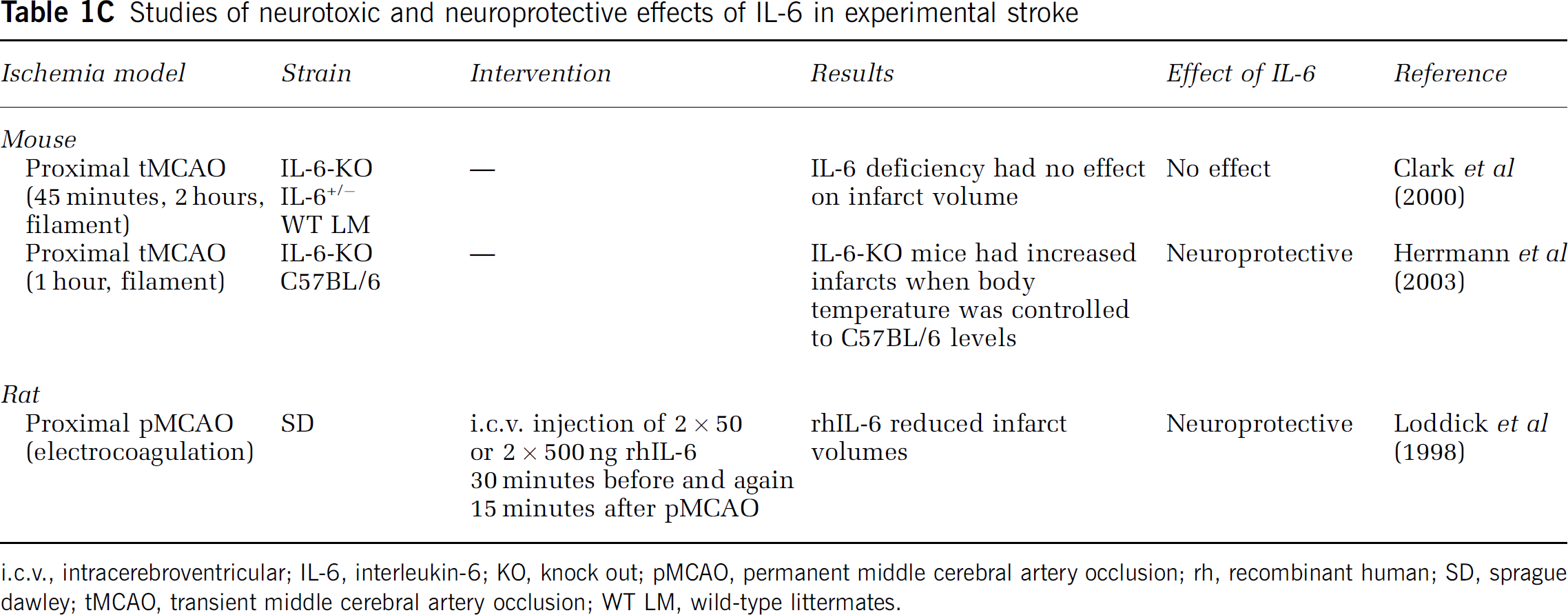
i.c.v., intracerebroventricular; IL-6, interleukin-6; KO, knock out; pMCAO, permanent middle cerebral artery occlusion; rh, recombinant human; SD, sprague dawley; tMCAO, transient middle cerebral artery occlusion; WT LM, wild-type littermates.
Unlike TNF, IL-1 has a clearly defined pathologic effect (Figure 2 and Table 1B). Interleukin-1 occurs in two forms, IL-1α and IL-1β, of which IL-1β, which is a secreted protein, is the one that is best investigated in experimental stroke (Allan et al, 2005). Both forms of IL-1 signal through the IL-1 receptor type I (IL-1RI), an effect, which is competitively blocked by the naturally occurring IL-1 receptor antagonist (IL-1Ra; Dinarello, 1994). Several independent studies have shown that administration of recombinant human IL-1Ra (rhIL-1Ra), neutralizing antibodies against IL-1 (Yamasaki et al, 1995; Loddick and Rothwell, 1996; Boutin et al, 2001; Touzani et al, 2002) or therapeutic inhibition of IL-1-converting enzyme (or caspase-1), which cleaves pro-IL-1β into the secreted bioactive IL-1β (Dinarello, 1994; Schielke et al, 1998), results in large decreases in infarct volume (up to 60% to 70%) after MCAO in mice and rats (Figure 2 and Table 1B). Similarly, mice deficient in IL-1 or in IL-1-converting enzyme show large decreases in infarct size, whereas intracerebroventricular (i.c.v.) injection of IL-1β increases infarct size (Figure 2 and Table 1B). In combination, these studies have paved the way for safety trials of treatment with rhIL-1Ra (Anakinra (Kineret)) in stroke patients (Emsley et al, 2005). Ongoing pharmacokinetic studies show that experimentally effective concentrations of rhIL-1Ra (Anakinra) given within the therapeutic window can be achieved in stroke patients (Galea et al, 2011).
There are only few reports on the role of IL-6 in experimental stroke (Figure 2 and Table 1C). However, although i.c.v. injection of rhIL-6 reduces infarct size in rats and gerbils (Matsuda et al, 1996; Loddick et al, 1998), initial studies on IL-6-deficient mice suggested that IL-6 has no impact on infarct evolution after neither severe nor mild tMCAO (Clark et al, 2000). This lack of effect on infarct size, however, was most likely due to hypothermia in IL-6-deficient mice as a later study showed increased infarct size and reduced survival in IL-6-deficient mice when controlled for hypothermia (Figure 2 and Table 1C). These results suggest that IL-6 is neuroprotective, as also shown in in vitro studies (Biber et al, 2008), and that infarct evolution is modulated by the IL-6 present in the neural tissue at stroke onset, as is the case for TNF.
Cytokines in Experimental Stroke
As mentioned in the introduction, TNF, IL-1, and IL-6 are extremely potent molecules and are present, along with their pro-forms and encoding mRNAs, in low levels also in normal brain. This is important, especially in the case of mRNA level changes, which, when using quantitative PCR analysis, typically are reported as fold-changes compared with baseline. The in situ hybridization technique, which is used to localize the ‘cellular site of’ the cytokine mRNA synthesis, and the immunocytochemical technique, which indicates the ‘location’ of the actual cytokine, unfortunately often have insufficient sensitivity to detect the baseline expression of cytokines. We have in Tables 2A, 2B, 2C and Figure 3 only included studies in which the cytokine of interest has been detected either using more than one technique and/or using appropriate specificity controls (Saper and Sawchenko, 2003; Lambertsen et al, 2009). Furthermore, if we found that some studies were covered by other studies, these were not included. In addition, a few studies not highlighted in the tables have been included in the text if, e.g., special dissections have been used.
Temporal profile of TNF mRNA and protein in experimental stroke
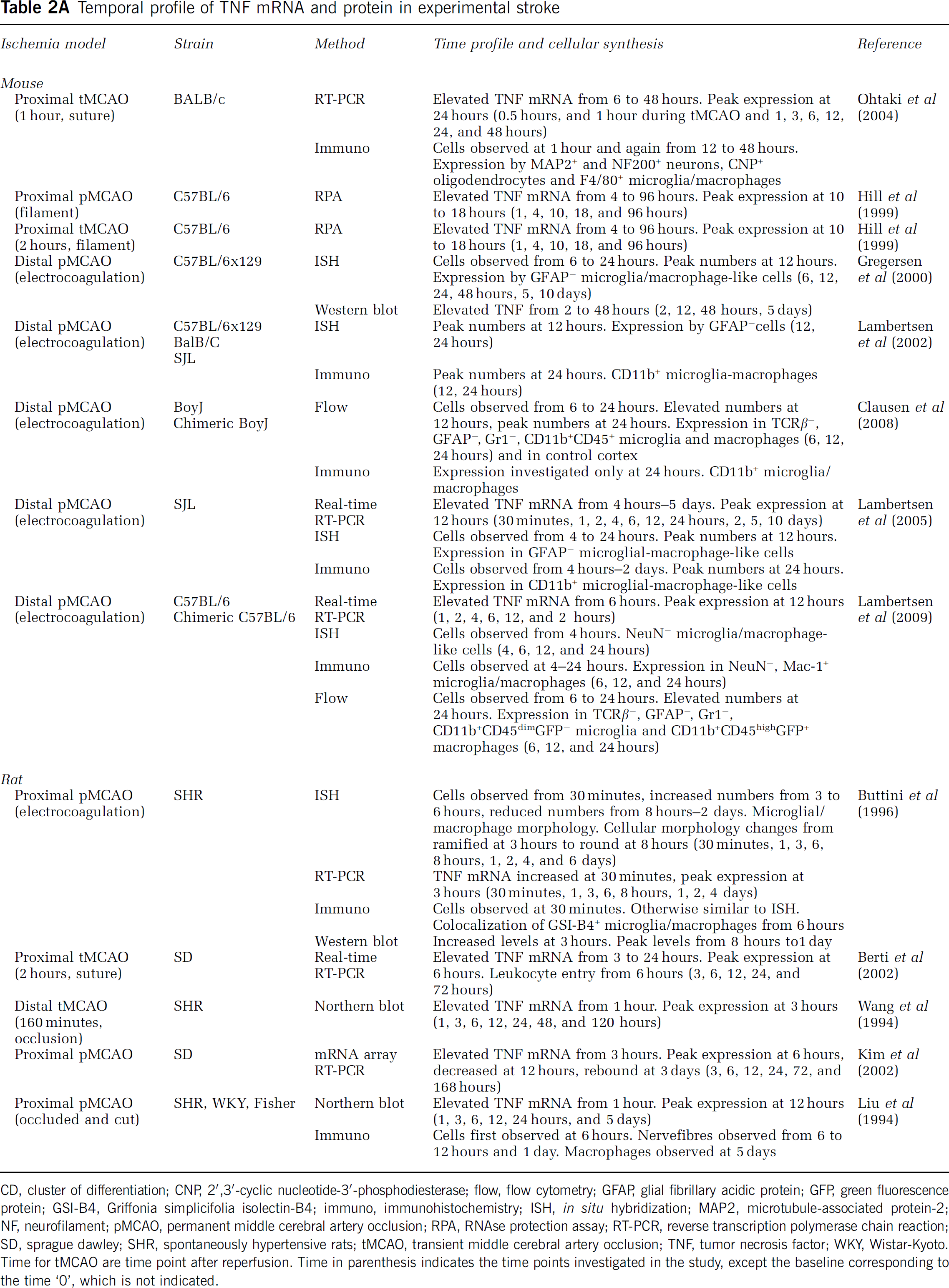
CD, cluster of differentiation; CNP, 2′,3′-cyclic nucleotide-3′-phosphodiesterase; flow, flow cytometry; GFAP, glial fibrillary acidic protein; GFP, green fluorescence protein; GSI-B4, Griffonia simplicifolia isolectin-B4; immuno, immunohistochemistry; ISH, in situ hybridization; MAP2, microtubule-associated protein-2; NF, neurofilament; pMCAO, permanent middle cerebral artery occlusion; RPA, RNAse protection assay; RT-PCR, reverse transcription polymerase chain reaction; SD, sprague dawley; SHR, spontaneously hypertensive rats; tMCAO, transient middle cerebral artery occlusion; TNF, tumor necrosis factor; WKY, Wistar-Kyoto.
Time for tMCAO are time point after reperfusion. Time in parenthesis indicates the time points investigated in the study, except the baseline corresponding to the time ‘0’, which is not indicated.
Temporal profile of IL-1β mRNA and protein in experimental stroke
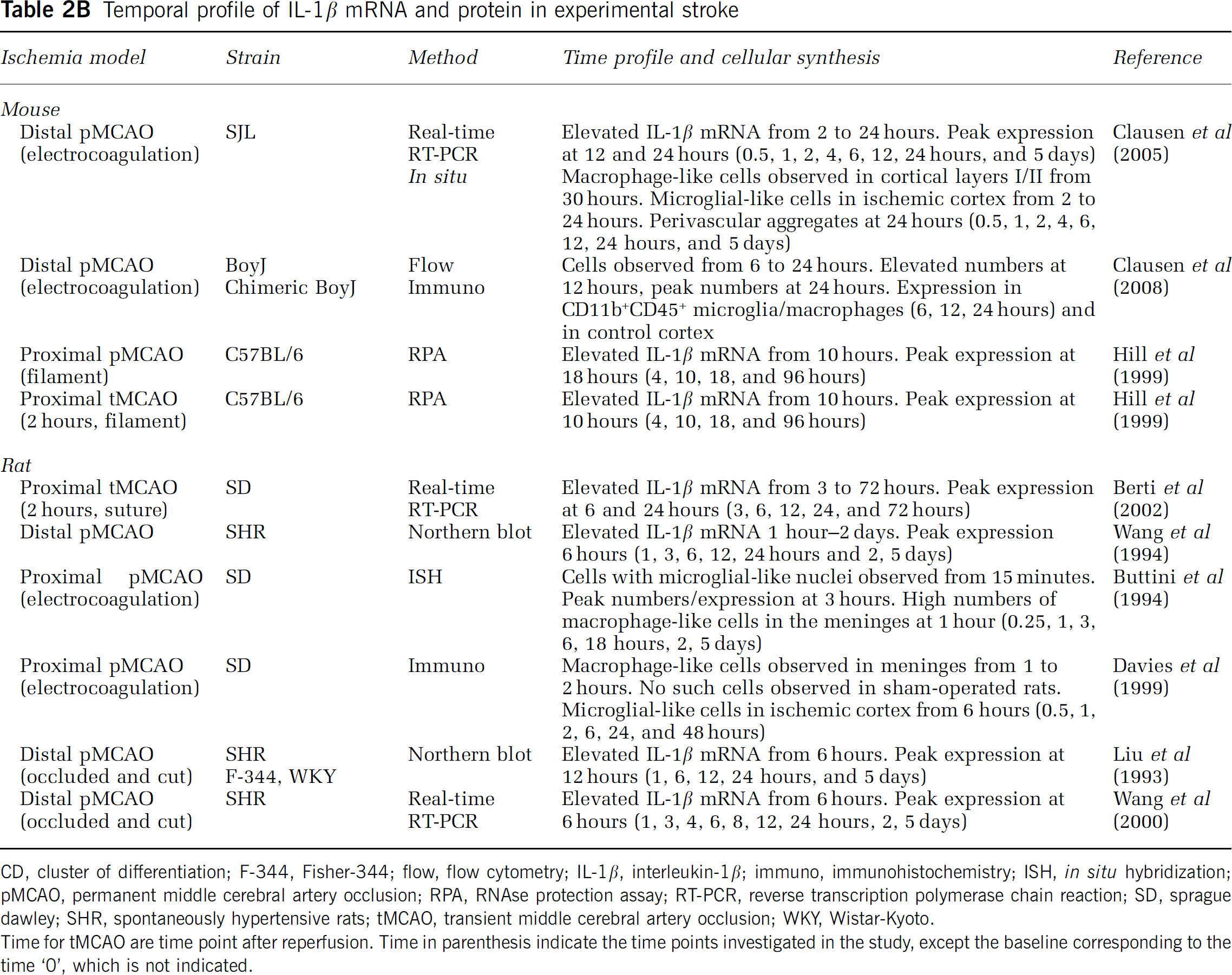
CD, cluster of differentiation; F-344, Fisher-344; flow, flow cytometry; IL-1β, interleukin-1β; immuno, immunohistochemistry; ISH, in situ hybridization; pMCAO, permanent middle cerebral artery occlusion; RPA, RNAse protection assay; RT-PCR, reverse transcription polymerase chain reaction; SD, sprague dawley; SHR, spontaneously hypertensive rats; tMCAO, transient middle cerebral artery occlusion; WKY, Wistar-Kyoto.
Time for tMCAO are time point after reperfusion. Time in parenthesis indicate the time points investigated in the study, except the baseline corresponding to the time ‘0’, which is not indicated.
Temporal profile of IL-6 mRNA and protein in experimental stroke
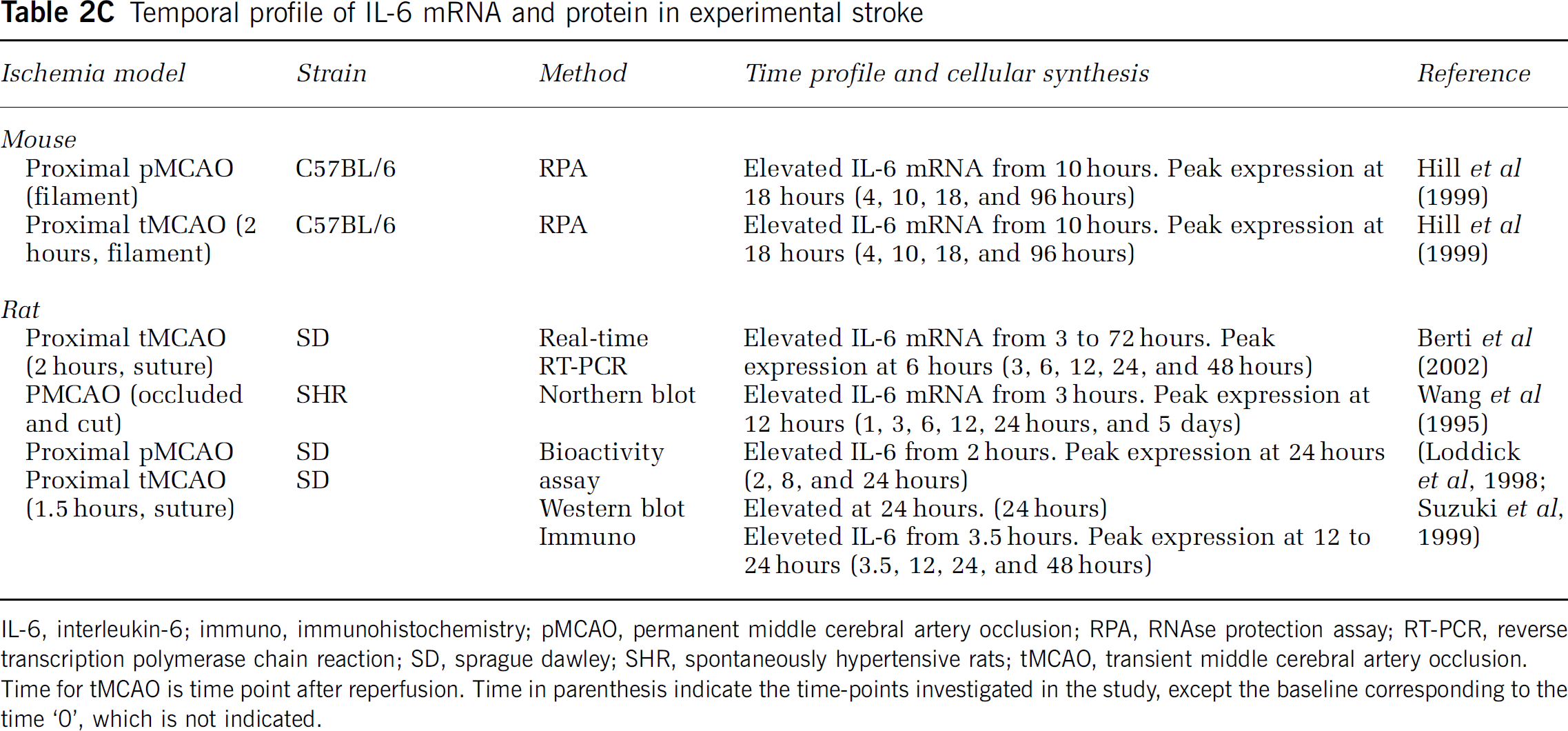
IL-6, interleukin-6; immuno, immunohistochemistry; pMCAO, permanent middle cerebral artery occlusion; RPA, RNAse protection assay; RT-PCR, reverse transcription polymerase chain reaction; SD, sprague dawley; SHR, spontaneously hypertensive rats; tMCAO, transient middle cerebral artery occlusion.
Time for tMCAO is time point after reperfusion. Time in parenthesis indicate the time-points investigated in the study, except the baseline corresponding to the time ‘0’, which is not indicated.
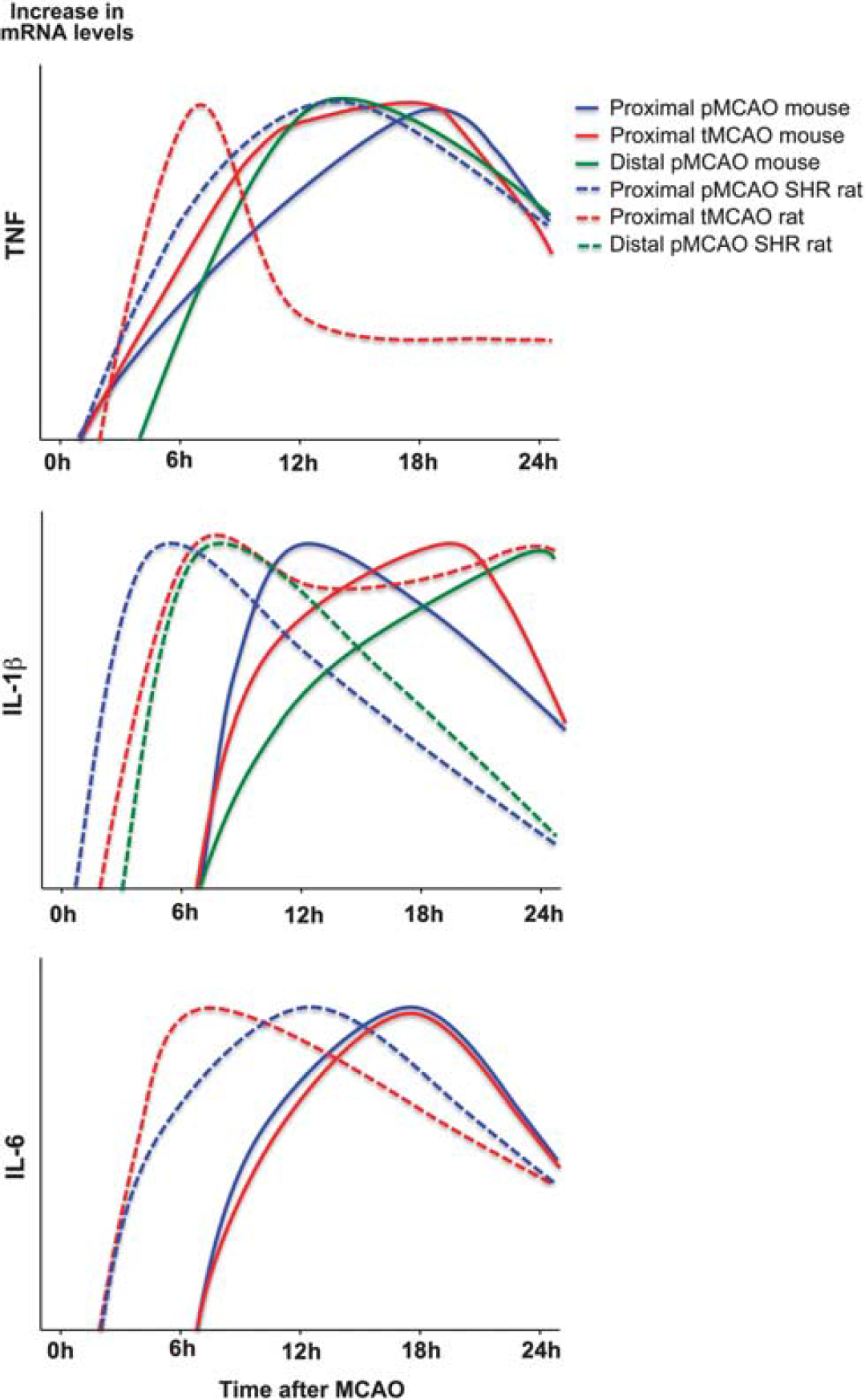
Temporal cytokine messenger RNA (mRNA) profile in experimental stroke. Graphic presentations of the temporal mRNA profiles of tumor necrosis factor (TNF), interleukin (IL)-1β, and IL-6 mRNA in the acute phase (<4 to 6 hours) and up till 24 hours after permanent MCAO (pMCAO) and transient MCAO (tMCAO) in mice (solid lines) and rats (dotted lines). Data are presented as relative increases in mRNA levels on an abitrary y axis and the peak time of expression after surgery on the x axis. Data for TNF were obtained from: solid blue and red (Hill et al, 1999); solid green (Lambertsen et al, 2009); and dotted blue (Liu et al, 1994) and red (Berti et al, 2002). Data for IL-1β were obtained from: solid blue and red (Hill et al, 1999); solid green (Clausen et al, 2005); dotted blue (Wang et al, 1994); dotted red (Berti et al, 2002); and dotted green (Wang et al, 2000). Data for IL-6 were from: solid blue and red (Hill et al, 1999), dotted blue (Wang et al, 1995); and dotted red (Berti et al, 2002).
Cytokine Synthesis in the Ischemic Territory
TNF is induced in low levels (up till four- to fivefold compared with baseline) within the early phase after stroke onset (<4 to 6 hours), with the earliest increase in mRNA expression observed from 30 minutes to 6 hours after both tMCAO and pMCAO in mice and rats (Figure 3, Table 2A and Supplementary Table 1). In the mouse, increased TNF protein is observed 4 hours after pMCAO, when single TNF immunoreactive cells are detected in the peri-infarct (Table 2A; Lambertsen et al, 2005). This is corroborated by ELISA data reporting on increased TNF in samples of cortex and striatum 1 hour after tMCAO in spontaneously hypertensive rats (Adibhatla and Hatcher, 2007). Peak levels of TNF mRNA and protein are reached from 12 to 24 hours, and TNF remains elevated for days after both tMCAO and pMCAO in the mouse (Figure 3, Table 2A, and Supplementary Table 1). In the rat, however, there is evidence that peak levels of TNF mRNA are reached earlier after tMCAO than after pMCAO (Figure 3; Liu et al, 1994; Wang et al, 1994; Berti et al, 2002), and TNF mRNA remains elevated for hours and days after stroke (Figure 3 and Table 2A). However, interpretations are additionally complicated by the use of different rat strains in different studies (Table 2A). Based on their results, Buttini et al (1996) and Wang et al (1994) suggested that spontaneously hypertensive rats have a TNF time profile different from normotensive rats. This was, however, not supported by findings by Liu et al (1994), who reported on comparable TNF profiles after pMCAO in spontaneously hypertensive rats, Wistar-Kyoto and Fisher-344 rats (Table 2A). Comparable time profiles of TNF production have also been reported for C57BL/6, SJL, BALB/c, and C57x129 mice after pMCAO; however, the magnitude of the TNF response was different (Lambertsen et al, 2002). In all strains, the TNF mRNA+ and TNF+ cells were diffusely distributed in the peri-infarct at both 12 and 24 hours after stroke onset (Table 2A; Lambertsen et al, 2002).
Small increases in IL-1β have been observed as soon as 1 to 2 hours after tMCAO and pMCAO in the rat, where it remains elevated up till 24 hours after tMCAO but not after pMCAO (Figure 3 and Table 2B). The ischemia-induced increase in IL-1β mRNA appears slightly delayed compared with the increase in TNF mRNA after both tMCAO and pMCAO in the mouse (Figures 3 and 4), but continues to increase through 12 and 24 hours after tMCAO and distal pMCAO, to decline toward baseline levels at day 5 (Table 2B and Supplementary Table 1). Very interestingly, in situ hybridization studies performed on tissues from mice and rats after pMCAO (Buttini et al, 1994; Clausen et al, 2005) have detected scattered IL-1β mRNA+ cells in the peri-infarct down to 1 to 2 hours after stroke onset. Of additional interest, the same studies reported on aggregates and perivascular infiltrates of intensely labeled IL-1β mRNA+ cells in the peri-infarct from 12 hours and even more so at 24 hours after the stroke. Both studies also reported on IL-1β mRNA+ macrophage-like cells in the meninges and the superficial cortical layers corresponding to the ischemic territory, during the first post-surgical hour. A similar phenomenon was reported by Davies et al (1999), who, in addition to the IL-1β+ macrophage-like cells in the meninges 1 to 2 hours after pMCAO in the rat, reported on numerous IL-1β+ microglial-like cells in the ischemic territory from 6 to 24 and 48 hours (Table 2B). Although, tempting to speculate that the meningeal infiltration might be ascribed to the craniectomy, which was performed in all three studies, it was specifically reported that IL-1β mRNA+ and IL-1β+ cells were absent from the meninges in the sham-operated animals (Buttini et al, 1994; Davies et al, 1999; Clausen et al, 2005).
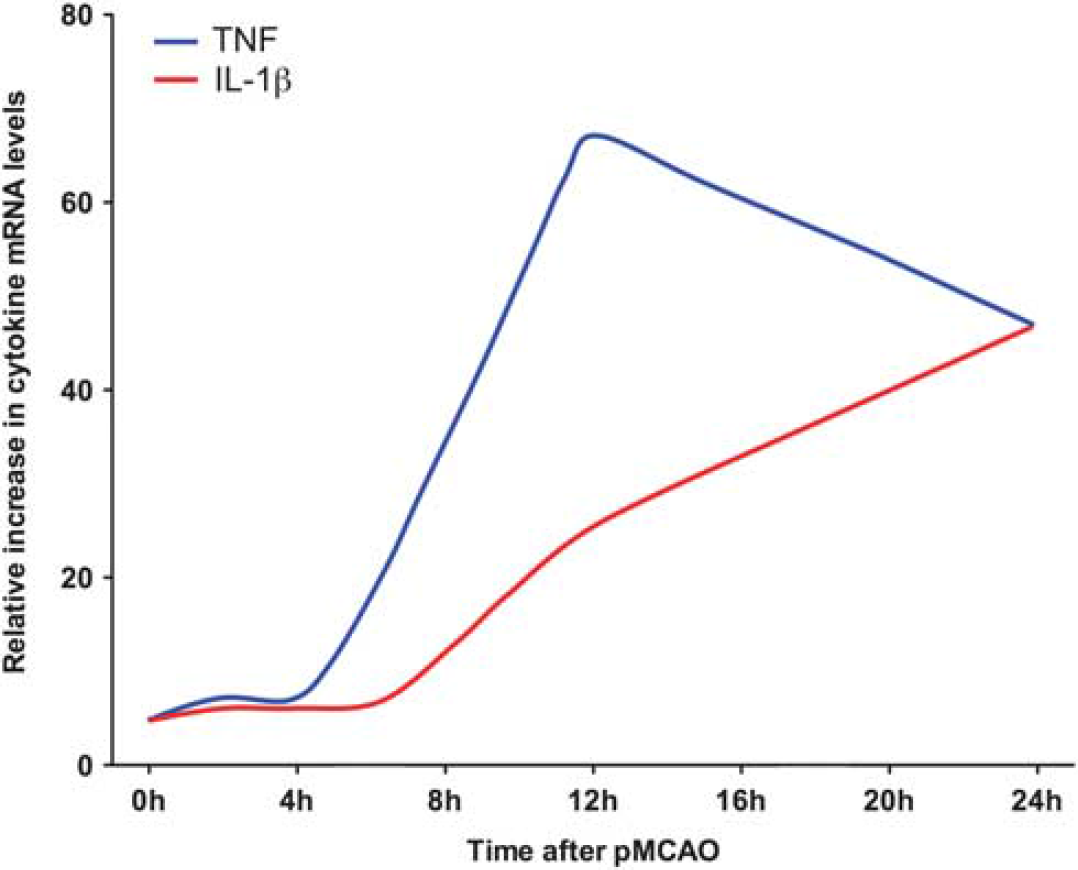
Temporal profile of tumor necrosis factor (TNF) and interleukin (IL)-1β messenger RNA (mRNA) upregulation after permanent MCAO (pMCAO) in mice. Graphic presentation of the temporal mRNA profile of TNF and IL-1β in the same ischemic hemispheres from mice subjected to pMCAO. Data are presented as relative increases in cytokine mRNA levels compared with unmanipulated controls, and have been obtained from Clausen et al (2005) and Lambertsen et al (2009). Note both increase in cytokine levels in the early phase (<4 to 6 hours) after MCAO, and the different profiles of TNF mRNA and IL-1β mRNA expression through 12 and 24 hours after MCAO. Although, only the TNF mRNA level, but not the IL-1β mRNA level, was significantly elevated compared with unmanipulated control and sham-operated mice in the early phase, cells expressing low levels of IL-1β mRNA were observed to be present the ischemic territory from 2 hours after MCAO (Clausen et al, 2005).
There are only few studies on the time profile and cellular production of IL-6 after experimental stroke, and to our knowledge no studies exploring the expression of both mRNA and protein in the same studies. Interleukin-6 mRNA has been shown to be significantly increased as soon as 3 hours and to peak 12 hours after pMCAO in the rat, and remains elevated at 24 hours (Figure 3 and Table 2C).
The striking finding from reviewing the above literature is that the increases in cytokine mRNA and protein levels are at most moderate in the therapeutic window (<4.5 hours; Figures 3 and 4). Even small increases in cytokine levels may, however, have profound effects on infarct evolution if released at the site of the ischemic neurons.
Microglia Produce TNF and IL-1β in the Early Phase and at the Time of MCAO
Studies of the cellular source(s) of TNF in the earliest phase, from minutes to 4 to 6 hours, after MCAO are limited to a few studies (Table 2A), one of them being Buttini et al (1996), who reported on a primarily microglial origin of TNF mRNA 30 minutes after pMCAO in the rat and a mixed microglial-macrophage origin in the later phase (Table 2A). These findings were subsequently reproduced in the mouse, in a series of studies showing a predominantly microglial origin of TNF in the early phase after pMCAO, and a more mixed microglial-macrophage origin from 12 to 24 hours after pMCAO in the mouse (Gregersen et al, 2000; Lambertsen et al, 2002, 2005, 2009; Clausen et al, 2008), although other studies have pointed to an ependymal, astroglial, and even neuronal TNF synthesis (Table 2A and Supplementary Table 1). The repeated lack of observation of neuronal TNF mRNA in situ hybridization signal in several independent well-controlled studies from our laboratory emphasizes that neuronal TNF production is negliable compared with the amount of TNF produced by microglia and macrophages after pMCAO in the mouse (Gregersen et al, 2000; Lambertsen et al, 2002, 2005, 2009). The predominant microglial-macrophage origin of TNF receives support from flow cytometry studies, showing that both microglia and macrophages, but not granulocytes, produce TNF after pMCAO in the stroke-lesioned mouse brain (Clausen et al, 2008; Lambertsen et al, 2009). Importantly, these studies also point to microglia as the source of the TNF present in the brain at stroke onset (Table 2A).
Regarding IL-1β, Buttini et al (1994), Clausen et al (2005, 2008), and Davies et al (1999) have convincingly shown that the main sources of IL-1β in rats and mice subjected to MCAO are microglia and macrophages (Table 2A), but apparently different subsets of microglia and macrophages than those producing TNF as demonstrated in flow cytometry studies (Clausen et al, 2008). Further, it appears that the microglia respond to the ischemic event by increasing their production of both IL-1β and TNF. This goes well hand-in-hand with flow cytometry results, that also point to a microglial origin of the IL-1β and TNF present in the normal brain (Clausen et al, 2008; Lambertsen et al, 2009; Tables 2A and 2B). In agreement with the histologic demonstration of perivascular infiltrates of IL-1β, but not TNF, producing cells in the later phase after MCAO, the proportion of IL-1β-producing macrophages is higher than of TNF-producing macrophages (Clausen et al, 2008). Further, as is the case for TNF, there is no evidence that granulocytes should be major producers of IL-1β (Clausen et al, 2008). In our laboratory, it has not been possible to reproduce reports on IL-1β immunoreactivity in astrocytes and endothelial cells in stroke-lesioned rats (Supplementary Table 1) and in our mouse model (Clausen et al, 2005, 2008).
There is, so far, very little information on IL-1α expression after experimental stroke. Interleukin-1α was recently shown to be upregulated in microglia in the peri-infarct 4 and 24 hours after tMCAO in the mouse (Luheshi et al, 2012). Further, there are, to our knowledge, no in situ hybridization studies of the expression of IL-6 mRNA after MCAO. Interleukin-6 immunoreactivity has been shown to increase in microglia and cortical neurons from 3.5 hours after pMCAO in rats (Suzuki et al, 1999; Table 2C).
Cytokines in Human Stroke
In humans, acute stroke has been shown to result in increased levels of TNF, IL-1β, and IL-6 in CSF and blood (Maas and Furie, 2009) and, so far, in the case of TNF also in the brain (Sairanen et al, 2001; Dziewulska and Mossakowski, 2003). These cytokines can be measured not only in serum or plasma but also are associated with a changed expression of numerous inflammatory genes in blood cells and, as shown recently, also in cells of the bone marrow (Denes et al, 2011; Sharp et al, 2011). These findings have lead to studies of stroke markers with the aim to improve diagnosis, prognosis, and therapy in stroke patients (Jickling and Sharp, 2011; Sharp et al, 2011). As TNF, IL-1β, and IL-6 can modulate infarct evolution in experimental stroke these cytokines have also attracted considerable interest as putative markers of stroke severity and neurologic outcome (Emsley et al, 2003, 2007; Jickling and Sharp, 2011).
Postmortem Studies
Because of the constraints with brain tissue from patients that did not survive an ischemic condition, the studies performed rely exclusively on the use of immunohistochemical methods. In two independent studies it was shown that TNF immunoreactivity is rapidly (<1 day) induced by stroke also in humans (Sairanen et al, 2001; Dziewulska and Mossakowski, 2003), thereby confirming animal studies (Table 3). However, the studies are contradictory with respect to the cellular source of TNF. Whereas Sairanen et al (2001) predominantly describe early TNF immunoreactivity in neurons and at later time points in astrocytes and infiltrating leukocytes, Dziewulska and Mossakowski (2003) show the most intense TNF immunoreactivity in microglia and conclude that these cells are the major source of TNF in human stroke. In an earlier study, TNF immunoreactivity was investigated in brains of stroke patients who died at later time points (up to around 30 days; Tomimoto et al, 1996). In this study, TNF immunoreactivity was predominantly found in microglia, with very few TNF+ astrocytes. The reason for these discrepancies are unclear, the results by Dziewulska and Mossakowski (2003), however, are consistent with the reports that limit early TNF production in stroke-lesioned rodent brain to microglia. According to our knowledge, TNF is yet the only cytokine that has been studied by immunohistochemical methods in post-mortem human stroke tissue.
Time profile and cellular synthesis of TNF, IL-1β, and IL-6 in human stroke
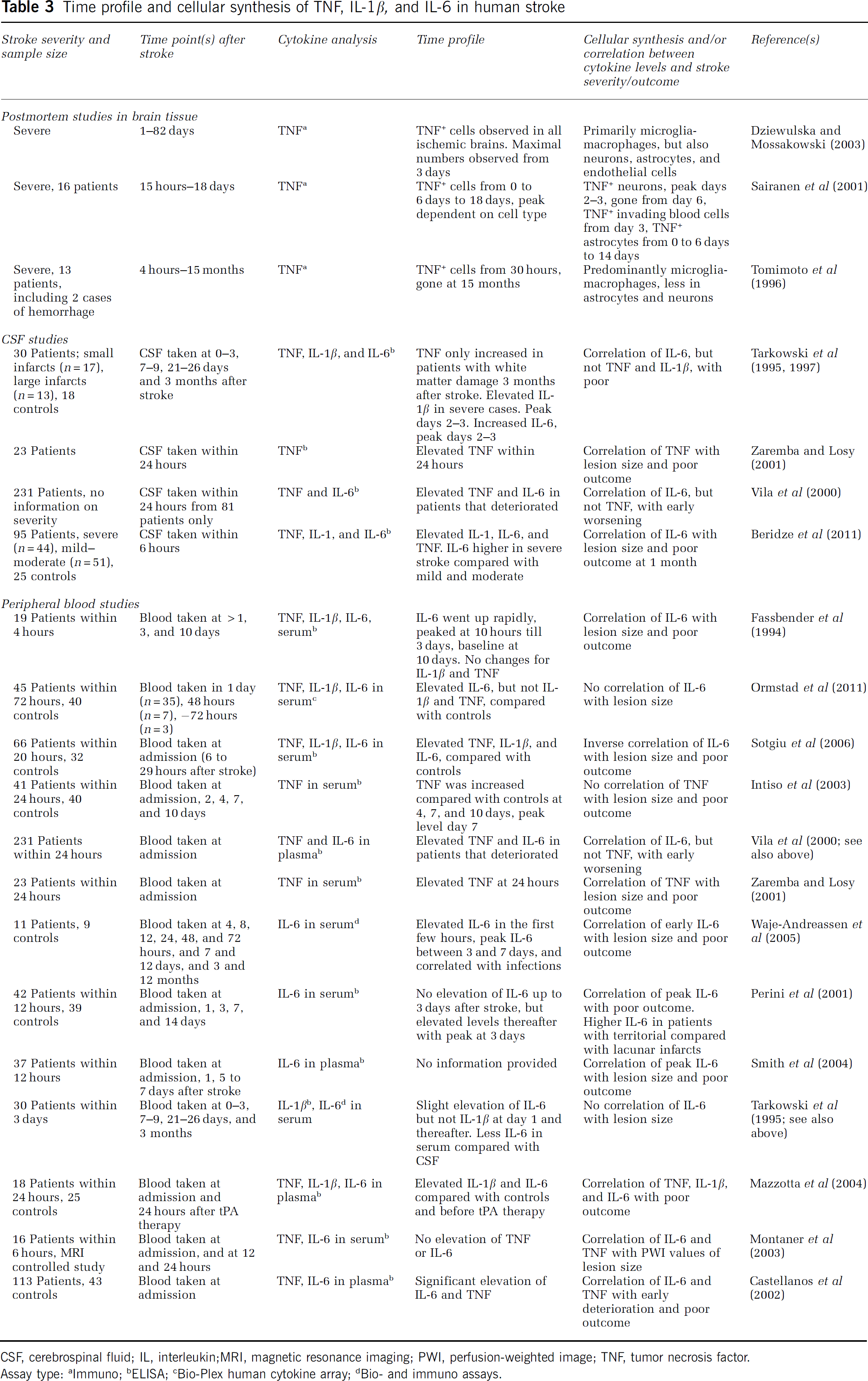
CSF, cerebrospinal fluid; IL, interleukin;MRI, magnetic resonance imaging; PWI, perfusion-weighted image; TNF, tumor necrosis factor.
Assay type:
Immuno;
ELISA;
Bio-Plex human cytokine array;
Bio- and immuno assays.
Cytokines in cerebrospinal fluid
More studies have measured cytokine levels in CSF of stroke patients assuming a central origin of these cytokines and little leakage from blood into CSF (Table 3). In an elegant prospective study by Tarkowski et al (1997), TNF levels (next to IL-8, granulocyte-macrophage colony-stimulating factor, and IL-10) were measured in CSF from stroke patients 0 to 3, 7 to 9, 21 to 26 days, and 3 months after stroke. In this study, TNF levels were only increased compared with neurologically normal controls when looking at stroke patients with white matter damage 3 months after the insult (Tarkowski et al, 1997). These results have been challenged by Zaremba and Losy (2001), who detected elevated levels of TNF in CSF that was sampled within 24 hours after stroke, and further showed that CSF levels of TNF correlated with stroke severity and neurologic outcome (Zaremba and Losy, 2001). Knowing whether or not this study only consisted of patients with white matter lesions might be instrumental to understand the reason for the differences in TNF levels in CSF (Zaremba and Losy, 2001). Even though elevated TNF levels were found in CSF of stroke patients that deterioated also by Vila et al (2000), TNF levels did not correlate with early worsening, thereby challenging the results by Zaremba and Losy (2001; Table 3). This study, however, did not include healthy controls, which hamper the direct comparison with the other studies (Vila et al, 2000). Recent studies have shown that TNF levels were elevated in CSF 6 hours after stroke in patients compared with controls, irrespective of stroke severity, however, TNF neither correlated with lesion size or neurologic outcome (Beridze et al, 2011).
To our knowledge, there are only three reports on IL-1β levels in CSF from stroke patients (Table 3). Whereas Tarkowski et al (1995) attribute increased IL-1β to major stroke cases, elevated IL-1β levels in CSF have also been observed in patients with small infarcts (Sun et al, 2009). Beridze et al (2011) also detected elevated IL-1β levels in CSF 6 hours after the stroke irrespective of its severity but here IL-1β levels did not correlate with lesion size or outcome (Table 3). In comparison, more studies have investigated IL-6 levels in stroke patients (Table 3). In several studies, IL-6 levels were elevated in CSF of stroke patients, and further correlated with stroke severity (Table 3). This gains support from very recent studies showing a positive correlation between IL-6 levels in CSF at 6 hours and the lesion size at 24 hours (Beridze et al, 2011). The levels of IL-6 increases within 24 hours after stroke and increases are more pronounced in the case of gray matter lesions (Table 3 and Supplementary Table 2). In a more recent study, IL-6 levels were not changed in a population of patients with small infarcts, which again might indicate that IL-6 in the CSF is a marker of severe lesions (Sun et al, 2009) of especially the gray matter (Beridze et al, 2011).
Although the reviewed studies are mostly restricted to one time point, it is potentially interesting that the cytokine that is most consistently found to be increased in the CSF from stroke patients is IL-6, which may be produced by both microglia and neurons in stroke.
Cytokines in Blood
The analysis of cytokines in peripheral samples may or may not reflect central cytokine production. In comparison to the situation in the CSF, there are contrasting reports on stroke-induced changes in the level of TNF in serum and plasma. Some studies have reported that TNF levels remain unchanged after stroke, whereas others report on increased TNF levels albeit with different peak times and contradictory results with regard to a correlation between peripheral TNF levels and stroke severity (Table 3). In one magnetic resonance imaging-based study, no significant elevations of serum TNF levels were observed in the first 24 hours, although a correlation between TNF levels and the PWI signal was observed (Montaner et al, 2003).
There is a general agreement that IL-6 levels increase in serum or plasma from stroke patients during the first week after the insult (Table 3 and Supplementary Table 2), i.e., in all studies where peripheral IL-6 levels were measured IL-6 levels were elevated compared with control subjects (Supplementary Table 2). Less agreement, however, has been reached on the time point of peak IL-6 levels and the correlation between IL-6 levels and the final infarct volume. High IL-6 levels, which range from a few hours till 1 day after the insult, have been described in Table 3. Others describe IL-6 peak levels at 3 days (Perini et al, 2001) or some time during the first week (Smith et al, 2004). It appears that early IL-6 levels correlate with stroke severity (lesion size), PWI signal, and patient outcome (Fassbender et al, 1994; Perini et al, 2001; Castellanos et al, 2002; Montaner et al, 2003; Smith et al, 2004; Waje-Andreassen et al, 2005), although one study observed the opposite (Sotgiu et al, 2006). In 66 stroke patients, an inverse relationship between early IL-6 levels, lesion size, and final patient outcome was observed, indicating that early IL-6 is neuroprotective rather than a marker of disease progression (Sotgiu et al, 2006).
Although the importance of IL-1β in experimental stroke is undisputed, it generally is not observed at elevated levels in serum or plasma from stroke patients (Fassbender et al, 1994; Tarkowski et al, 1995; Emsley et al, 2007), which is likely due to its localized role (Emsley et al, 2007). The only exception is one report that shows modestly elevated serum levels of IL-1β in a small group of patients compared with healthy controls (Mazzotta et al, 2004). The importance of IL-1β in stroke, however, is underlined by a rapid upregulation of its natural antagonist IL-1Ra in brain ischemia (Emsley et al, 2007) and the potential use of IL-1Ra as a new treatment for stroke patients (Mulcahy et al, 2003; Emsley et al, 2005).
Taken together, there is little homogeneity in the data on peripheral cytokines in human stroke. A lack of homogeneity also exists between studies of lipopolysaccharide-induced release of cytokines (IL-1β, TNF, and IL-6) from whole blood cells drawn from stroke patients. Whereas Ferrarese et al (1999) found that blood cells from stroke patients released significantly higher cytokine levels, the opposite was described by Emsley et al (2007), who observed less cytokine release from blood cells from stroke patients. Moreover, it is becoming clear that systemic inflammation and thereby cytokine levels may be elevated before and influence stroke susceptability and outcome (McColl et al, 2009). Further, the large heterogeneity of human stroke should be taken into consideration when evaluating findings of stroke-induced changes in peripheral cytokines (Maas and Furie, 2009). In conclusion, significant information about peripheral cytokine responses in human stroke is still lacking, especially in the early phase (<4 to 6 hours) after the stroke (Table 3 and Supplementary Table 2).
Comparing Systemic Cytokine Responses in Human and Experimental Stroke
Our understanding of the role of systemic cytokines in human stroke is furthermore hampered by the fact that experimental stroke studies almost exclusively investigated the central cytokine response, leaving a gap of information concerning the systemic immune response in animals (Herrmann et al, 2003; Offner et al, 2009). There are only few studies that have in detail analyzed the systemic cytokine profile in response to experimental stroke. Increased IL-6 levels have been observed in plasma starting from 2 hours of reperfusion (3 hours after stroke onset) and in the brain from 24 hours (Chapman et al, 2009), whereas other studies have shown an increase of TNF levels in serum at 6 and 24 hours after MCAO (Chang et al, 2011), and provided evidence of higher release of IL-6 and TNF from stimulated splenocytes and blood cells at 6 and 24 hours, respectively (Chang et al, 2011; Offner et al, 2009). Interestingly, IL-1β was not detected systemically suggesting that this cytokine was only expressed in the ischemic brain (Offner et al, 2009). Chang and colleagues furthermore showed that the treatment with cocaine- and amphetamine-regulated transcript peptides reduced the infarct size and the cytokine response both centrally and systemically, indicating that the systemic cytokine response parallels the events in the brain (Chang et al, 2011). Although there are results pointing toward the systemic cytokines as potential target in stroke therapy, it should not be forgotten that one of the major clinical problems is post-stroke immunosuppression. A further dampening of systemic immune functions may therefore not be advisable (Denes et al, 2011).
Cytokine Receptors and Cytokine Signaling in Experimental Stroke
A detailed understanding of how cytokines influence the balance of the ischemic neurons between neuroprotection and neurotoxicity is still missing. As there are several excellent recent reviews on the presumed signaling mechanisms (Allan et al, 2005; McCoy and Tansey, 2008; Suzuki et al, 2009), we will mainly focus on neuronal expression of cytokine receptors in brain ischemia. Neurons express both TNF-Rs and IL-1Rs, and IL-6R (Allan et al, 2005; McCoy and Tansey, 2008; Suzuki et al, 2009), however, baseline expression may, as has been estimated for the IL-1RI, be limited to very few receptors per cell (Dinarello, 1994), which makes it difficult to study their expression in tissue sections. Nevertheless, it appears that the TNF-RI is increased in both neurons and non-neuronal cells from 4 to 6 hours and up till 5 days after pMCAO (Botchkina et al, 1997; Lambertsen et al, 2007) and tMCAO (Yin et al, 2004) in mice and rats. The TNF-RII is upregulated from 24 hours after pMCAO in the rat (Botchkina et al, 1997), at which time point it in the mouse is expressed in high levels by microglia and macrophages, consistent with that also the increase of TNF-RII mRNA is higher than that of TNF-RI mRNA (Lambertsen et al, 2007). This could reflect that the TNF-RI, besides a role in normal neuronal function (Botchkina et al, 1997), have a role for neuroprotection after experimental stroke (Figure 2). The TNF-RII to a larger extent appears to be involved in inflammatory responses, although neuroprotection though TNF-RII has been reported (Fontaine et al, 2002; Marchetti et al, 2004).
Both TNF-Rs can be cleaved from the cell surface as sTNF-RI and sTNF-RII and function as decoy molecules for TNF (Kohno et al, 1990; Nophar et al, 1990). Thus, increasing the cleavage of TNF-RI is an important step in reducing activation of TNF-RI-mediated apoptotic signaling in spinal cord neurons (Bartsch et al, 2010). Although cleavage is mediated by TNF alpha converting enzyme (TACE) in normal central nervous system, it is mediated by ADAM8 under pathologic conditions (Bartsch et al, 2010). Interestingly, ischemic tolerance induction in vitro depends on TACE-mediated TNF-release, which leads to upregulation of glutamate transporters (Romera et al, 2004), whereas, ischemic preconditioning involves upregulation of TACE and TNF-signaling through TNF-RI (Cardenas et al, 2002; Pradillo et al, 2005). Tumor necrosis factor signaling in ischemic neurons in vivo is still unclear. However, it appears that the pleiotrophic effect of TNF in experimental stroke (Figure 2 and Table 1A) can mainly be attributed to the binding of TNF to the TNF-RI, which through stepwise recruitment and activation of signaling molecules, leads to neuroprotection or apoptosis (McCoy and Tansey, 2008). Neuroprotection via TNF-RI can also be attributed to increases in calcium-binding proteins and Mn-superoxide dismutase in neurons (Mattson et al, 1997; Ginis et al, 2002).
Binding of IL-1 to IL-1RI leads to recruitment of a co-receptor accessory protein, which is essential to IL-1RI signaling (Greenfeder et al, 1995; Cullinan et al, 1998; Lang et al, 1998). Interestingly, N-methyl-
IL-6 exerts its function through binding to the IL-6R, which has no signaling properties by itself. However, ligand binding allows the formation of gp130/gp130 homodimers that associates with Janus-associated kinases, activates signal transducer and activator of transcription and mitogen-activated protein kinase signaling pathways (Heinrich et al, 2003; Kamimura et al, 2003; Ernst and Jenkins, 2004; Bauer et al, 2007). Unlike sTNF-R and sIL-1R, the sIL-6R does not have any decoy properties, but instead activates the ubiquitously expressed gp130 like the membrane-bound form of IL-6R (Scheller and Rose-John, 2006). This mechanism is called trans-signaling and contributes to explain that IL-6 has numerous effects in many different cell types, despite the limited expression of the IL-6R (Scheller and Rose-John, 2006). Brain neurons express IL-6R, whereas glial cells, including microglia, require sIL-6R to respond to IL-6 (Suzuki et al, 2009). In contrast to the IL-6 there is so far no evidence that the levels of IL-6R change after MCAO (Suzuki et al, 2005). Although the mechanisms behind the apparently neuroprotective effect of IL-6 in stroke (Figure 2 and Table 1C) have not yet been ressolved, we have shown that IL-6 upregulates the expression and function of the neuronal adenosine A1 receptor (Biber et al, 2008). This upregulation appears to be crucial for the protective effect of IL-6, as IL-6 is not protective in neurons lacking adenosine A1 receptors (Moidunny et al, 2010). Furthermore, as IL-6 inhibits IL-l synthesis (Schindler et al, 1990) and stimulates the production of the IL-1Ra and sTNF-RI (Tilg et al, 1994), these actions may also contribute to the neuroprotective effects of IL-6.
Discussion
From the literature reviewed here, it is clear that modulating the function of TNF, IL-1, or IL-6 in experimental stroke has significant effects on infarct evolution, after both pMCAO and tMCAO, and across different species and strains (Figure 2). This finding, and in particular the robustness of the finding, indicates that these cytokines also influence infarct evolution in human stroke. Due to the narrow therapeutic window, the few-fold increases in cytokine levels in the early phase after stroke onset (<4 to 6 hours), and even the low baseline levels of cytokines present at stroke onset, are the cytokines that can directly act on the ischemic neurons, and must therefore be the cytokines that are important for the evolution of the infarct. This is particularly clear in the case of pMCAO where the penumbra is recruited into the infarct within the first few hours after stroke onset (Figures 1A and 1B). Evidence that the low baseline levels of cytokines influence infarct evolution is also provided by the findings of increased infarct sizes in TNF- and TNF-RI-deficient mice (Bruce et al, 1996; Gary et al, 1998; Taoufik et al, 2007; Lambertsen et al, 2009) and IL-6-deficient mice (Herrmann et al, 2003), and of reduced infarct sizes in IL-1α/β-deficient mice (Boutin et al, 2001), findings obtained by several independent laboratories (Figure 2). Nevertheless, as these results have been obtained using transgenics with embryonic deletions, which could lead to gestational adaptations of neurons, and compensatory changes of other cytokine systems, key findings should be re-investigated using mice with cell-specific or conditional cytokine deletions.
An obvious question is if the effect of cytokines in general, or of individual cytokines, depends on the model used, pMCAO or tMCAO? At present there is no clear answer to this question. Using the wording by Hossmann (2012), ‘In promptly reversed transient ischemia as after mechanical occlusion’, blood-borne cells and cytokines have fast access to the infarct core and the penumbra. Although, there are no studies that have directly compared leukocyte kinetics after pMCAO and tMCAO, it is obvious from the literature that larger numbers of leukocytes accumulate faster in the ischemic territory after tMCAO (Gelderblom et al, 2009) than after pMCAO (Clausen et al, 2008). Grossly, 1% to 2% of myeloid cells were macrophages in the hemispheres from perfused, unmanipulated central nervous system in these studies. This percentage had at 12 hours increased to, respectively, 5% and 20%, and at 24 hours to, respectively, 25% and close to 30%, of the total number of myeloid cells, after pMCAO (Clausen et al, 2008) and tMCAO (Gelderblom et al, 2009). Although, the model used by Gelderblom et al (2009) is a model for proximal tMCAO compared with the distal pMCAO model used by Clausen et al (2008), these percentages appear representative for previously published work on leukocyte entry into the stroke-lesioned brain (Zhang et al, 1997; Lehrmann et al, 1997; Gregersen et al, 2000; Lambertsen et al, 2009). Consistent with that macrophages infiltrate the ischemic territory with a delay after pMCAO, we in our recent studies of the effect of microglial- versus macrophage-derived TNF, found that the neuroprotective TNF in pMCAO is produced by microglia and not macrophages (Lambertsen et al, 2009). However, this finding clearly leaves open the possibility that macrophage-derived TNF might influence infarct evolution after tMCAO. Indeed, such effect will depend on the cytokine and on its production by the macrophages after they have infiltrated the neural parenchyma. The best evidence that systemic cytokines influence infarct evolution is provided by experimental stroke studies, showing that i.v., subcutaneous, or intraperitoneal treatment of the experimental animal with cytokine or cytokine antagonist at the time of, or after MCAO, reduces the size of the infarct (Dawson et al, 1996; Relton et al, 1996; Nawashiro et al, 1997a, 1997b; Lavine et al, 1998; Wang et al, 2004) (Figure 2). Recently, it was also shown that IL-1α is contained in platelets, and that it modifies endothelial cell adhesion molecule expression in vitro, thereby putatively facilitating leukocyte extravasation (Thornton et al, 2010). Taken together, systemic administration of cytokines or cytokine-antagonists still holds promise as a treatment option in acute stroke therapy, taking into consideration the therapeutic window.
Although the available information on IL-6 in experimental stroke, argue against a pathogenic role of IL-6 in stroke (Suzuki et al, 2009), it was nevertheless surprising that there was so little data on its production in experimental stroke. The importance of this question is emphasized by the finding that IL-6 is the cytokine, which is most consistently found to be increased in the CSF from stroke patients. Although, there is data suggesting that IL-6 is produced in both neurons and non-neuronal cells after MCAO in rats (Suzuki et al, 1999, 2009), IL-6 is also produced by microglia in other models of neurologic disease (Nakanishi et al, 2007; Cho et al, 2011). At present, it is therefore difficult to know whether the increased IL-6 in CSF after a stroke in the human, reflects release of IL-6 into the CSF from dying neurons or if IL-6 is produced by microglia. In the lack of technology that allows molecular modeling (e.g., imaging, nuclear magnetic resonance spectroscopy) of cytokines and their receptors in human stroke, we still depend on information from studies of postmortem human brain tissue. Although, studies of postmortem human brain tissue are far from trivial, and may not necessarily reflect the situation in stroke survivors, we therefore still need well-controlled studies to be performed on the in situ production of IL-6. In fact, we would argue the same for IL-1, whose cellular production also remains undetermined in postmortem human stroke tissue, and even for TNF, as the results from the available immunohistochemical reports are contradictory (Table 3).
In the peripheral nervous system, it is generally accepted that cytokines released from inflammatory cells regulate neuronal sensitivity to stimulation. Any tissue damage and the subsequent release of inflammatory mediators cause a transient hypersensitivity of the pain-sensing system of the affected area, a phenomenon called inflammatory pain. An injection of TNF, IL-1, or IL-6 causes rapidly (within minutes) hyperalgesia in numerous animal models (Thacker et al, 2007). This rapid effect is explained by numerous direct effects of these cytokines on nociceptive neurons, such as changes in ion-currents, enhancement of transient receptor potential channel activity, and increases in glutamatergic effects (St Pierre et al, 2009). Several lines of evidence indicate that, at least for IL-1 and TNF, similar effects are also observed in central nervous system neurons (Viviani et al, 2003; Gardoni et al, 2011). These and other results have led to the hypothesis that the direct sensitization by IL-1 of cortical neurons to glutamatergic input may contribute to the known detrimental effects of IL-1 in stroke (Fogal and Hewett, 2008). In comparison, cortical neurons respond to IL-6 with a rapid upregulation of adenosine A1 receptors, which results in decreased electrical excitability and decreased sensitivity toward glutamate (Biber et al, 2008; Moidunny et al, 2010). This could potentially also occur in the stroke-lesioned brain where an upregulation of adenosine A1 receptors would most likely increase survival of penumbral neurons.
The findings discussed here show the limitations of focussing on one cytokine only, which is part of a dynamic network. Upcoming reports based on the use of transgenics with conditional and cell-specific deletion of cytokines will provide insight into these questions. Furthermore, as we now have evidence that microglia is a diverse cell population with regard to cytokine production (Clausen et al, 2008), it should be clarified what regulates microglial production of early stroke cytokines or if different subsets microglia are destined to respond by producing different cytokines. Possibly, the more general issue about cellular cytokine production can be reduced to that microglia just produce much higher levels of, e.g., TNF and IL-1 than any other cell in the neural parenchyma, while sub-threshold produced cytokines still matters, and modulate neuronal sensitivity to, in this context, ischemia. What should also be considered is that cytokines, despite their importance, only constitute a single stratum of a multi-layered immune system. Removing a single cytokine may therefore not solely modify the production of other cytokines, but also modify the production of other immune mediators and alter the sensitivity of neuronal and non-neuronal cells to these mediators. To model cytokine effects on the neuronal network at the genomic and proteomic level, in the penumbra in the early phase after experimental stroke, using models for both pMCAO and tMCAO, could be instrumental to pave the way for new strategies for inhibiting detrimental effects and promoting beneficial effects of key inflammatory cytokines in stroke.
Footnotes
Acknowledgements
Disclosure/conflict of interest
The authors declare no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.