
Abstract
Halothane is a strong inhibitor of potassium evoked spreading depression (SD) in cats. In the current study, we investigate halothane effects on induction of perifocal SD-like depolarizations, CBF, and infarct evolution in focal ischemia. Calomel and platinum electrodes measured cortical direct current potential and CBF in ectosylvian, suprasylvian, and marginal gyri. Left middle cerebral artery occlusion (MCAO) induced permanent focal ischemia for 16 hours in artificially ventilated cats (30% oxygen, 70% nitrous oxide) under halothane (0.75%, n = 8) or α-chloralose anesthesia (60 mg/kg intravenously, n = 7). Under α-chloralose, MCAO induced severe ischemia in ectosylvian and suprasylvian gyri(mean CBF <10 mL/100 g/min), and direct current potentials turned immediately into terminal depolarization. In marginal gyri, CBF reduction was mild (more than 20 mL/100 g/min), and in six of seven animals, frequent SD-like depolarizations turned into terminal depolarization at a later stage of the experiments. Under halothane, MCAO induced severe ischemia (less than 10 mL/100 g/min) and immediate terminal depolarization only in ectosylvian gyrus. In suprasylvian gyrus, residual CBF remained significantly higher (more than 10 mL/100 g/min) than under α-chloralose, whereas in marginal gyri, CBF did not differ between groups. Compared with chloralose, the number of transient depolarizations was significantly reduced in marginal gyrus, and in suprasylvian gyrus transient but significantly longer depolarizations than in marginal gyrus were recorded. Except for one animal, transient depolarizations did not turn into terminal depolarization under halothane, and infarct volume reduction was particularly seen in suprasylvian gyrus. We conclude that halothane, the most commonly used anesthetic in studies of experimental brain ischemia, has protective properties, which may depend on both cerebrovascular and electrophysiologic influences.
Spreading depression (SD) of Leão (Leão, 1944) is characterized by a transient suppression of the electrocorticogram and a negative deflection of the cortical direct current (DC) potential. Extracellular rise of potassium or glutamate has been regarded as an important factor for SD generation (Grafstein, 1956; Van Harreveld, 1959). In normal brain, SD does not injure neuronal tissue (Nedergaard and Hansen, 1988). On the other hand, focal ischemia induces SD-like depolarizations in border zones of the ischemic focus (Nedergaard and Astrup, 1986). Each of these ischemic transient depolarizations is associated with a stepwise deterioration of tissue conditions, as documented by a decrease of regional tissue PO2, probably resulting from a lack of compensatory CBF enhancement (Back et al., 1994) that accompanies SD in nonischemic tissue (Hansen et al., 1980). In a final stage, transient ischemic DC deflections may turn into terminal depolarizations (Graf et al., 1995).
We demonstrate that halothane strongly inhibits potassium-evoked SD in cats and suggest that factors other than potassium or glutamate release are involved in induction and propagation of SD waves (Saito et al., 1995a). Among those factors, intercellular communication through gap junctions may be of particular interest, since halothane has been shown to be an excellent gap junction blocker (Mantz et al., 1993). We therefore investigated whether halothane anesthesia inhibits SD-like cortical DC deflections in cat focal ischemia compared with α-chloralose anesthesia, and whether it modifies infarct evolution. A preliminary report has been published elsewhere (Saito et al., 1995b).
METHODS
In 15 cats of either sex weighing 2.5 to 4 kg, anesthesia was initialized with ketamine hydrochloride (25 mg/kg intramuscularly). The left femoral artery and vein were catheterized, and the animals were tracheotomized and immobilized with pancuronium bromide (0.2. mg/kg intravenously). Thereafter, artificial ventilation (0.75% halothane in 70% nitrous oxide and 30% oxygen) was started. Immobilization was kept throughout the experiment by continuous intravenous infusion of Ringer's solution containing 5 mg/kg gallamine triethiodide at a rate of 2 mL/kg. Mean arterial blood pressures were continuously recorded. Arterial blood gases were intermittently monitored, and PaO2 and PaCO2 were controlled within normal range (Herbert and Mitchell, 1971). Rectal temperature and skull temperature were independently controlled using a heating blanket and heat lamps, respectively. The set point temperature was 37°C. The left middle cerebral artery was prepared transorbitally, and a device for remote occlusion was implanted (Graf et al., 1986). Cats were placed in a stereotaxic holder. They received five small craniotomies, 3 mm in diameter, according to a stereotactic atlas (Reinoso-Suarez, 1961), one above the left ectosylvian gyrus (8 mm anterior/15 mm lateral), two above the left suprasylvian gyrus (4 mm and 11 mm anterior/8 mm lateral), and two above the left marginal gyrus (4 mm and 11 mm anterior/3 mm lateral). After careful dura incision, a set of electrodes for DC recording (glass micropipette, tip diameter 2 to 4 µm, filled with sodium chloride solution inserted into a calomel electrode to avoid electrode polarization; Shibata et al., 1977) and for measurement of CBF using hydrogen clearance (etched platinum iridium wires 250 µm in diameter, glass insulated up to 1 mm of the tip) were inserted with a tip-to-tip distance of 1.5 mm into each of the five regions below the craniotomies. The depth of insertion was adjusted to 1.5 mm.
Two experimental groups were studied according to the anesthetic used. In the halothane group (n = 8), halothane anesthesia was kept as described throughout the entire experimental protocol. In the chloralose group (n = 7), a bolus of α-chloralose was administered (60 mg/kg intravenously) after preparation of the animals, halothane was switched off and artificial ventilation was kept at 70% nitrous oxide/30% oxygen. To keep α-chloralose anesthesia throughout the experiment, a continuous α-chloralose infusion (5 mg/kg/hour) was started about 3 hours after the initial bolus had been injected. In this group, experimental protocols were not started before at least 2 hours after the initial α-chloralose injection had passed. Effects of ketamine on SD induction (Gorelova et al., 1987) were negligible in both groups, since measurements and experimental procedures were started at the earliest 6 hours after ketamine injection.
The experimental protocol started with control measurements of the DC potential and of regional CBF. Thereafter, the middle cerebral artery was occluded for about 16 hours. The DC potentials were recorded continuously. The CBF was measured 30 to 60 minutes and again 14 hours after middle cerebral artery occlusion (MCAO). Inclusion criterion for experimental animals was the severity of ischemia in the ectosylvian gyrus (core of ischemia): only animals showing severe CBF reduction and continued (terminal) negative shifts of the DC potential in ectosylvian gyrus were further analyzed. Experiments were terminated by perfusion fixation with 4% formalin buffer solution and removal of the brains. After paraffin embedding, 7-µm coronal sections were cut and Nissl stained. Infarct areas were evaluated using an image analyzer (Gesotec, Germany) under microscopic control. Infarct areas per section were calculated using the following formula (Swanson et al., 1990):

The volume of infarction was calculated by summing the infarct areas of consecutive sections multiplied by the distance between sections. Accordingly, separate evaluation of infarct areas and volumes of the three investigated gyri were based on the assessment of the volume of noninfarcted tissue in gyri of the left hemisphere and the volume of respective gyri in the right hemisphere. Data are expressed as means ± standard deviation. Statistical evaluation (STATISTICA, StatSoft, Tulsa, OK, U.S.A.) was performed using parametric (t test, two-way analysis of variance with Newman-Keuls post-hoc comparison) or nonparametric methods (Kruskal-Wallis, Fisher exact) to test significance (<0.05) within and between groups.
RESULTS
Physiologic variables
The experimental protocol, including MCAO, did not significantly alter physiologic variables such as blood gases, blood glucose concentration, and mean arterial blood pressure. Similarly, treatment with the two different anesthetics did not cause significant alterations of these variables (Table 1).
Physiological variables
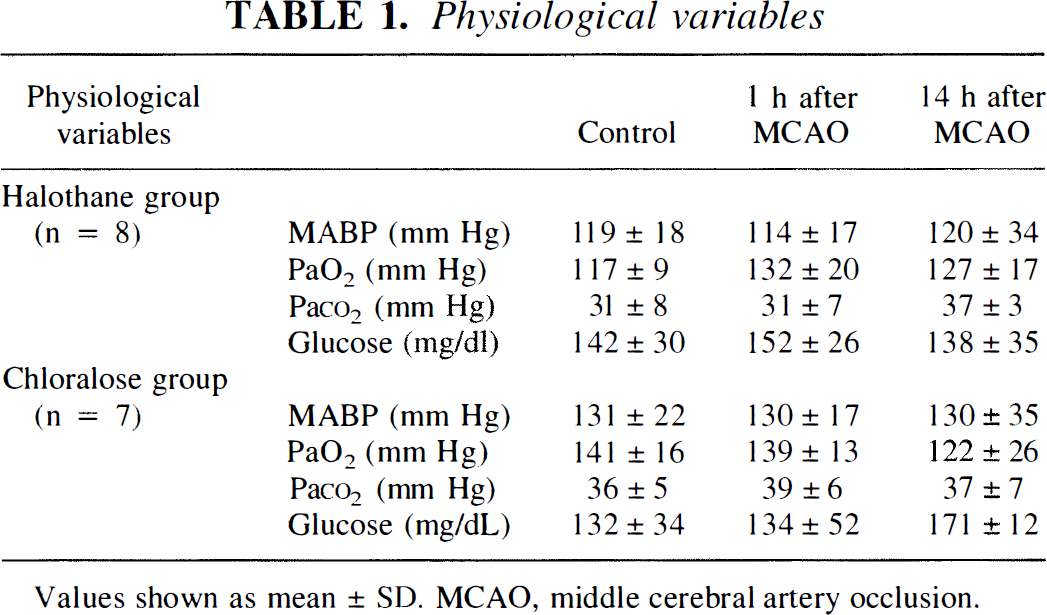
Values shown as mean ± SD, MCAO, middle cerebral artery occlusion.
Cerebral blood flow
Both during preischemic control and after MCAO, CBF was influenced by the type of anesthesia (Fig. 1). Chloralose anesthesia decreased CBF considerably during control. At the locations in marginal gyrus and suprasylvian gyrus, this decrease was significant. In the early phase (1 hour) after MCAO, CBF reduction was similar among the two groups in ectosylvian gyrus and marginal gyrus. In the suprasylvian gyrus, in contrast, chloralose reduced CBF to a deeper level than halothane. The magnitude of reduction was comparable with that observed in the ectosylvian gyrus. In the late phase (14 hours) after MCAO, the difference between the halothane and the chloralose group became more apparent: under chloralose, a further secondary CBF decrease was particularly observed in marginal gyrus, whereas under halothane, CBF values remained stable throughout the experiments.
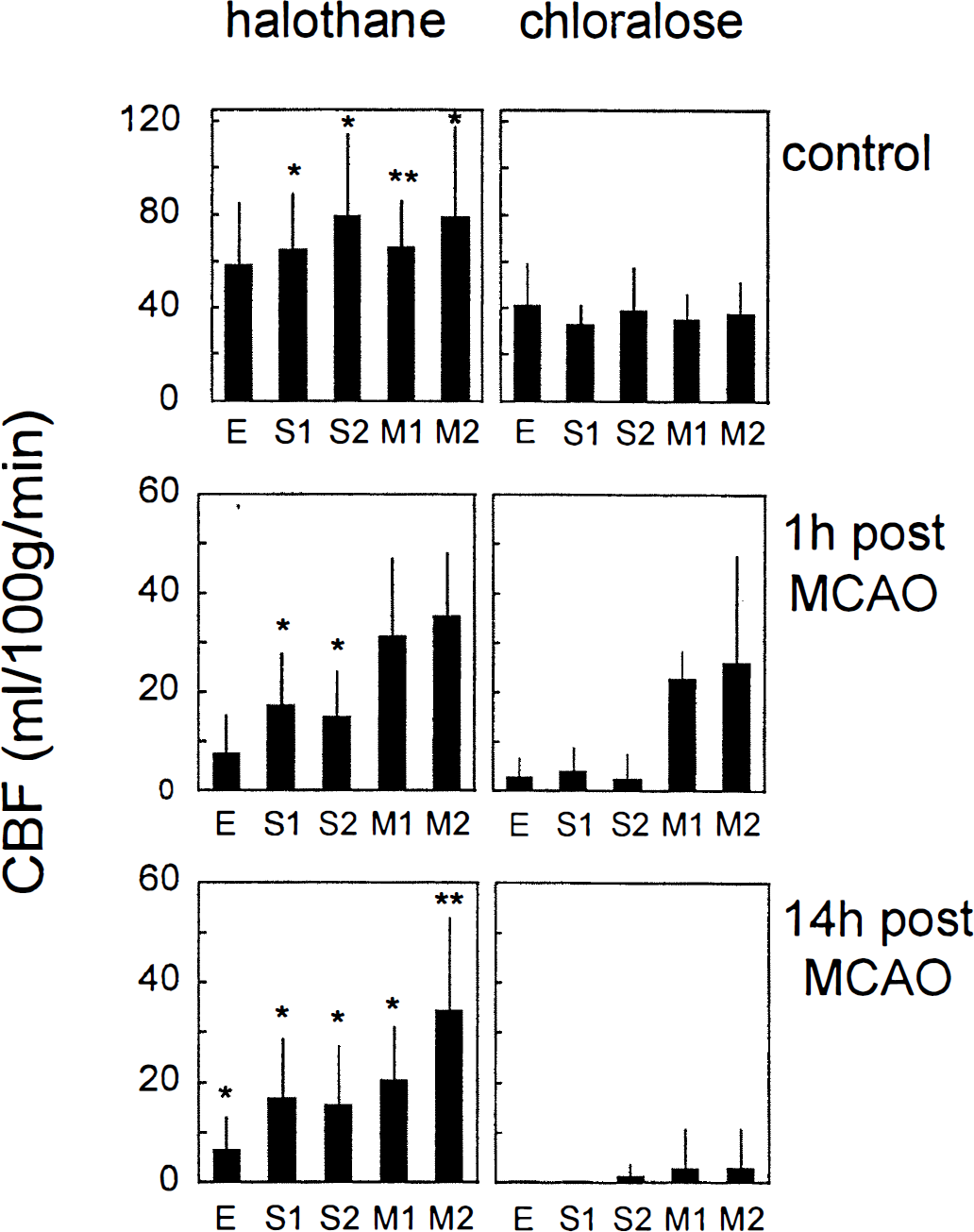
Graph of CBF before, 1 hour after, and 14 hours after middle cerebral artery occlusion in halothane- and chloralose-anesthetized animals. Electrodes were positioned in ectosylvian gyrus (E), suprasylvian gyrus (S1, S2), and marginal gyrus (M1, M2). Means ± SD are shown. *, **Significant differences between groups (Kruskall-Wallis test; P < 0.05 and P < 0.01, respectively). Notice the different scales before and after middle cerebral artery occlusion.
Direct current potentials
Under control conditions, the change from halothane to chloralose anesthesia did not influence cortical DC potentials. On MCAO, long-lasting negative DC potential shifts (terminal depolarizations) were recorded in gyri with most severe ischemia; however, the spatial pattern differed among groups (Fig. 2). In the chloralose group, terminal depolarization was seen in ectosylvian and suprasylvian gyri (electrodes E, S1, S2), whereas in the halothane group, these long-lasting DC deflections were restricted to the ectosylvian gyrus (electrode E). At locations outside of the ischemic core, transient depolarizations were recorded. in the chloralose group, frequent SD-like depolarizations appeared in marginal gyrus. In six of seven animals of the chloralose group, these transient depolarizations turned into terminal depolarization within 4.5 to 14 hours after MCAO (Fig. 2, recordings at electrodes M1 and M2 of the chloralose-anesthetized animal). In the halothane group, SD-like depolarizations were less frequently observed in marginal gyrus, and only in one of eight animals was delayed permanent depolarization observed within the observation period. The induction rate of SD-like depolarizations measured in marginal gyrus during the first 4 hours after onset of ischemia was significantly higher under chloralose than under halothane (Fig. 3). Mean duration and amplitude of transient SD-like deflections did not differ significantly among groups and amounted to 2.0 ± 0.7 minutes (mean ± standard deviation) and 13.6 ± 10.3 V, respectively, in the halothane compared with 2.8 ± 0.9 minutes and 14.0 ± 6.2 V in the chloralose group. Interestingly, recordings in the suprasylvian gyrus of halothane-anesthetized animals revealed another type of transient depolarizations (Fig. 2, recordings at electrode M1 and M2 of the halothane-anesthetized animal) with a similar amplitude but much longer duration than DC deflections recorded in both groups in the marginal gyrus. This type of prolonged depolarization was observed in four animals of the halothane group. A comparison of DC recordings in this group revealed a significantly longer duration of transient depolarizations in suprasylvian compared with marginal gyrus (Fig. 4). The amplitude varied considerably but did not show a significant difference between gyri. Similarly, the number of induced DC deflections was almost the same in the two gyri of these halothane-anesthetized animals (1.9 ± 2.8 in the suprasylvian gyrus compared with 2.4 ± 4.4 in marginal gyrus during first 4 hours after arterial occlusion). In two animals of the halothane group, immediate terminal depolarization was recorded on MCAO in the suprasylvian gyrus, and no DC alterations were observed in one animal.
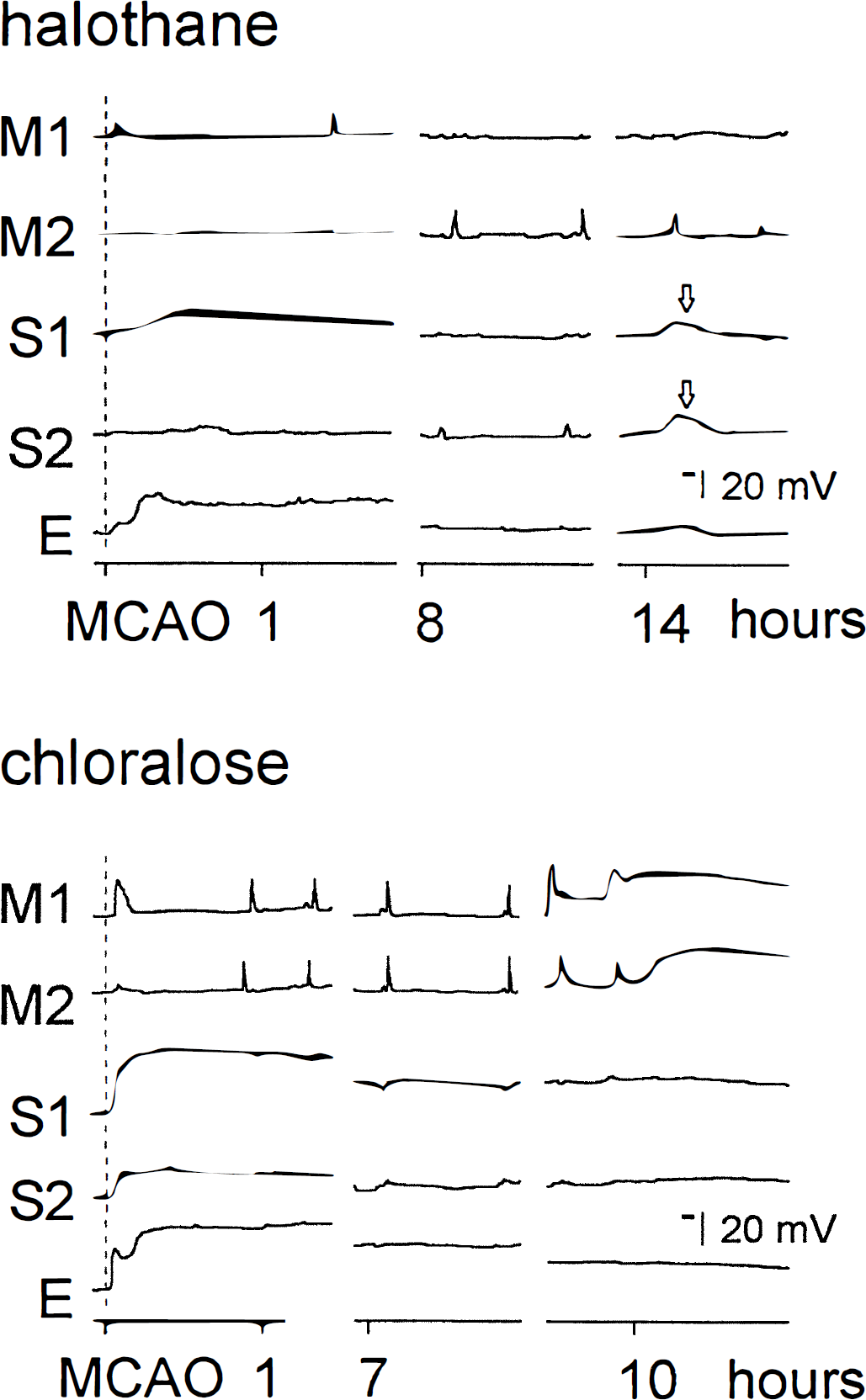
Plots of original recordings of direct current (DC) potential after middle cerebral artery occlusion (MCAO) obtained with five electrodes in the cortex of two individual cats under halothane and chloralose anesthesia, respectively. Electrodes were positioned in marginal gyrus (M1 and M2), suprasylvian gyrus (S1 and S2), and in ectosylvian gyrus. Notice the immediate negative direct current shifts (terminal depolarization) in suprasylvian gyrus (S1 and S2) of the chloralose-anesthetized cat compared with prolonged but transient longer depolarizations (arrows) in suprasylvian gyrus under halothane anesthesia. Also notice the high induction rate of recurrent spreading depression-like depolarizations and the delayed transition to terminal depolarization in marginal gyrus (M1 and M2) of the chloralose-anesthetized cat.
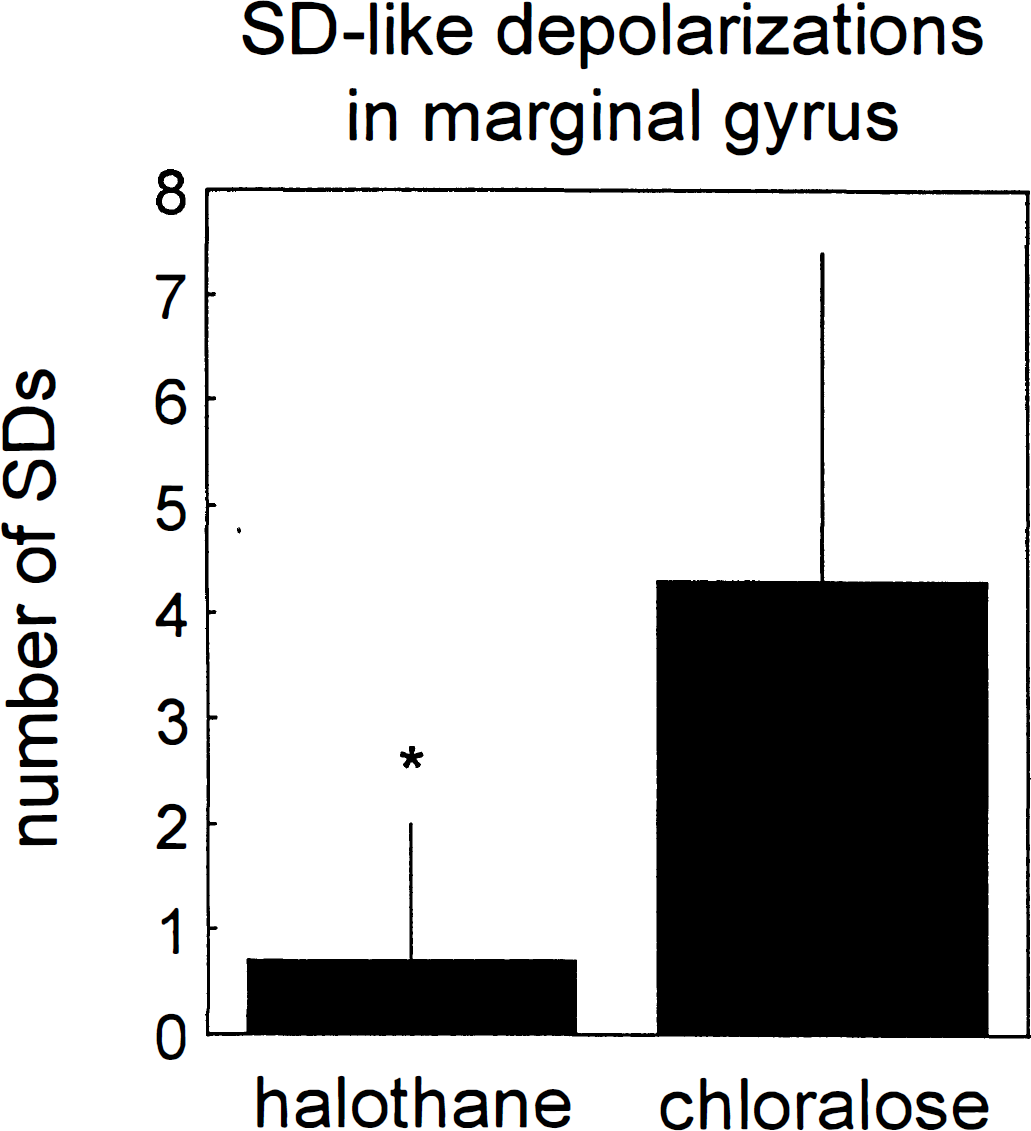
Number of transient spreading depression-like depolarizations after middle cerebral artery in marginal gyrus (M) of halothane- and chloralose-anesthetized cats. Shown are means ± SD in the first 4 hours after onset of ischemia. *Significant difference between groups (t test; P < 0.05).
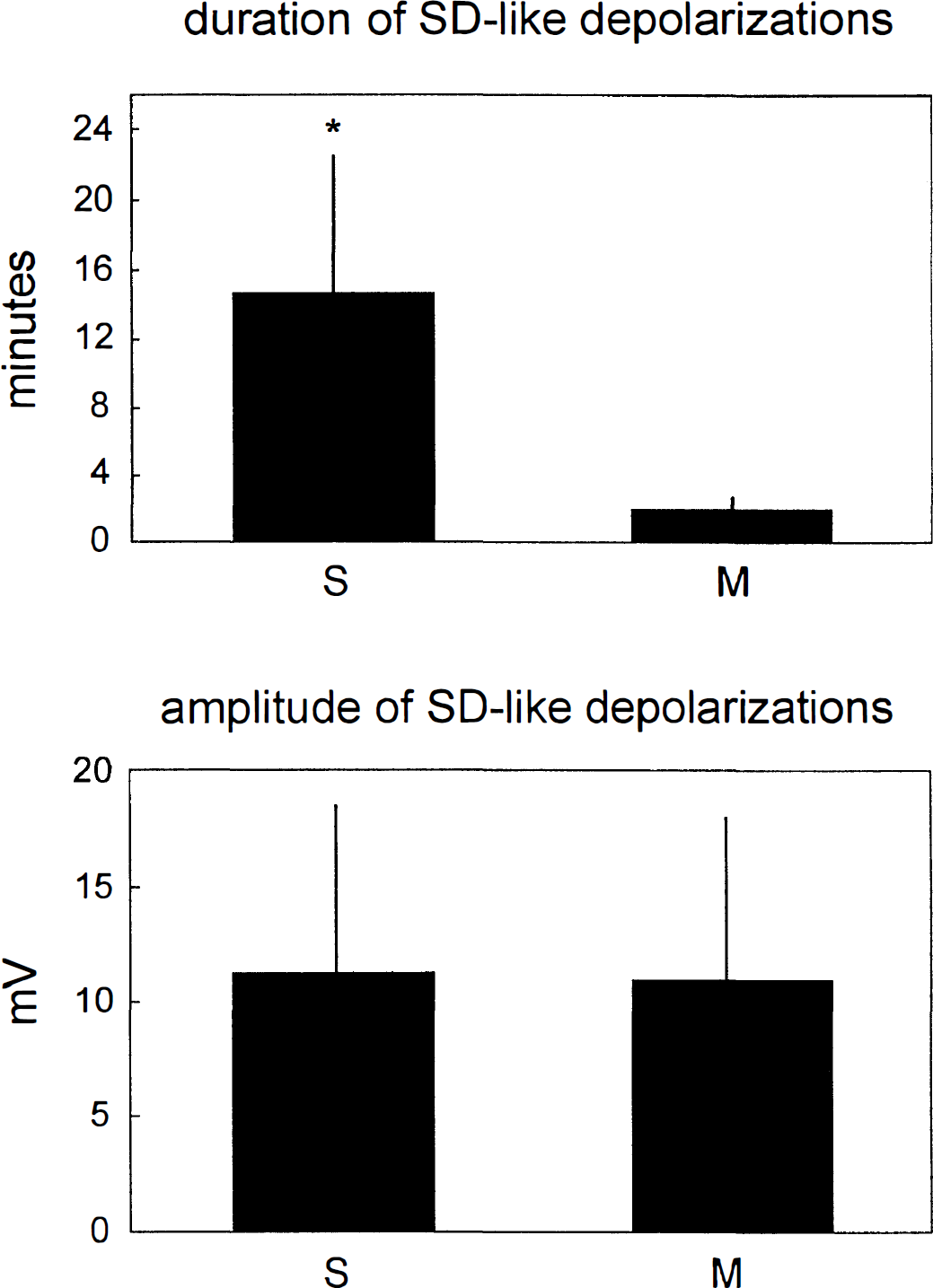
Duration and amplitude of transient depolarizations after middle cerebral artery occlusion in marginal (M) and suprasylvian gyrus (S) of halothane-anesthetized cats. Shown are means ± SD. *Significant difference between gyri (t test; P < 0.05).
Histologic damage
One animal of the chloralose group showing the earliest transition from SD-like into terminal depolarization about 4.5 hours after MCAO was excluded from histologic examination because infarct areas had not yet been sufficiently demarcated. In the other animals, analysis of infarct areas at eight stereotaxic levels and subsequent calculation of infarct volumes revealed that infarcts generally were smaller under halothane than under chloralose (Fig. 5, total infarct volume), and that this effect was specifically seen in the suprasylvian gyrus (Fig. 5, infarct volume in cortical gyri), whereas in ectosylvian gyrus and marginal gyri, no significant difference was observed.
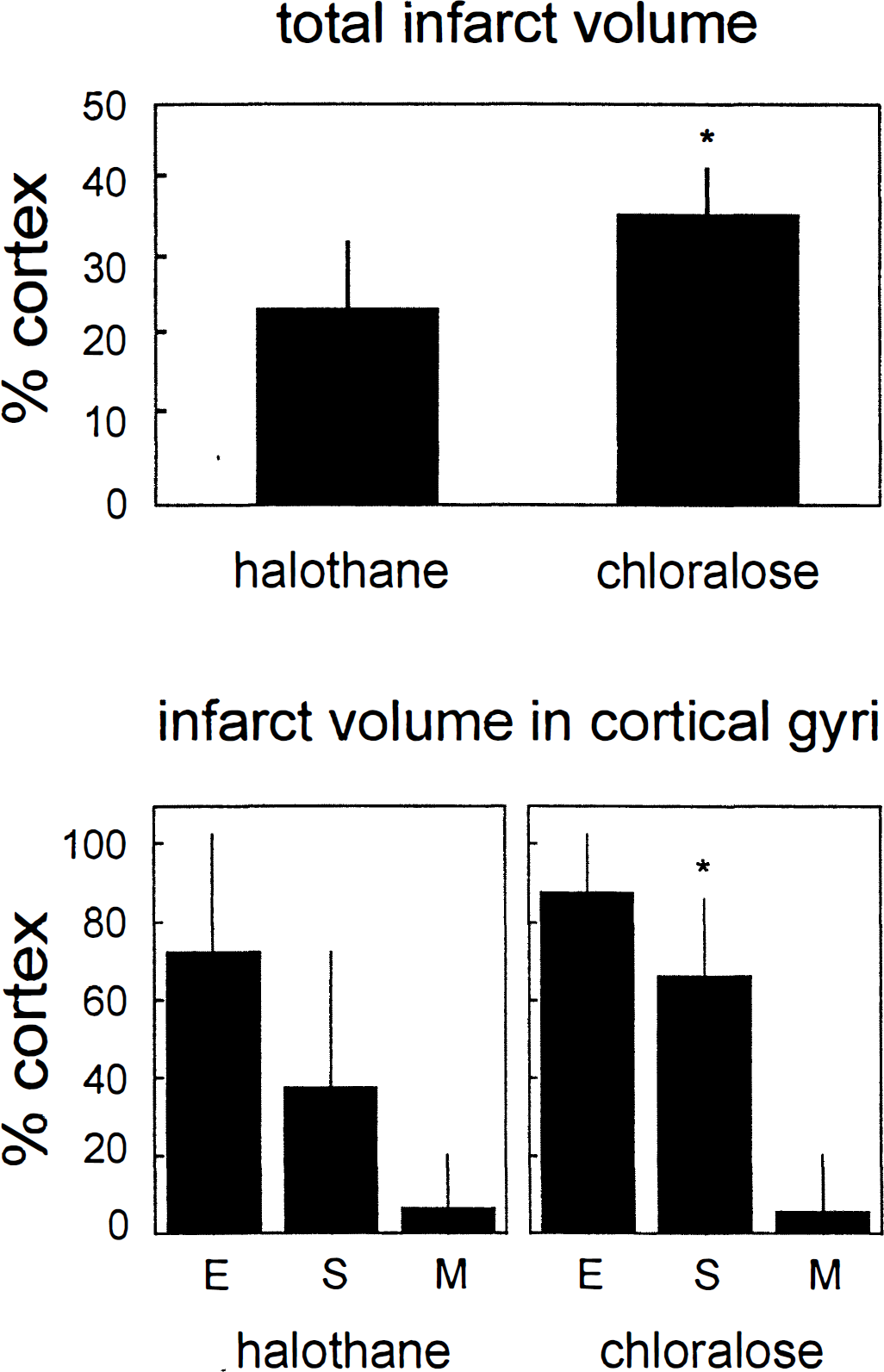
Graph of total cortical infarct volume (top) and of infarct volumes in three electrophysiologically investigated cortical gyri (bottom) evaluated about 16 hours after middle cerebral artery occlusion in halothane- and chloralose-anesthetized animals. E, ectosylvian gyrus; S, suprasylvian gyrus; M, marginal gyrus. Means ± SD are shown. *Significant difference between anesthesia groups (t test for total infarct volume and two-way ANOVA for infarct volume in cortical gyri, respectively; P < 0.05).
DISCUSSION
These results demonstrate that halothane, compared with α-chloralose, increases perifocal residual CBF in the suprasylvian gyrus and almost prevents secondary perifocal CBF reduction in the later phase of the experiments. Furthermore, halothane reduces the expansion of areas undergoing immediate terminal depolarization after MCAO. In part of the cats, it rather produces a “sub-critical” electrophysiologic state in the suprasylvian gyrus characterized by prolonged transient depolarizations. It also reduces the number of perifocal SD-like depolarizations in the-in relation to the middle cerebral artery territory-more peripheral marginal gyrus, and it almost abolishes the occurrence of secondary, delayed terminal depolarization seen regularly in α-chloralose-anesthetized animals. Finally, halothane reduces infarct volume, an effect seen most clearly in the suprasylvian gyrus.
Compared with the awake state, volatile anesthetics, including halothane and sevoflurane, reduce brain damage in animals subjected to transient focal cerebral ischemia (Warner et al., 1993). Protection by halothane persists even if brain temperature is controlled at normothermic levels (Warner et al., 1995). Since other anesthetics such as barbiturates seem to possess higher protective properties than halothane (Smith et al., 1974; Michenfelder and Milde, 1975), an agent-to-agent comparison may obscure such benefit in studies comparing the effects of different anesthetics. In case of our study, protection by halothane became apparent because of the choice of α-chloralose as a second treatment, and therefore, our findings confirm the results obtained in rats. Since halothane is not ranked high on the scale of neuroprotectants, one might look on the less effective α-chloralose to closely mimic the awake state in studies of brain ischemia.
The question rises as to whether the shown circulatory and electrophysiologic improvements account for the protective effect of halothane. Anesthetics, as other agents, may counteract ischemic damage by various routes (Todd and Warner, 1992) including reduced energy depletion from suppression of electrical activity, enhanced oxygen and glucose supply caused by increased intraischemic CBF, interference with pathophysiologic processes such as disturbance of ion homeostasis, or accumulation of excitotoxins during ischemia and postischemic free radical formation.
In the early phase immediately after onset of ischemia, suppression of neuronal activity by anesthesia may delay depletion of energy phosphates and thus terminal depolarization, but this prolongation of a “time window” probably is only in the minute range and therefore irrelevant for effective brain protection in severe ischemia (Todd and Warner, 1992). The scene changes apparently if substrate supply is simultaneously enhanced. Halothane has long been known to increase basal CBF (Wollman et al., 1964). As demonstrated in transient focal ischemia in cats by a study of Helfaer and others (1990), it seems to affect ischemic CBF to a lesser degree, even though recalculation of absolute from relative CBF values given in the study reveals a tendency for enhancement of residual CBF during halothane if compared with α-chloralose or pentobarbital anesthesia. This result is in good agreement with our findings, particularly because the authors of the aforementioned study report on significant but regionally restricted CBF elevations, which is similar to our finding of regional CBF enhancement during ischemia in the suprasylvian gyrus under halothane. Since the effect lasted for the full observation period of about 16 hours permanent ischemia, we suspect it to prevent the secondary decrease of perifocal CBF and thus perhaps the occurrence of secondary, delayed terminal depolarization seen regularly under α-chloralose (Graf et al., 1995) but rarely under halothane. We conclude, therefore, that perifocal CBF elevation by halothane contributes to a reduction of cerebral infarct evolution.
Does the decline of infarct volume result from CBF amelioration through preventing the induction or propagation of periinfarct SD-like depolarizations? It is impossible to conclusively answer this question with the given results, since circulatory and electrophysiologic effects, namely CBF enhancement and inhibition of transient perifocal depolarizations, occurred concomitantly. To approach the question, we differentiate between two observed electrophysiologic phenomena: (1) short transient depolarizations seen in marginal gyrus both under halothane and α-chloralose, which we denominate SD-like depolarizations, and (2) prolonged transient depolarizations seen exclusively in the suprasylvian gyrus under halothane, which we denominate transient ischemic depolarizations, following the rationale of Nedergaard and Hansen (1993).
As to SD-like depolarizations in marginal gyrus, we demonstrated their inhibition by halothane. Back and associates (1994) have shown that SD-like depolarizations in perifocal areas may enhance tissue hypoxia caused by lack of compensatory CBF elevations typical for potassium-evoked SD in normal brain, thereby worsening infarct evolution. The induction of perifocal SD-like depolarizations was inhibited, and infarct volume was reduced by use of MK-801 (Iijima et al., 1992) or application of mild hypothermia (Chen et al., 1993). Furthermore, Mies and coworkers (1993) have shown that the number of SD in perifocal areas correlates with size of infarcts, and that induction of perifocal SD-like depolarizations increases in parallel to augmentation of infarct volumes. Since in the current study infarction in marginal gyri was not significantly affected by halothane, we assume that reduction of infarct volume from increased perifocal CBF provides one good explanation for limitation of this type of transient depolarizations by halothane.
Blockade of perifocal SD-like depolarizations, however, also resembles the inhibition of potassium-evoked SD by halothane found in cats under physiologic conditions (Saito et al., 1995a; Piper and Lambert, 1996). It has been speculated that SD are generated by simultaneous depolarization of many cells including neurons and glia (Higashida et al., 1974; Sugaya et al., 1975). Such simultaneous depolarization might be facilitated by cortical synchronization (e.g., under the influence of α-chloralose). It is well known that electrical activity of the brain is differently affected by the two anesthetics used. Unlike halothane, α-chloralose produces an electroencephalogram characterized by intermittent high-amplitude waves, documenting electrical synchronization (Winters and Spooner, 1966). Interestingly, hippocampal neurons, which are known to exhibit a high degree of synchronization, seem to be particularly susceptible to SD (MacVicar and Dudek, 1980; Somjen et al., 1992). From in vitro studies, evidence also exists for the involvement of astrocytic gap junction opening in SD propagation (Somjen et al., 1992; Nedergaard et al., 1995). Intercellular communication through gap junctions controls the exchange of electrolytes and metabolites between cells and may be involved in neuronal synchronization and signaling from astrocytes to neurons (Dermictzel and Spray, 1993; Nedergaard, 1994). Since in cat cortex the portion of glia cells is large compared with that of neurons (Tower, 1954), and since glial activity protects against SD induction (Szerb, 1991), synchronous depolarization of a large population of cells including glia and glial gap junction opening might be a prerequisite for SD induction. Our observation of a facilitating influence of synchronizing agents such as α-chloralose on SD induction is in line with this hypothesis. We propose that halothane, a well-known gap junction blocker (Mantz et al., 1993), inhibits SD in cats through the closure of gap junctions (Saito et al., 1995a). This may be true not only under physiologic conditions but also in perifocal areas of ischemia. It would be interesting to investigate how these findings relate to the awake state.
An additional explanation for ameliorative effects of halothane may be provided by a reduction of ischemia-induced glutamate accumulation, which has been reported for another volatile anesthetic isoflurane (Patel et al., 1995). Ischemic glutamate elevation may be responsible for induction of recurrent SD-like depolarizations in perifocal ischemic areas and thereby produces its devastating influence (Hossmann, 1994). Decreased ischemic glutamate elevation by halothane, in turn, thus could be responsible for the reduction of SD-like depolarizations found in our study and for infarct volume reduction.
In rats, halothane does not strongly inhibit SD (Verhaegen et al., 1992). The smaller portion of glia in rat compared with cat cortex may provide a reasonable explanation for halothane being a less effective SD blocker in this species. As to our knowledge, a study of halothane's influences on perifocal SD in rats has not been performed. The fact, however, that halothane is capable of reducing infarct volume in rats (Warner et al., 1993) leads to the assumption that other than electrophysiologic influences may be more relevant for brain protection by this anesthetic. Among those factors, amelioration of residual CBF and reduction of ischemic glutamate accumulation may be equally important.
As to prolonged transient DC deflections (ischemic transient depolarizations; see earlier) seen in the suprasylvian gyrus under halothane, we were not able to investigate differential anesthetic influences because we did not see this type of depolarization under α-chloralose. Its occurrence under halothane, however, may underline the importance of this anesthetic for ischemic residual circulation. In our opinion, halothane favors collateralization, thereby producing less steep blood flow gradients from the ischemic core to the ischemic periphery. The described elevation of residual CBF in suprasylvian gyrus probably is a correlate of this phenomenon, as is the lack of immediate terminal depolarization, the persistence of transient deflections over the relatively long observation period of 16 hours, and finally, the reduction of damage in this gyrus. We believe that the investigated region in the suprasylvian gyrus precisely reflects a subcritical state typically referred to as the ischemic penumbra. From recent positron emission tomography and nuclear magnetic resonance studies, evidence exists that such a region will deteriorate in most instances in the further course of infarct evolution (Heiss et al., 1994; Kohno et al., 1995). Halothane may have some capacity to delay this process. In our study, this fact is best documented by the prevention of delayed terminal depolarization in areas adjacent to the ischemic focus such as the marginal gyrus and by the reduction of infarct volume. Whether this ameliorative capacity of halothane has implications for clinical use cannot be decided by the current investigation. However, our study demonstrates that effects of anesthetics have to be carefully considered for the design of experimental study of ischemic penumbra.