
Abstract
We investigated the effect of hyperglycemia on the initiation and propagation of spreading depression-like peri-infarct ischemic depolarization (SD) induced by focal cerebral ischemia in rats. Peri-infarct SD were monitored during the initial 15 minutes after remotely induced middle cerebral artery occlusion (MCAO) using serial diffusion weighted magnetic resonance imaging. Maps of the apparent diffusion coefficient (ADC) were calculated and ADC decreases were monitored over time. Hyperglycemic rats (n = 6) had a significant prolongation of the time from induction of MCAO to the start of the ADC decrease as compared with normoglycemic control rats. The time to the maximal ADC decrease was significantly delayed and recovery of transient ADC declines in the area adjacent to the ischemic core was significantly faster in hyperglycemic rats. We conclude that hyperglycemia delays the terminal depolarization in the ischemic core and supports a faster repolarization in severely mal-perfused penumbral tissue after SD, which reflects the increased availability of energy substrates in the state of hyperglycemia.
Keywords
The restoration of the extracellular and intracellular ion homeostasis after spreading depression (SD) is a process that demands high energy (Csiba et al., 1985; Kocher, 1990). The additional need for energy can be met by a higher perfusion rate in normal tissue (Lauritzen et al., 1987). In contrast, the peri-infarct tissue, which is already critically mal-perfused, is not able to cope with the increased energy demand (Nedergaard and Astrup, 1986; Back et al., 1994). Consequently, SD-like transient depolarization in the peri-infarct zone can increase the final infarct volume (Iijima et al., 1992; Busch et al., 1996). However, depolarization during ischemia can be delayed (presumably because of a postponement of ion pump failure), and repolarization enhanced, if the brain has improved access to energy stores such as those available in hyperglycemic animals (Nedergaard and Astrup, 1986; Hansen, 1978).
Diffusion-weighted magnetic resonance imaging is able to detect transient declines of the apparent diffusion coefficient (ADC) that represent SD-associated disturbances of the ion homeostasis and the concomitant water shift in the peri-infarct tissue (Latour et al., 1994; Röther et al., 1996a, b ). We investigated the dynamics of the initial SD-related ADC transients in hyperglycemic and normoglycemic rats after remote middle cerebral artery occlusion (MCAO) to test the hypothesis that an increased blood glucose level will result in a faster recovery of SD-like transients in the peri-infarct tissue. Dynamic contrast-enhanced susceptibility magnetic resonance imaging was performed immediately after the serial ADC mapping in order to correlate the ADC waveforms with the degree of perfusion deficit.
METHODS
Male Sprague-Dawley rats (weighting 280 to 380 g) were rendered diabetic (n = 6) by a single intraperitoneal injection of streptozotocin (60 mg/kg/body weight) 36 hours before the investigation. An intravenous injection of 50% glucose (1 mL) was administered one hour before MCAO. Normoglycemic rats from a previously published study (Röther et al., 1996b) served as the control group (n = 7) and were re-evaluated according to a standardized protocol. Physiological status was monitored during the experiment. Focal cerebral ischemia was induced remotely in the magnet using a modified intraluminal suture model (Röther et al., 1996a).
Ultrafast diffusion-weighted MR imaging (2 Tesla GE CSI spectrometer) was performed using a spin-echo echo planar imaging technique (repetition time 2 seconds, echo time 88 milliseconds, field of view 40 mm, 2.5 mm slice thickness, 64 × 64 pixel matrix, 1 average, 3 coronal slices, 6 b-factors varying from 0 to 1780 s/mm2) covering an imaging time of 17 minutes. The suture was advanced to occlude the middle cerebral artery 2 minutes after the start of the echo planar imaging diffusion scans. Perfusion imaging was performed immediately after the diffusion-weighted magnetic resonance imaging using the same multi-slice echo planar imaging technique, but with repetition time = 1.5 seconds, to follow a bolus injection of 0.3 mmol/kg Gd-DTPA (Magnevist, Schering, Berlin, Germany).
Data evaluation was performed as detailed recently (Röther et al., 1996b). Briefly, all pixels without ADC recovery after MCAO were integrated into one region of interest (ROI 1) and were defined as ischemic core. Multiple (5 to 7) adjacent ROI, each containing 2 pixels, were defined starting from the cortical region immediately next to the ischemic core and ending with the cortical region next to the hemispheric sulcus (Fig. 1). Parameters were evaluated according to a standardized protocol (Fig. 2). Normalized perfusion ratios of the signal intensity decline and the bolus-to-peak time were calculated based on the perfusion raw data.
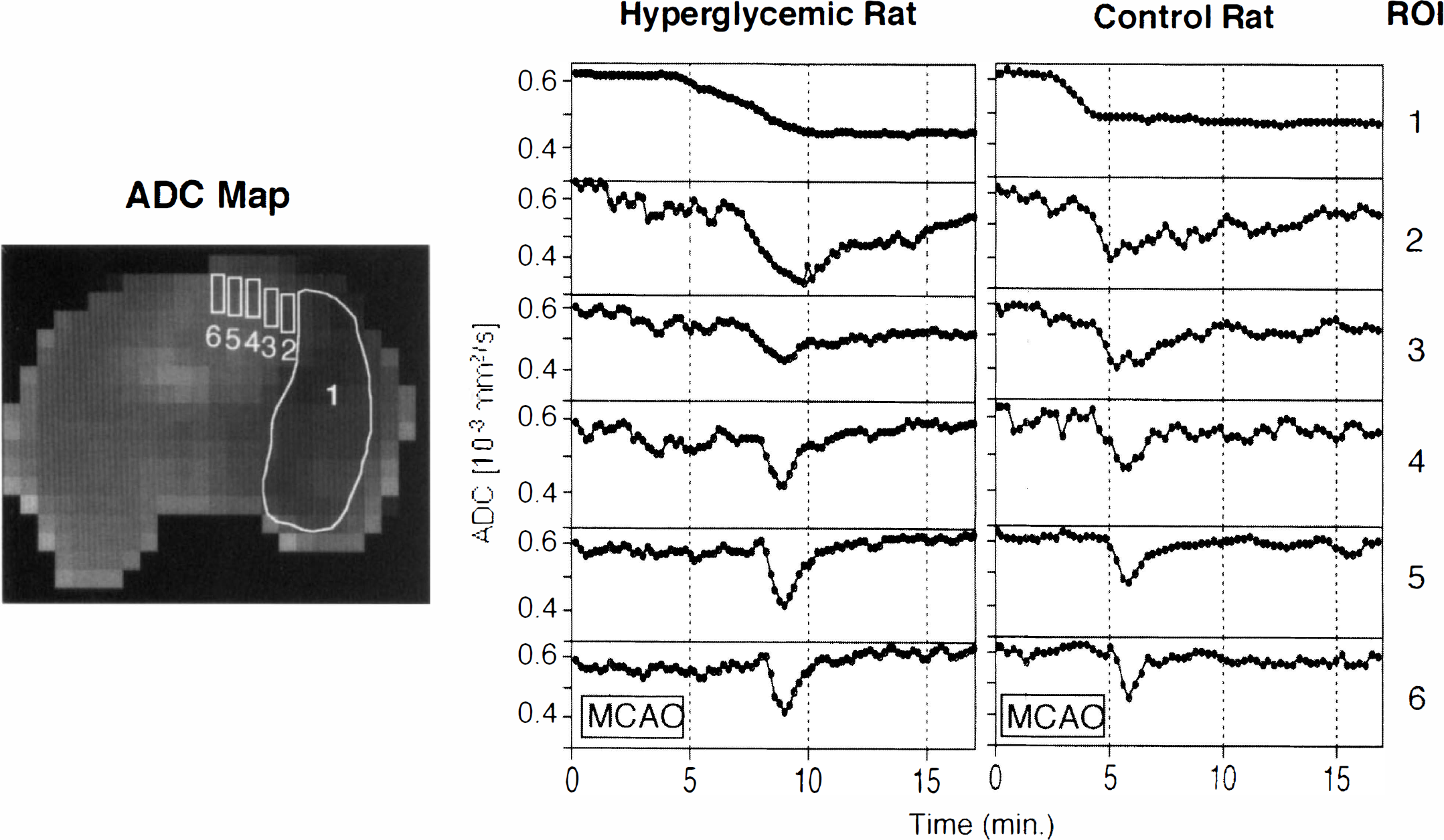
The standardized evaluation of ADC changes over time is shown. The infarct core was defined as the region with no ADC recovery (ROI 1). To assess this region, the ADC time series was evaluated for each slice on a pixel-by-pixel basis and all pixels with no ADC recovery were integrated. Using this ischemic core as a reference, multiple cortical ROI (2 to 6 in this case), each consisting of two pixels, were defined adjacent to each other until the hemispheric sulcus. Relative to a normoglycemic rat, the hyperglycemic animals show a longer latency time from MCAO to the start of the ADC decrease, a longer duration between the start of the ADC decline to the maximum ADC decline, and a faster recovery of the ADC after SD in ROI 2 and 3 that may represent the ischemic penumbra. ADC, apparent diffusion coefficient; MCAO, middle cerebral artery occlusion; ROI, region of interest.
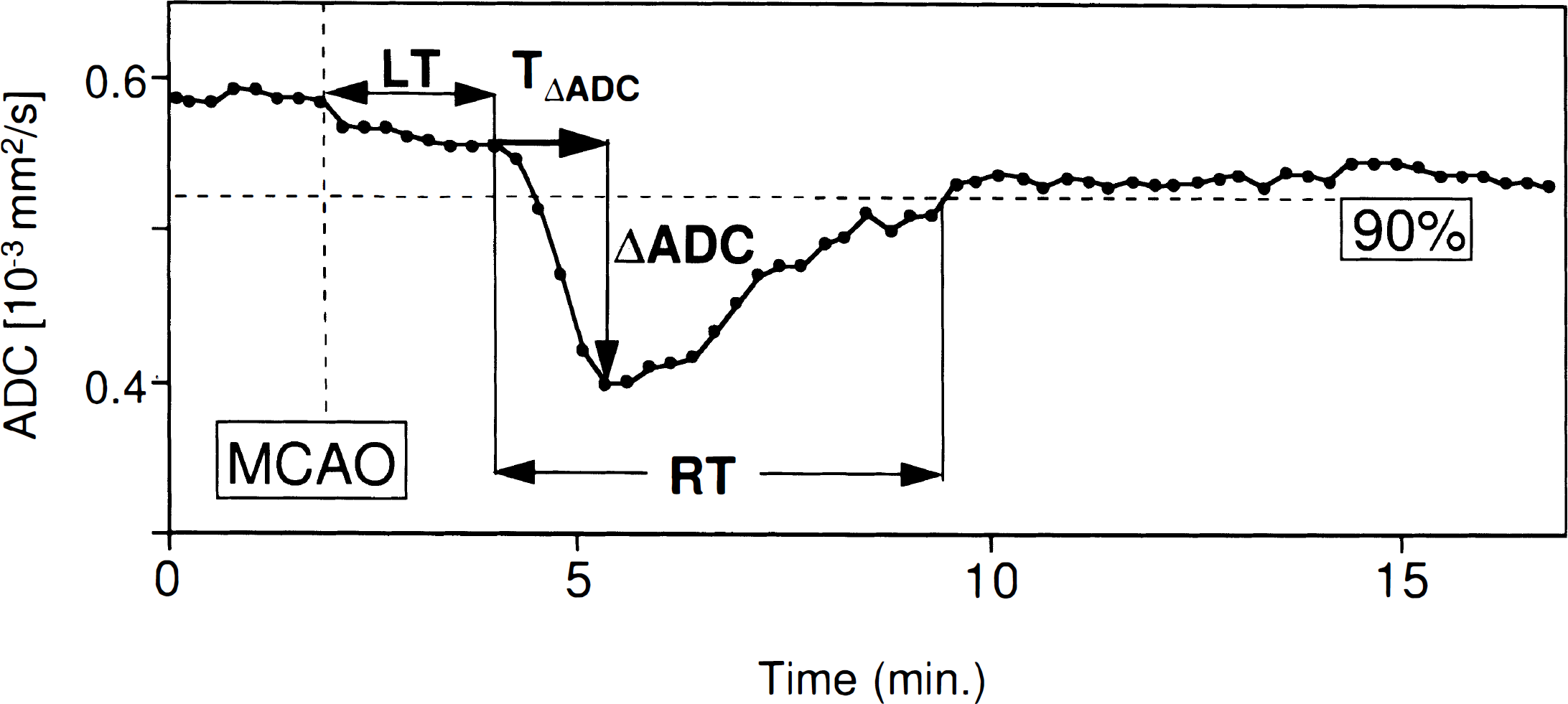
Parameters characterizing the temporal evolution of ADC changes were evaluated for the time from MCAO to the onset of the ADC decrease (latency time), time from the start of the ADC decrease to the maximal ADC decline, time from the start of the ADC decrease to the ADC recovery (in case of transient changes; RT), and the maximal ADC decrease (ΔADC). A return of the ADC value to 90% of control was considered as full recovery. ADC, apparent diffusion coefficient; LT, latency time; MCAO, middle cerebral artery occlusion; RT, recovery time; TΔADC, time from the start of the ADC decrease to the maximal ADC decline.
STATISTICS
Analysis of variance was used for the statistical analysis of repeated measurements of the ROI Nos. 1 to 7. Statistical significance was accepted when P < 0.05. All values are expressed as mean and standard deviation.
RESULTS
All physiological variables were kept within the physiological range (Table 1). Five hyperglycemic and five normoglycemic control animals showed an area without ADC recovery (ROI 1) that could be differentiated from areas with transient ADC decreases in the periphery (ROI 2 to 7). The baseline ADC value before MCAO and the maximal ADC decrease after MCAO were not significantly different in all ROI between the hypoglycemia and control rats (P = 0.38 and P = 0.48, respectively). The latency time between MCAO and the start of the ADC decrease was significantly prolonged in hyperglycemic animals as compared with controls (P = 0.0001). This prolongation was most pronounced in the ischemic core of hyperglycemic rats. The time from the start of the ADC decrease to the maximal ADC decline (TΔADC) was significantly prolonged in hyperglycemic animals in all ROI as compared with controls (P = 0.0035). This delay was most marked in the ischemic core and was less pronounced in ROI further away from the core (Table 2).
Physiological parameters
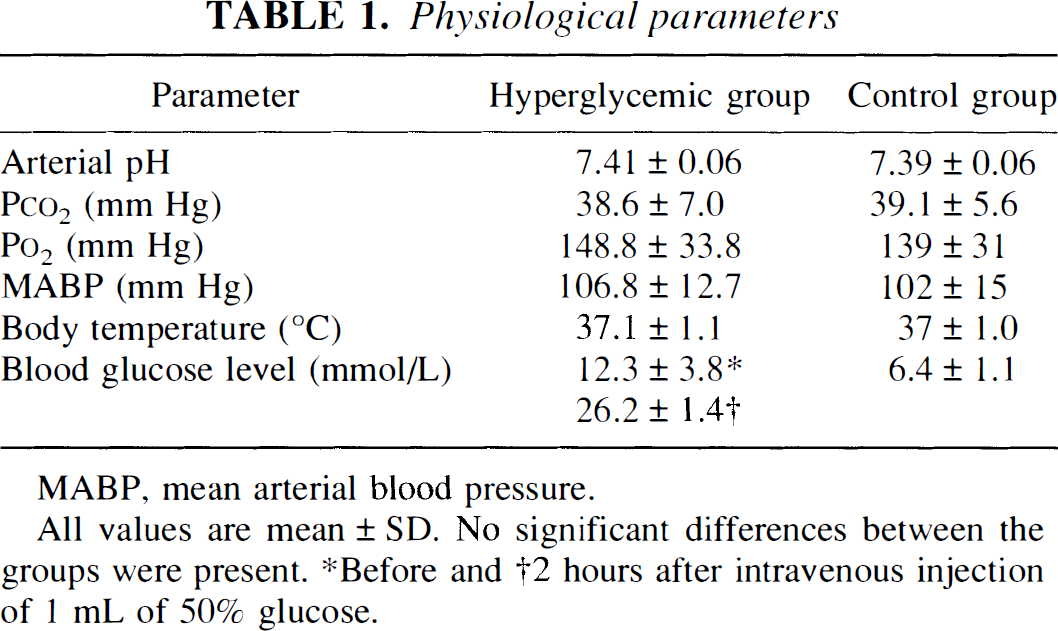
MABP, mean arterial blood pressure.
All values are mean ± SD. No significant differences between the groups were present.
Before and †2 hours after intravenous injection of 1 mL of 50% glucose.
Summary of the temporal evolution of the apparent diffusion coefficient of water in hyper- and normoglycemic rats
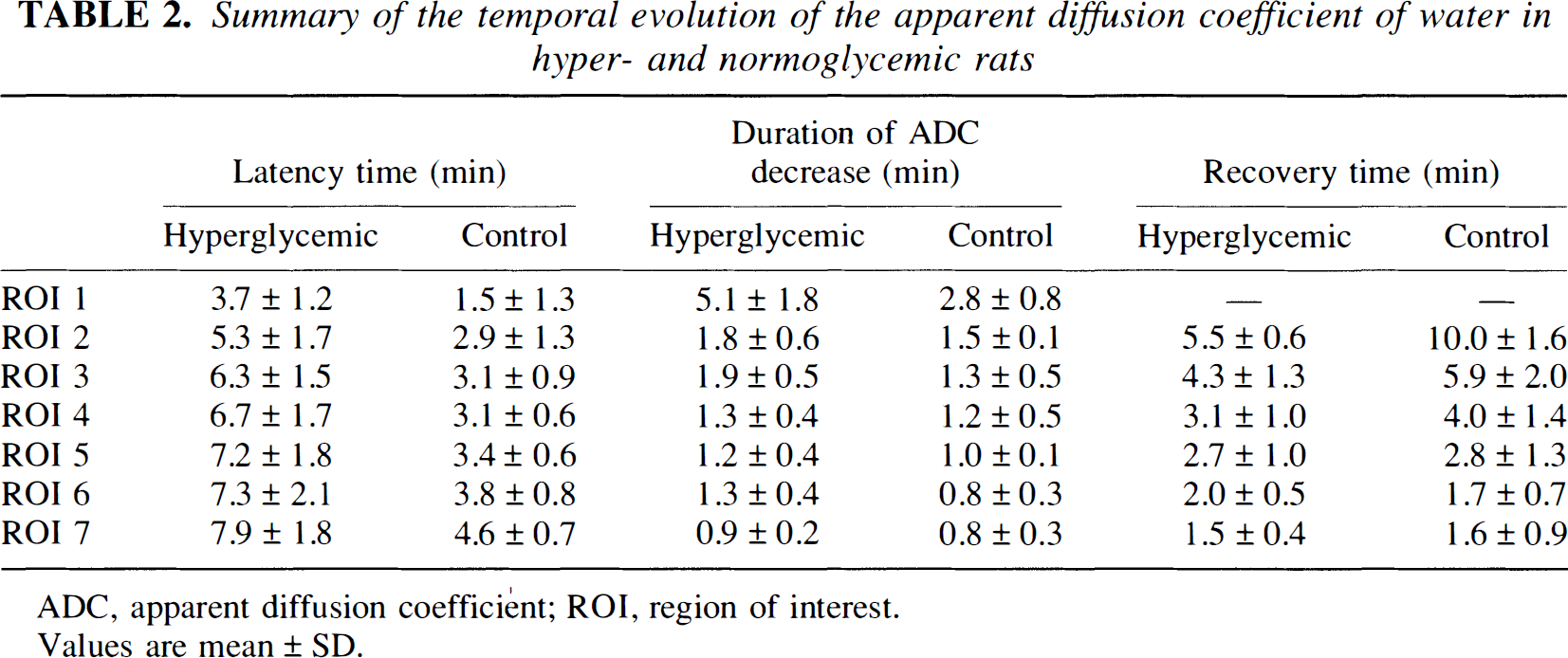
ADC, apparent diffusion coefficient; ROI, region of interest.
Values are mean ± SD.
In hyperglycemic animals, ADC waveforms in tissue adjacent to the ischemic core region (ROI 2) recovered significantly faster to 90% of the baseline as compared with the controls (P = 0.009). Very short-lasting ADC declines were observed in the remote peri-infarct tissue of the anterior and posterior cerebral artery territory in both groups (ROI 6 and 7).
Severe perfusion deficits were found in the ischemic core and the adjacent ROI, whereas only moderate perfusion deficits were found in ROI located in the periphery. There was no difference between both groups in signal intensity ratio (P = 0.87) and bolus peak arrival (P = 0.43). The reader is referred to Röther et al., 1996b for more details about the correlation of the ADC recovery time with the tissue perfusion.
DISCUSSION
Apparent diffusion coefficient decreases in the ischemic core
The scope of the present study was to investigate the influence of acute hyperglycemia on the initial development of focal cerebral ischemia and SD-like ADC transients in the peri-infarct tissue. Tissue with acute ischemia undergoes an efflux of potassium and an influx of sodium and calcium (Astrup et al., 1977; Harris et al., 1981) and it is thought that the ADC decrease in ischemic tissue reflects the associated water shifts that go along with the ionic imbalance (Moseley et al., 1990). Similarly to the ischemic threshold for the release of potassium, the ADC decrease in ischemic tissue depends on a critical threshold of cerebral blood flow and on the amount of energy available to maintain ion homeostasis (Hoehn-Berlage et al., 1995).
Therefore, the most likely explanation for an increased latency time from MCAO to the start of the ADC decrease is a delay of the breakdown of transmembrane ion gradients due to increased energy stores in hyperglycemic rats. This view is supported by studies from Hansen who found a delay of the time from cardiac arrest to the start of the anoxic depolarization in hyperglycemic rats (Hansen, 1978). Furthermore, an increase of blood glucose was shown to maintain intracellular levels of ATP for a prolonged time after ischemia (Wagner et al., 1992; Hsu et al., 1994) and therefore is available to maintain the function of the energy dependent ion channels.
Whereas our interpretation of preservation of cerebral metabolism in hyperglycemic rats holds true for the ischemic core (ROI 1), the increased latency time in the peri-infarct tissue with transient ADC decreases (ROI 2 to 7) is thought to be the result of the delayed release of trigger factors like potassium and glutamate from the core region.
Our finding of a prolonged time to reach ADC baseline (TΔADC), which is most pronounced in the ischemic core of the hyperglycemic animals, seems to indicate that not only the start of the terminal depolarization is delayed, but that the process of the membrane potential breakdown is spread over a longer time period than in normoglycemic animals. It is known that the release of intracellular potassium during ischemia does not follow an all-or-none law but occurs in different stages depending on the severity of the perfusion deficit (Branston et al., 1977). Astrup et al. have shown that the rate of the rise of the extracellular potassium can be modulated by the preischemic metabolic rate (Astrup et al., 1980). This leads us to conclude that the exhaustion of the energy stores and the concomitant shift of fluid from the extracellular to the intracellular compartment is delayed in hyperglycemia and is mirrored in the longer TΔADC.
Transient apparent diffusion coefficient decreases in the peri-infarct tissue
In normal brain tissue, the repolarization after SD is coupled to an immense increase of glucose metabolism and blood flow (Mies and Paschen, 1984; Lauritzen, 1987; Mayevsky and Weiss, 1991). It therefore seems plausible to assume that the depletion of the intracellular energy stores and a reduced delivery of energy substrates, as is the case in the penumbra, severely delays the restoration of the membrane potential after SD. Indeed, the metabolic stress that is imposed on the peri-infarct tissue by SD is reflected by a marked delay of the ADC recovery in severely mal-perfused tissue, thus emphasizing that the increased energy demand after SD in the peri-infarct tissue is not followed by a sufficient supply of energy substrates (Röther et al., 1996b).
An increased duration of SD in the peri-infarct tissue as compared with KCl-induced SD has been previously reported and emphasizes the delayed repolarization after SD in tissue with a restricted energy availability (Nedergaard and Astrup, 1986; Back et al., 1994). Conversely, a faster recovery after SD has been observed in the infarct rim of hyperglycemic rats (Nedergaard and Astrup, 1986). These aforementioned studies of SD are in agreement with our own MR-derived temporal profiles of SD in normoglycemic and hyperglycemic rats.
We therefore conclude that our observation of shorter ADC recovery times in ROI close to the ischemic core in hyperglycemic rats, as compared with normoglycemic rats, reflects a faster restoration of the membrane potential because of a higher level of glucose. This interpretation is further corroborated by identical perfusion gradients in both experimental groups with severely impaired perfusion in the ischemic core and gradually improving perfusion in the periphery. Assuming that the ADC recovery reflects the membrane repolarization (de Crespigny et al., 1996), the elevated brain glucose concentration level favors the restoration of the ion homeostasis in the severely mal-perfused region close to the ischemic core. In contrast, only minor differences of the ADC recovery times between both groups are observed farther away in the ischemic periphery (ROI 4 to 7). In these peripheral ROI with only moderate to mild perfusion deficits, the tissue perfusion seems to be sufficient to restore the transmembrane ion gradients and the elevated level of brain glucose in hyperglycemic animals does not evidently accelerate the process of repolarization.
Although a prolonged preservation of ATP by an extended anaerobic glycolysis in hyperglycemia delays the breakdown of the transmembrane ion gradients, the initial beneficial effect of elevated energy levels will be overcome by accentuated lactate accumulation and pH decrease in the long term (Wagner et al., 1992; Widmer et al., 1992).
Footnotes
Acknowledgement
We gratefully acknowledge statistical support from J. Schulte-Moenting, Institute for Biomathematics and Statistics, University of Freiburg, FRG.