
Abstract
The suture model for middle cerebral artery occlusion (MCAO) was used to induce acute ischemia in rats remotely within a magnetic resonance (MRI) scanner. Serial MR diffusion weighted imaging (DWI) was performed during remote MCAO using an echo planar imaging technique. MR perfusion imaging was performed before and after occlusion using the bolus tracking technique. Transient apparent diffusion coefficient (ADC) changes were detected in six of seven rats as early as 2.7 ± 1.5 min post MCAO. ADC values declined transiently to 70.1 ± 6.0% of control and recovered to 95.5 ± 6.8% of control within 3.3 ± 2.9 min. These ADC changes propagated bidirectionally away from the ischemic core with a speed of 3.0 ± 1.1 mm/min. Transient ADC decreases only occurred in ischemic areas characterized by moderately decreased tissue perfusion. Propagation toward cortical regions with severe tissue perfusion deficits was not detected. DWI can detect the earliest dynamic, reversible ADC changes in the ischemic tissue. The speed of propagation of the decreasing ADC wave, the waveform characteristics, and the occurrence in moderately perturbated tissue are compatible with cortical spreading depression.
Keywords
Magnetic resonance (MR) techniques of perfusion (Rosen et al., 1990) and diffusion weighted imaging (Le Bihan et al., 1986) can provide information on the status of brain tissue with high spatial and temporal resolution. In animal models, in vivo MR measurements of the apparent diffusion coefficient (ADC) highlight regions of early ischemic damage within minutes after the stroke (Moseley et al., 1990; Minematsu et al., 1992; de Crespigny et al., 1993). The shift of water from the extracellular to the intracellular space occurring shortly after onset of ischemia is considered to be the cause of the ADC decrease (Sotak et al., 1992). Changes in diffusion-weighted MR imaging (DWI) after stroke have been correlated with cerebral blood flow measurements and the regional distribution of ATP, glucose, lactate, and pH. The measurement of water diffusion by means of DWI proved to be the method of choice in detecting the earliest changes of ischemic nerve cell injury (Hossmann et al., 1994).
Cortical spreading depression (CSD) is a pathophysiological event characterized by a short-lasting cell membrane depolarization (Leão, 1944). CSD can be induced by a variety of cortical stimuli including potassium chloride (KCl) or glutamate application, electrical or mechanical stimulation, and also occurs secondary to ischemia (Nedergaard and Astrup, 1986). Under normoxic conditions, CSD is not followed by permanent neuronal damage (Nedergaard and Hansen, 1988) and the depression of the neuronal activity is compensated by increased glucose metabolism and blood flow during the repolarization phase (Kocher, 1990). The periinfarct tissue, however, is in a marginal metabolic state so that repolarization and compensation after CSD is not easily achieved (Back et al., 1994). The increased demand for blood flow and glucose delivery to this tissue, which is at risk for infarction, is suspected to be one of the reasons for progressive neuronal injury (Nedergaard and Hansen, 1988). Recent studies have proved the capability of MR imaging to detect reversible metabolic events accompanying CSD after cortical application of KCl. Measurements of ADC changes (Latour et al., 1994; Hasegawa et al., 1995) as well as magnetic susceptibility variations due to probable changes in vascular deoxyhemoglobin concentration (Gardner-Medwin et al., 1994) have detected waves of CSD propagating along the cortex with velocities of 3–4 mm/min. Spontaneous, reversible ADC declines in the periinfarct tissue, resembling CSD, have been reported in two studies after middle cerebral artery occlusion (MCAO) in rats (Gyngell et al., 1994; Hasegawa et al., 1995). MR detection of the earliest episodes of periinfarct CSD after acute ischemia is handicapped because of the lack of an appropriate model to produce ischemia within the magnet of the MR scanner. We have developed a modification of the suture model for MCAO in rats (Longa et al., 1990) comparable to recently described models (Kohno et al., 1995), which enables us to visualize CSD in the very beginning of acute ischemia using high-speed ADC measurements made during and after the MCA occlusion.
METHODS
Animal preparation
Male Sprague–Dawley rats (300–380 g, n = 13) were anesthetized with ketamine (40 mg/kg i.m.) and xylazine (4 mg/kg i.m.). Catheters were placed into a femoral vein for contrast agent injection, and into a femoral artery to take samples for blood gas analysis. A tracheotomy was performed and the rats were ventilated; anesthesia was maintained with 0.75–1.5% halothane. SaO2 and heart rate were measured continuously via a pulse oximeter. Core temperature was monitored and maintained at 37 ± 1.0°C using a warm-air circulation system. Physiological parameters were kept within the normal range of pH 7.25–7.45, Paco2 35–50 mm Hg and Pao2 80–150 mm Hg. Six rats were excluded from further evaluation because they developed no lesion (n = 3), only small basal ganglia lesions (n = 2), or exhibited an ischemic lesion before the occluding device was advanced (n = 1). Branches of the right carotid artery were exposed and the external carotid and pterygopalatine arteries were tied off. A PE-50 polyethylene tube (37 cm long) containing the occluding device was prepared. The occluding device consisted of a monofilament line (Mono Line, 10 LB., Fitec International, Memphis, TN, U.S.A.) with a 4–0 nylon thread (Ethilon 4–0, Ethicon, Inc., U.S.A.) glued to its end (Fig. 1). The tip of the suture was rounded by heating. After ligation of the proximal common carotid artery, the extracranial internal carotid artery was temporarily occluded with a vascular clip. The PE-50 tube containing the occluding device was then introduced into the common carotid artery and was attached by ligations to the chest wall. The tip of the occluding device was positioned at the skull base by advancing it through the tube before the animal was fixed in a Plexiglas holder with ear and tooth bars. MCAO was achieved in the magnet by further advancing the tip of the suture 9–12 mm into the intracranial internal carotid artery (Fig. 2).
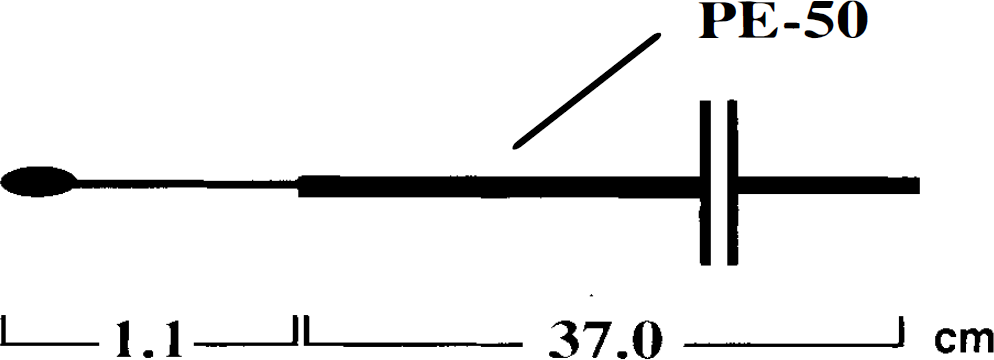
Occluding device for remote middle cerebral artery occlusion.
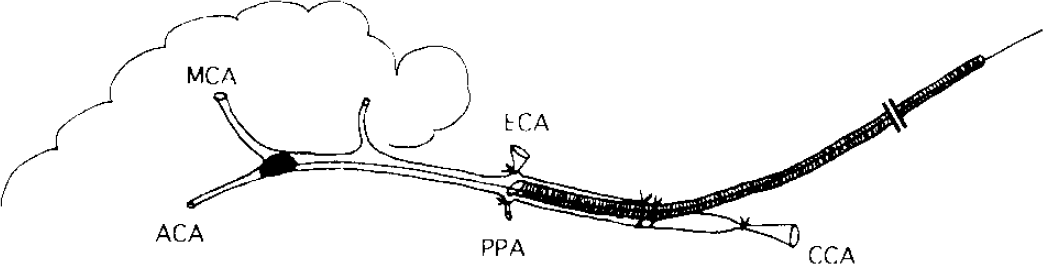
Schematic drawing of the rat model of remote middle cerebral artery (MCA) occlusion. CCA, common carotid artery; ACA, anterior communicating artery; PPA, pterygopalatine artery; ECA, external carotid artery.
MR measurements
Imaging was performed at 2 T using a spin-echo echo planar (EPI) technique (TE 88 ms, field of view 40 mm, 2.5-mm slice thickness, 64 × 64 pixel matrix, one average). Serial DWI was performed using a repeated sequence of six or eight diffusion gradient amplitudes with b factors varied from 0 to 1,780 s/mm2. Diffusion weighting was applied along the Z direction (long axis of the brain). Each image was acquired in 100 ms with a recycle time (TR) of 2 s, so that data for each complete ADC calculation was acquired in 12–16 s. One (n = 4) or three adjacent (n = 3) slices were applied. Perfusion imaging was performed using the same multislice EPI technique, but with TR = 1.5 s and without diffusion weighting, to follow a bolus injection of 0.3 mmol/kg Gd-DOTMA (Ninon Medi-Physics Co. Ltd., Chiba, Japan). The imaging protocol began with a DWI scan to screen for animals with preocclusion ischemia, followed by a perfusion examination to map the baseline perfusion pattern. Serial DWI with 512 EPI images and a total imaging time of 17 min followed. The suture was advanced 1–2 min after the start of the EPI diffusion scan. To ascertain whether successful MCA occlusion had been achieved, a second perfusion experiment and a high-resolution spin-echo diffusion experiment (slice thickness = 1 mm) was performed immediately after the end of the 512 EPI images to more accurately map the ischemic area.
Data processing
The 512 EPI diffusion images were processed off-line using customized image display software (MRVision Co., Menlo Park, CA, U.S.A.) to yield serial ADC maps, from which various regions of interest (ROIs) in the brain were manually defined. Control ADC measurements were made in the contralateral cortex. For the localization of the ischemic core and transient, propagating ADC changes, serial ADC maps were displayed in a cine loop.
ROI measurements were performed in three to nine studied cortical areas that showed transient ADC changes. The temporal changes of the ADC values in different ROIs were graphically analyzed. From these graphs, the time from MCAO to the onset of the ADC decrease, time to the point of maximum decrease, and time to the point of ADC recovery (in case of transient changes) was evaluated for each ROI. The velocity of propagation was calculated from the temporal shift of the point of the maximum ADC decrease in correlation to the spatial distance of the center of the ROI. Areas of permanent and transient ADC decrease were expressed as a percentage of the ischemic hemisphere.
The perfusion data were processed to generate relative blood volume (rCBV) (integral of the ΔR2 transit curve) and bolus arrival time maps for each slice (de Crespigny et al., 1993). The ROIs previously defined on the ADC maps were transferred to the rCBV, and arrival time images and regional perfusion were classified qualitatively as normal (rCBV and arrival time comparable to control hemisphere), moderately reduced (normal or reduced rCBV, delayed bolus arrival), or severely reduced (low rCBV, delayed or absent bolus peak).
RESULTS
The physiological variables remained within the normal range during the entire experiment and are summarized in Table 1.
Physiological variables

Table 2 presents a comprehensive summary of the ADC changes. Control ADC values (in units of 10−3 mm2/s) measured in the cortex of the contralateral hemisphere remained stable throughout the experiment. These values were pooled, resulting in a mean ADC of 0.67 ± 0.05 (mean ± SD). Serial ADC measurements exhibited a permanent decline of ADC values in the basal ganglia, the subcortical white matter, and in cortical areas in six of seven animals. At the end of the 17-min experiment time, ADC values had declined to 71.1 ± 7.8% (0.44 ± 0.08) of the initial ADC value measured before MCAO. The area of permanent ADC decline measured as the percentage of the ipsilateral hemisphere was 31.5 ± 14.3%. These areas showed a corresponding severe perfusion deficit as depicted by black areas on the rCBV maps and severely delayed or absent bolus peak (displayed on the bolus arrival time maps as bright areas).
Characterization of apparent diffusion coefficient (ADC) changes in the ischemic core and the periphery
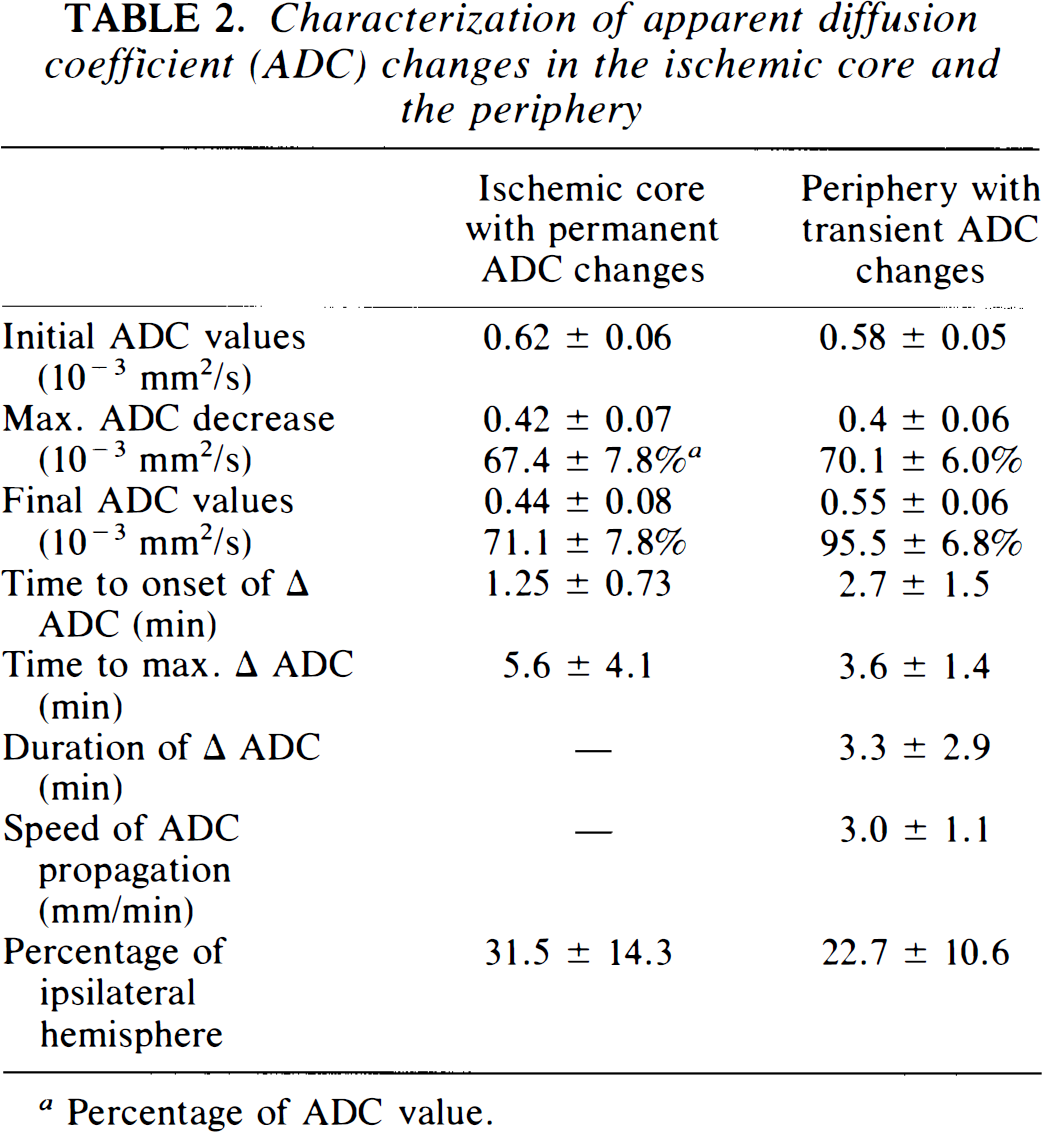
Percentage of ADC value.
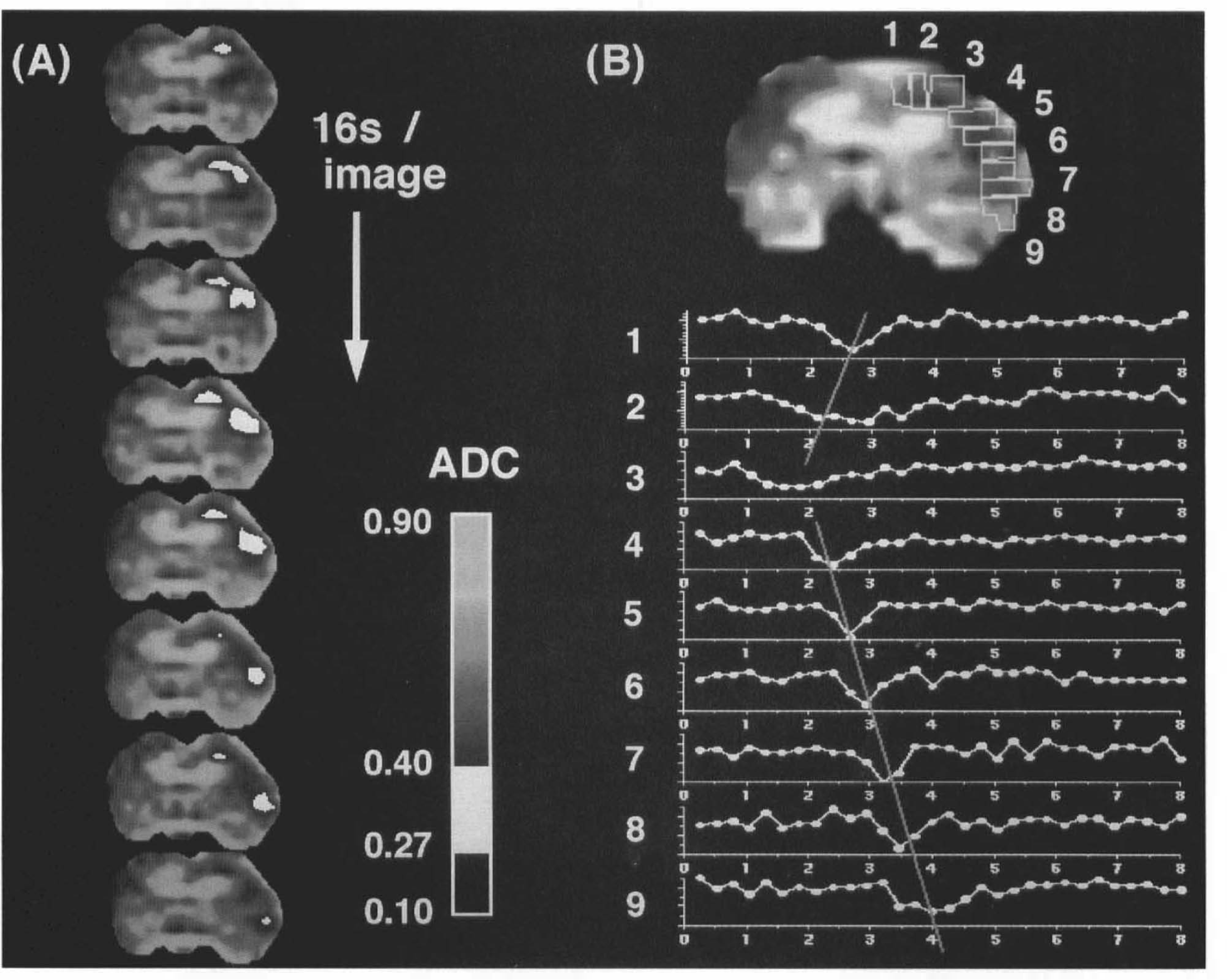
Transient ADC changes in cortical ROIs were detected in six of seven animals. These ADC changes were restricted to the cortex and did not occur within the deep white matter. ADC values declined to 70.1 ± 6.0% (0.4 ± 0.06) of the preocclusion level and then recovered to 95.5 ± 6.8% (0.55 ± 0.06).
The duration of these transient changes varied strongly with the distance of the ROI from the ischemic core, with more gradual changes adjacent to the lesion and more rapid changes further away. The mean duration was measured as 3.3 ± 2.9 min. The latency time, from MCAO until the onset of the transient ADC decrease, was 2.7 ± 1.5 min and the time to the maximum ADC change was 3.6 ± 1.4 min. The wave of ADC decrease propagated over the cortex with an estimated speed of 3.0 ± 1.1 mm/min. Only one wave of spreading ADC decrease was observed in each animal during the 17-min experiment time. Different modes of propagation were detected: (a) In two animals the transient ADC decrease started in the ischemic tissue and moved bidirectionally along the cortex. This mode was only observed in animals that did not present with a large cortical ischemic involvement in that particular slice, (b) In three animals with extended cortical ischemia the ADC decline started in the ischemic tissue and propagated towards viable regions near the midline (Fig. 3).
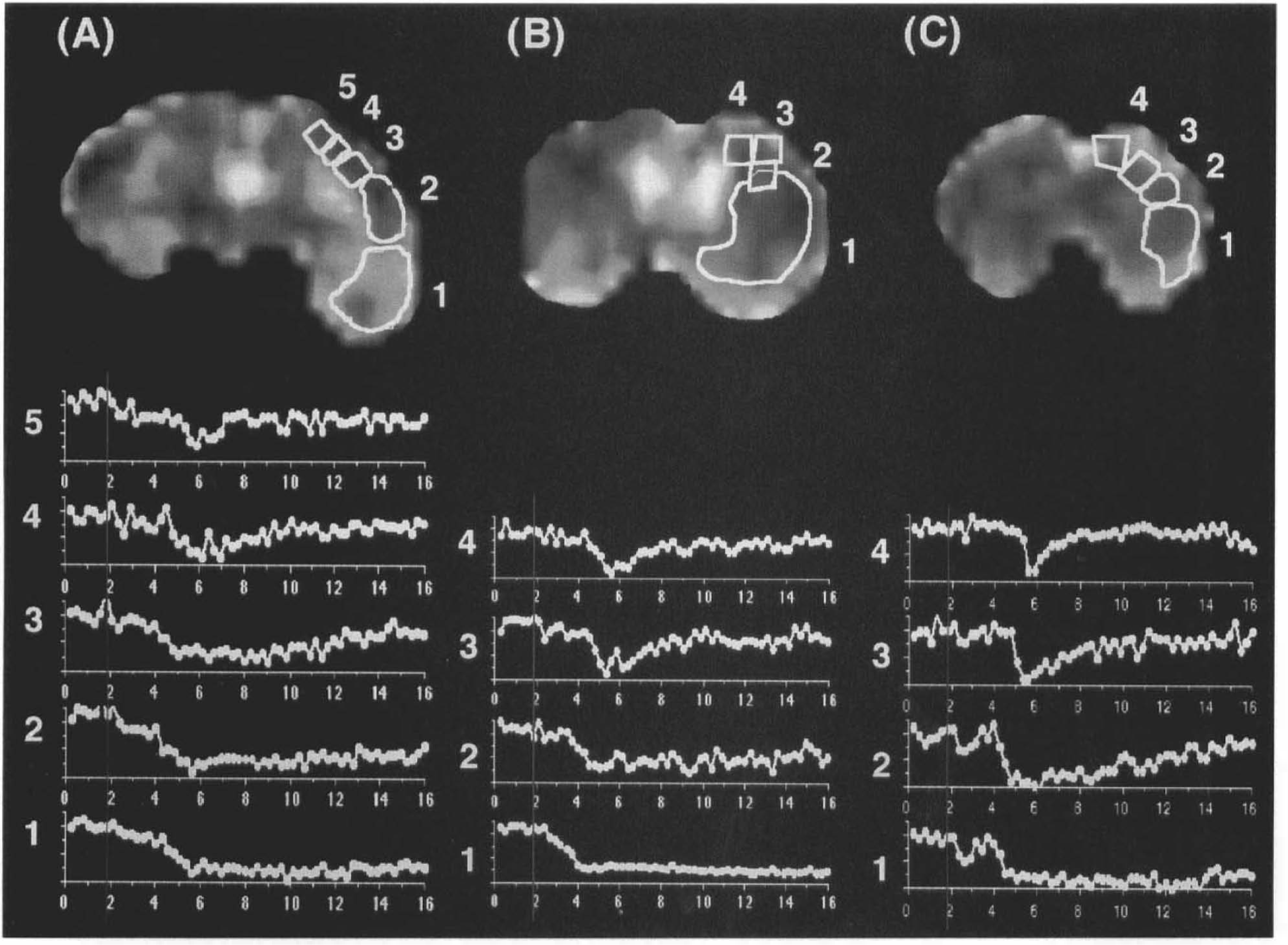
Apparent diffusion coefficient (ADC) maps from three slices of a rat brain: posterior
The area that displayed transient ADC changes was calculated as 22.7 ± 10.6% of the ipsilateral hemisphere. Areas with transient ADC decrease occurred in the periphery of the ischemic core characterized by only moderately decreased tissue perfusion. To match areas of transient ADC decrease in the ADC maps with the information provided by the perfusion data, ROIs of the cortex exhibiting transient ADC decrease were transferred to rCBV and bolus delay maps. This was possible because correlating slices for diffusion and perfusion measurements were acquired and perfusion imaging was performed immediately after the end of the 17-min diffusion experiment. The transferred ROIs were then compared with the contralateral normal hemisphere. As shown in Fig. 4, areas with transient ADC changes were characterized by a delayed contrast bolus transit (indicated as bright cortical areas) and by reduced rCBV (indicated as less bright cortical areas).
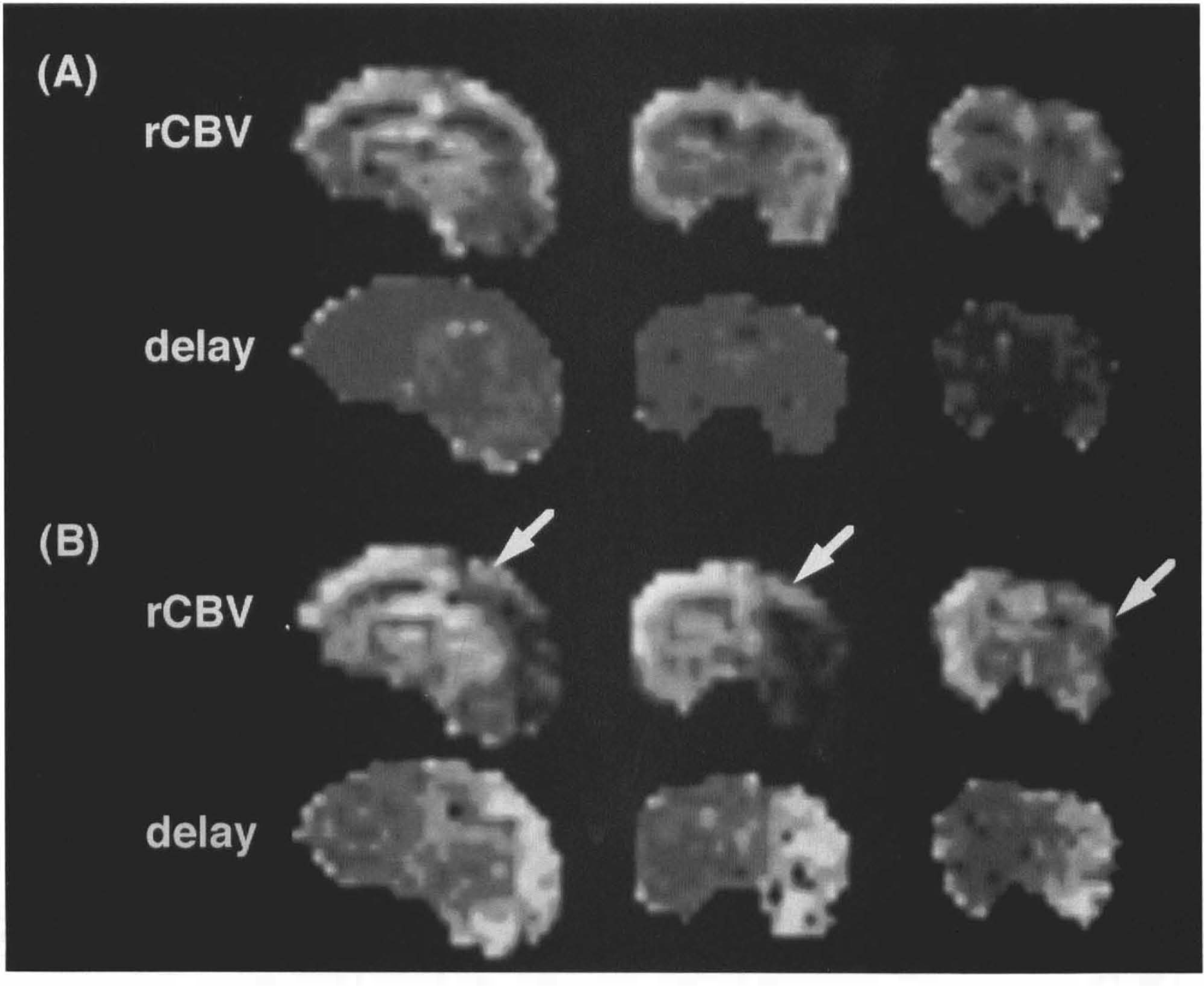
Synthesized relative blood volume (rCBV) and bolus arrival time (delay) maps acquired pre-
One animal was included in the study evaluation even though it did not present a permanent ADC drop after the initial suture occlusion. Transient ADC changes migrated bidirectionally covering the majority of the ipsilateral cortex in the coronal plane (Fig. 5). The perfusion data showed a moderate impairment in the maps of rCBV and bolus arrival time when compared with the contralateral hemisphere. Areas of permanent ADC decrease were observed, when the suture was subsequently advanced further.
Bidirectional propagation of the wave of transient apparent diffusion coefficient (ADC) decrease. This animal did not exhibit a permanent ADC drop, due to incomplete middle cerebral artery occlusion after advancing the suture (at the 1-min timepoint).
One animal with a permanent ADC decrease in the basal ganglia and the cortex exhibited no transient ADC changes within the 17-min imaging period.
DISCUSSION
Using echo-planar DWI in a remote MCAO suture model, we detected very early transient ADC decreases originating in the ischemic tissue and propagating along the cortex with a velocity of 3 mm/min. Although we did not perform concomitant electrophysiological recordings, the characteristics of the observed transient ADC declines make its coincidence with CSD most likely and our observations refer to recent MR detection of CSD (Gardner-Medwin et al., 1994; Latour et al., 1994; Hasegawa et al., 1995). The probable mechanism of transient ADC decrease during CSD is the swelling of cells and shrinkage of the extracellular space due to severe disturbance of the extracellular ion homeostasis. Since Nedergaard and Astrup (1986) first reported CSD in the periinfarct tissue by recording deflections of the DC potential, numerous studies were addressed to the pathomechanism of spreading depression–like DC shifts in periinfarct tissue. Recent studies characterized the impact of CSD on ischemic tissue, showing that it represents not simply an accompanying phenomenon of ischemia but considerably increases the infarct volume (Mies et al., 1993).
The recording of direct current potential (DC) shifts for the characterization of CSD has several disadvantages, one being that it is unable to provide an accurate spatial analysis of the propagating waves due to its limitation to one or two electrodes. As shown in our study, the origination in the ischemic tissue and the propagation of CSD can be directly visualized by the means of noninvasive MR imaging. Different patterns of CSD propagation were observed that depend on the severity of the perfusion perturbation. In the majority of rats, the wave of decreased ADC originated from the border of the ischemic tissue identified by permanently decreased ADC values and traveled towards the normally perfused tissue near the midline. Correlating perfusion imaging data showed that the cortical tissue, where CSD initiated, presented a mild reduction of rCBV and a delay of the bolus arrival time if compared with the contralateral hemisphere. This observation fits well with recent findings that the periinfarct tissue from which CSD originates shows a 50% decrease of cerebral blood flow but still presents electroencephalographic activity (Back et al., 1994) and that CSD is expected to occur only in tissue that still has an intact membrane metabolism (Iijima et al., 1992).
Propagation of transient ADC changes toward tissue characterized by severe rCBV reduction was not observed; reasoning that repolarization after CSD is an energy-requiring process with markedly increased glucose and oxygen consumption (Kocher, 1990), it seems plausible that CSD is restricted to tissue still able to cope with the increased metabolic requirements of repolarization. More evidence to support the finding that CSD is restricted to moderately affected tissue derives from the observation that the duration of the transient ADC changes is longer in ROIs close to the ischemic core than in the more peripheral areas. Again, one might hypothesize that the tissue closer to the ischemic core takes a longer time to recover from ionic disturbances and to restore membrane polarization due to impaired energy delivery than does tissue in the periphery, where the perfusion deficit is less severe. This interpretation is supported by electrophysiological recordings of CSD in periinfarct tissue that show longer durations of DC deflections the closer the electrode is located to the ischemic core (Gill et al., 1992).
Due to an incomplete MCAO, one animal presented with a temporary ADC decrease in the absence of a permanent ADC drop. Transient ADC changes migrated bidirectionally over the cortex in the coronal plane (Fig. 5). This case is interesting because it shows that the initiation of CSD is not necessarily coupled to ischemic areas developing into a permanent ADC decrease. Obviously, even a moderate reduction of the rCBV may be enough to stimulate propagating ADC transients. The occurrence of CSD in humans is still unclear; however, if CSD takes place in clinical stroke, this observation stimulates speculations on the role of CSD in various other conditions such as vascular dementia and transient ischemic attacks.
In conclusion, the noninvasive detection of CSD by means of serial, ultrafast DWI opens a new field for experimental designs on a fascinating and still poorly understood issue. The application of serial DWI to human stroke and migraine will prove whether CSD is a pathophysiological mechanism occurring primarily in rodents or whether it plays an important role as a stereotyped response of neurons to toxic or traumatic stimulation and metabolic disturbances.
Footnotes
Acknowledgment:
Joachim Röther is supported by a grant from the Deutsche Forschungsgemeinschaft.