
Abstract
We investigated the protective effect of hypothermia on ultra-early-type ischemic injury in the thalamic reticular nucleus of the rat. Cerebral ischemia was produced by 5 min of cardiac arrest followed by resuscitation. Rectal and cranial temperature during and after cardiac arrest was maintained at 37–38°C in the normothermic group and at 32–33°C in the hypothermic group. In the postischemic hypothermic group, temperature was maintained at 32–33°C starting 15 min after normothermic ischemia. Histological damage was evaluated quantitatively. While after 5 min of recirculation there was no difference in morphological changes in terms of neuronal halo formation, intraischemic hypothermia reduced the severity of the degenerative changes represented by vacuolated or dark neurons by 15 min. Postischemic hypothermia failed to show any evidence of protection by 30 min. The protective effect of intraischemic hypothermia remained significant when evaluated at 14 days after ischemia by volumetry of the lesion and neuronal density analysis, whereas postischemic hypothermia had no clear protective effect. These results suggest that the protective effect of intraischemic hypothermia applies to neurons susceptible to ultra-early-type injury, but the effect of postischemic hypothermia is limited because normothermic ischemia results in extensive degeneration in these neurons by 15 min.
While hypothermia has been known to have a protective effect against brain injury for almost four decades (Bigelow et al., 1950; Drake et al., 1964), the importance of brain temperature in ischemic insults was recently reevaluated experimentally, and the protective effect of even mild hypothermia was confirmed in animal global ischemia models (Busto et al., 1987; Minamisawa et al., 1990a; Welsh et al., 1990). Presently, increasing numbers of clinical facilities are undertaking trials of protective mild hypothermia using more sophisticated methods than in the past (Spetzler et al., 1988; Hayashi et al., 1992; Hayashi et al., 1993; Marion et al., 1993; Shiozaki et al., 1993).
One important aspect of hypothermia revealed by recent studies is differences in sensitivity to hypothermia among different brain regions (Minamisawa et al., 1990a). Thus, in the rat global ischemia model, the severity of damage in the hippocampal CA1 area diminished linearly as temperature decreased, whereas hypothermia had an all-or-nothing protective effect on the caudoputamen. Strikingly, however, hypothermia provided only equivocal protection of the nucleus reticularis thalami (NRT) after 5–15 min of global ischemia (Minamisawa et al., 1990b). Similarly, the NRT was not protected by hypothermia in a rat model of 15-min global cerebral ischemia produced by increasing intracranial pressure (Ross and Duhaime, 1989).
The NRT consists of the most lateral portion of the thalamus, and serves as a gateway for the thalamocortical and corticothalamic pathways. Thalamic reticular nucleus neurons, all of which are GABA-containing, receive excitatory afferent axon collaterals from both thalamocortical and reciprocal corticothalamic neurons, and in turn project inhibitory efferents to thalamic neurons (Steriade et al., 1990). Physiological studies of the NRT have suggested its involvement in a range of important brain functions such as selective attention or setting the excitation level of thalamic relay neurons in the sleep-wake cycle (Crick, 1984; Steriade and Llinás, 1988; Shosaku et al., 1989; Mitrofanis and Guillery, 1993; Steriade et al., 1993). The NRT, along with the hippocampal CA1 region, is also known as one of the regions most vulnerable to ischemic insult in various rat global ischemia models (Smith et al., 1984; Blomqvist and Wieloch, 1985; Ross and Duhaime, 1989; Kawai et al., 1992a) and in human cardiac arrest (Ross and Graham, 1993). One purpose of the present work was to obtain conclusive findings regarding the effect of hypothermia on ischemic injury in the NRT. Because ischemia of longer duration is associated with a less clear protective effect of hypothermia (Minamisawa, et al., 1990b), we speculated that the lack of a clear protective effect of hypothermia on the NRT in previous studies resulted from the severity of the ischemic injury. Therefore, we produced a less severe ischemic insult by using 5 min of cardiac arrest in the present study. We also used two sensitive methods recently reported (Kawai et al., 1995a; Ross et al., 1995) to histologically quantify damage in the NRT 14 days after cardiac arrest.
The other important aspect of ischemic injury in the NRT is its acuteness in the postischemic time course. The NRT is the most vulnerable region of the brain in the early phase of recirculation after transient global ischemia in the rat. Neurons in the NRT show severe degeneration accompanied by cytoplasmic vacuolation and calcium accumulation as early as within 1 hour of recirculation, and a reduction in number of neurons as early as 3 hours after recirculation (Kawai et al., 1992a; Kawai et al., 1992b; Kawai et al., 1995a). Whereas most protective hypothermia procedures against cerebral ischemia or trauma are carried out postischemically in clinical settings, as opposed to those performed in certain specific situations such as during surgery under general anesthesia, the effect of postischemic hypothermia in experimental settings is still controversial (Dietrich et al., 1993; Colbourne and Corbett, 1995). We are very interested in the effects of hypothermia, especially of postischemic hypothermia, on ischemic injury in the NRT, which manifests such acute degeneration. The effect of hypothermia on neurons that are susceptible to acute ischemic degeneration as well as on neurons that manifest postischemic delayed degeneration should be clarified before extensively applying protective hypothermia in clinical settings. Therefore, in the present study we evaluated acute and chronic damage in the NRT of rats subjected to intraischemic and postischemic mild hypothermia. This study is the first to show that a small difference in intraischemic temperature can result in a clear difference in morphological damage in the brain even in the early recirculation phase following transient global ischemia.
MATERIALS AND METHODS
Cardiac arrest was induced in 42 adult male Sprague-Dawley rats weighing 270–330 g using a method described previously (Kawai et al., 1992a). Briefly, animals were anesthetized with 1.5% halothane in 2:1 nitrogen/oxygen for 20 min before the onset of ischemia, and a catheter was inserted into the left femoral artery to monitor blood pressure. Rectal and cranial temperature was monitored continuously. Blood gases and glucose concentrations were measured in arterial blood drawn immediately before induction of cardiac arrest. Cardiac arrest was induced by compressing the major cardiac vessels with a bent wire inserted through the right second intercostal space. Five min after occlusion, resuscitation was initiated by closed-chest cardiac massage and artificial ventilation via a nasal catheter. Successful resuscitation was typically achieved with a return of spontaneous and effective cardiac output within 1.5 min, and the time at which the mean arterial blood pressure reached 50 mm Hg was recorded as the start of recirculation (Kawai et al., 1995b). The start of recirculation was within 9 min after initiation of cardiac arrest in most rats (Kawai et al., 1995b). However, in a small number of rats the start of recirculation was later than this, and these animals were excluded from the following comparative analysis.
Temperature control was achieved as follows. Rectal and cranial temperature in the 16 rats in the normothermic group was maintained between 37 and 38°C, by using a heating pad and lamp, from before the onset of cardiac arrest until killing for histological evaluation in the early recirculation phase, and until 60 min after the start of recirculation for histological evaluation in chronic phase. In the 16 rats in the intraischemic hypothermic group, rectal and cranial temperatures were maintained between 32 and 33°C with ice pads during the same period as in the normothermic group. In the 10 rats in the postischemic hypothermic group, rectal and cranial temperatures were maintained between 37 and 38°C during cardiac arrest with a heating pad and lamp, and then between 32 and 33°C from 15 min after recirculation until killing for early evaluation, and until 60 min after recirculation for chronic evaluation, by rapid cooling with ice pads started within 5 min after recirculation.
To evaluate early neuronal changes, 28 rats subjected to cardiac arrest were perfused with 100 ml of 0.9% heparinized saline via the ascending aorta followed by 400 ml of 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) in the early recirculation phase. Four rats in the normothermic group and 4 rats in the intraischemic hypothermic group were killed 5 min after recirculation. Four rats in the normothermic group and 4 rats in the intraischemic hypothermic group were killed 15 min after recirculation. Four rats in the normothermic group, 4 rats in the intraischemic hypothermic group, and 4 rats in the postischemic hypothermic group were killed 30 min after recirculation. The animals' brains were embedded in paraffin and sectioned coronally 5-μm thick at 100 μm intervals, and the sections were stained with cresyl violet. Morphological changes in the NRT, which was identified as the most lateral area of the thalamus, were examined by light microscopy. In the sections showing the most extensive changes in the NRT, which were located in the middle of the rostrocaudal extent of the nucleus (sections No. 11–14 in Figure 2 of Kawai et al., 1995a) and correspond to Figure 27 or 28 of the Paxinos and Watson (1986) rat brain atlas, quantitative evaluation was made by separately counting the number of neurons that displayed a pericytoplasmic halo and microvacuolation or dark change (Kawai et al., 1992a).
To evaluate chronic damage, 4 rats in the normothermic group, 4 rats in the intraischemic hypothermic group, and 6 rats in the postischemic hypothermic group, which had been subjected to cardiac arrest 14 days before, were anesthetized and perfused using the above-mentioned method. Their brains were equilibrated for 2 days at 4°C in phosphate-buffered saline containing 20% (weight to volume ratio) sucrose, and then cut coronally approximately at 0.5 mm and 5.0 mm posterior to the bregma. The middle blocks containing the thalamus were cut into 20-μm coronal sections in a cryostat at −15°C. Every fifth section was collected in phosphate-buffered saline for parvalbumin immunocytochemistry. Free-floating sections were incubated in 5% blocking serum for 1 h and then with monoclonal antiparvalbumin antibody (P3171, Sigma Chemical Co., St. Louis, MO, U.S.A.) in 5% blocking serum (1:5000) overnight at room temperature with shaking. Sections were washed in phosphate-buffered saline and incubated with secondary antibody that has undergone biotinylation (1:200 horse anti-mouse IgG, Vector Laboratories) at room temperature for 2 h and in avidin-biotin-horseradish peroxidase complex (Vectastatin Elite ABC kit, Vector Laboratories, Burlingame, CA, U.S.A.) for 2 h. Diaminobenzidine tetrahydrochloride was used as a chromogen for the immunoperoxidase reaction. Sections were mounted on gelatin-coated slides. Parvalbumin-stained sections collected at 100 μm intervals along the rostrocaudal extent of the NRT were analyzed by two methods: volumetry of the lesion (Kawai et al., 1995a) and neuronal density analysis (Ross et al., 1995). For volumetry of the lesion, the area (mm2) of the lesion which could be clearly identified as an immunostaining-negative region was measured using a computer-based image-processing system in serial sections at 100-μm intervals along the rostrocaudal extent of the entire NRT. A previous study showed that the loss of parvalbumin immunostaining can be interpreted as the loss of neuronal soma and neuropil by counterstaining (Kawai et al., 1995a). The volume of the lesion was approximated by using the following formula, which calculates the volume lying between two sections as a conical segment.
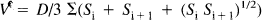
where V is the volume of the lesion, D is the interval between sections, and Si is the area of the lesion in each section. For neuronal density analysis, four serial sections at 200-μm intervals where the lesion areas were largest were selected for each NRT. The caudal end of the anterodorsal and anteroventral thalamic nuclei, which could be identified by parvalbuminpositive puncta, was also used as a landmark for selecting the four serial sections. The caudal end of these nuclei was always located between the first and the second or between the second and the third of the four sections. They correspond to Figures 26–29 of the Paxinos and Watson rat brain atlas (1986), and the most extensive damage of the NRT was always found in this area (corresponding to sections No. 10–16 in Figure 2 of Kawai et al., 1995a). In each section selected, the number of NRT neurons was counted using a 350 × 350 μm ocular grid from two adjacent grid-fields in the middle of the NRT in the dorsoventral dimension (Ross et al., 1995). The middle NRT was further divided into a medial quarter, a central half, and a lateral quarter, and counts were made separately in these three regions. The number of grid boxes in the 7 × 7 grid-field from which the counts were made was also counted. Neuronal density in each of the three mediolateral regions of each section was determined by dividing the number of NRT neurons by the sampling volume (= 50 μm × 50 μm × 20 μm × the number of grid boxes). Neuronal density was determined by averaging the neuronal densities from four sections for each region of each the NRT.
All values given are means ± SD. One-way analysis of variance followed by Scheffe's F test was used to compare numbers of degenerated neurons, lesion volume, and neuronal density in the three groups, and to analyze difference between physiological parameters. The unpaired t test was used to compare pattern of blood pressure changes immediately after resuscitation. Differences were considered to be significant when p values were less than 0.01.
RESULTS
Physiological parameters
The preischemic PaO2, PaCO2, and glucose concentration of arterial blood did not differ significantly among the groups. The preischemic arterial blood of hypothermic rats was slightly acidemic than that of normothermic rats although it was within normal limits. Resuscitation was initiated 5 min after induction of cardiac arrest, and the intervals from occlusion to successful recirculation were not significantly different among the groups (Table 1). Rectal and cranial temperature was for the most part maintained at the desired level although there were some variations (Table 2). There were no significant differences between rectal and cranial temperature before, during, and after cardiac arrest among the groups. Rapid systemic cooling with ice pads started within 5 min of recirculation and decreased rectal and cranial temperature to 32–33°C within 15 min of recirculation in the postischemic hypothermic group.
Preischemic physiological parameters
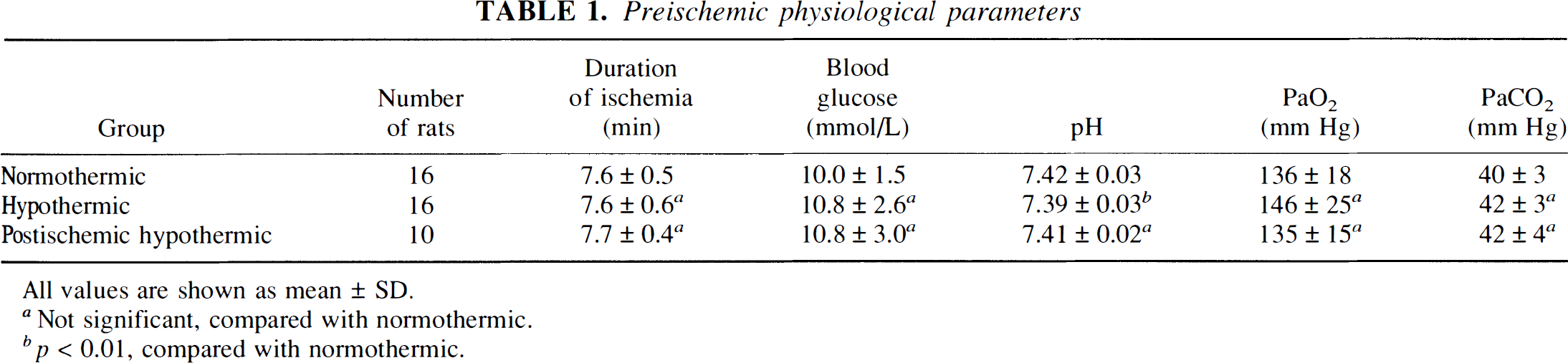
All values are shown as mean ± SD.
Not significant, compared with normothermic.
p < 0.01, compared with normothermic.
Rectal and cranial temperatures before, during, and 15 min after cardiac arrest
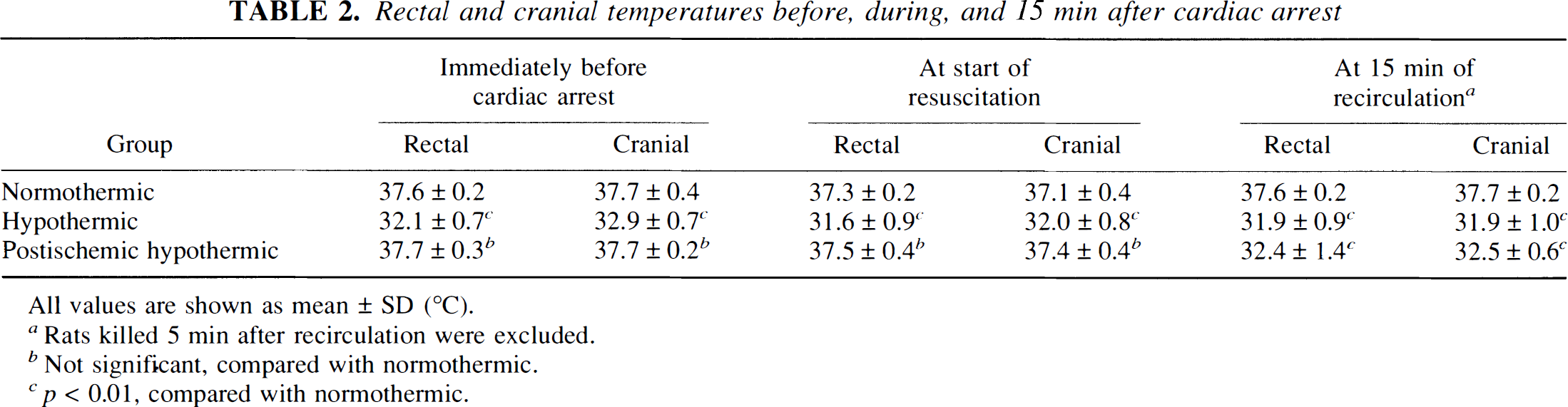
All values are shown as mean ± SD (°C).
Rats killed 5 min after recirculation were excluded.
Not significant, compared with normothermic.
p < 0.01, compared with normothermic.
The intraischemic differences in temperature resulted in marked differences in the pattern of arterial blood pressure changes immediately after resuscitation (Fig. 1, Table 3). In normothermic rats, arterial blood pressure increased steeply and formed a reactive hypertension phase that lasted about 5 min, whereas in hypothermic rats the increase in arterial blood pressure was slower and reactive hypertension was less prominent. The time from the point when mean arterial blood pressure recovered to 50 mm Hg to the point when it reached the maximum level was significantly longer in the hypothermic group. The maximal mean arterial blood pressure was also lower in hypothermic rats although this difference was not significant (p = 0.564). There was no significant difference in the rate of successful resuscitation among the groups.
Pattern of blood pressure changes immediately after resuscitation

All values are shown as mean ± SD.
Time during which mean arterial blood pressure increased from 50 mm Hg to the maximum.
Maximal mean arterial blood pressure.
p < 0.01
not significant.
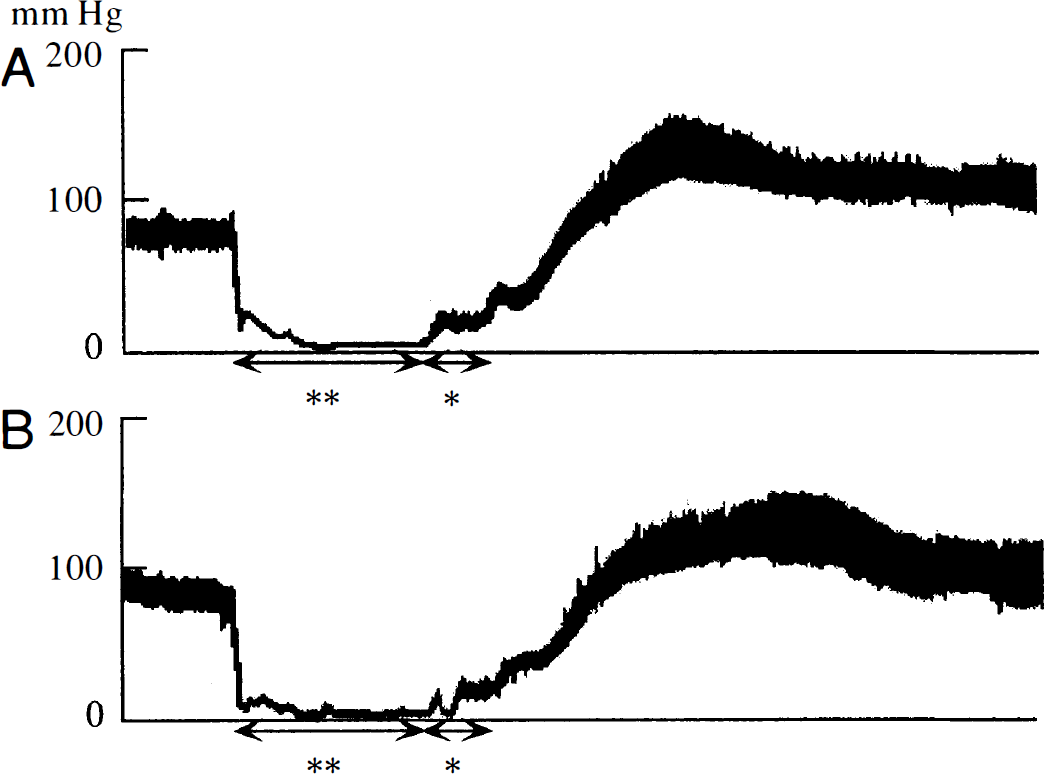
Differences between the recovery pattern of arterial blood pressure after cardiac arrest in normothermic and hypothermic rats. Two representative recordings are shown. The reactive hypertension phase is less marked in the hypothermic rat (
Early neuronal changes
Histological examination of neuronal changes in the early recirculation phase revealed that differences in the intraischemic temperature resulted in striking differences in early neuronal changes in the NRT (Figs. 2 and 3). Some neurons in the NRT showed definite postischemic changes, as early as after 5 min of recirculation, characterized by peripheral halos associated with condensation of the remaining cytoplasm around the nucleus (Figs. 3A and 3B). These changes were detected to a similar degree in both normothermic and hypothermic rats after 5 min of recirculation. No differences were detected in the numbers of halo-neurons or of vacuolated or dark neurons between the normothermic and hypothermic groups at this point (Fig. 2, left columns). While the differences within 5 min of recirculation were not marked, in many of the NRT neurons of normothermic rats, cytoplasmic halos became compartmentalized forming peripheral vacuoles within 15 min of recirculation, whereas vacuole formation was much less prominent in the hypothermic rats (Fig. 3). At 15 min after recirculation, the number of halo-neurons in the normothermic group was significantly smaller than in the hypothermic group, and the number of vacuolated or dark neurons was larger in the former group than in the latter group (Fig. 2, middle columns). Within 30 min of recirculation, the differences became more prominent. Most neurons in the NRT of normothermic rats showed severe vacuolation or dark pyknotic changes. The peripheral portion of some neurons appeared unclear, and they had lost their cellular boundaries. In contrast, progression of the degenerative process in the NRT neurons of the hypothermic rats seemed to stop. Only a moderate degree of cytoplasmic vacuolation occurred, although some scattered dark neurons that had undergone pyknosis were noted. Otherwise, the interstitial small vacuoles became more prominent. Damage to the NRT of postischemic hypothermic rats was basically the same as in the NRT of normothermic rats. The numbers of vacuolated or dark neurons in the postischemic hypothermic group were not significantly different from their numbers in the normothermic group, but their numbers in the intraischemic hypothermic group were significantly smaller than in the normothermic group (Fig. 2, right columns).
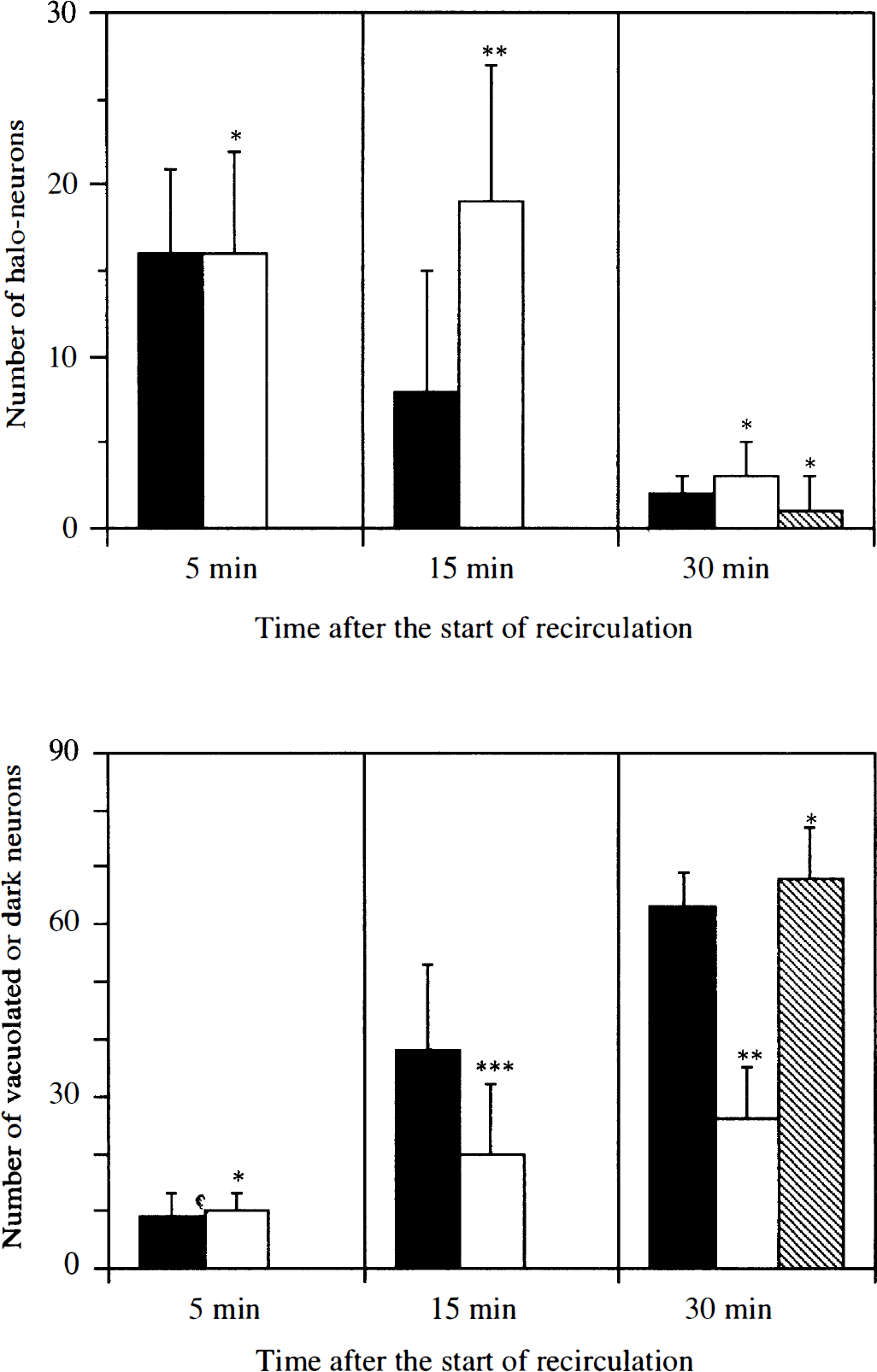
Chronological changes in the number of halo-neurons (
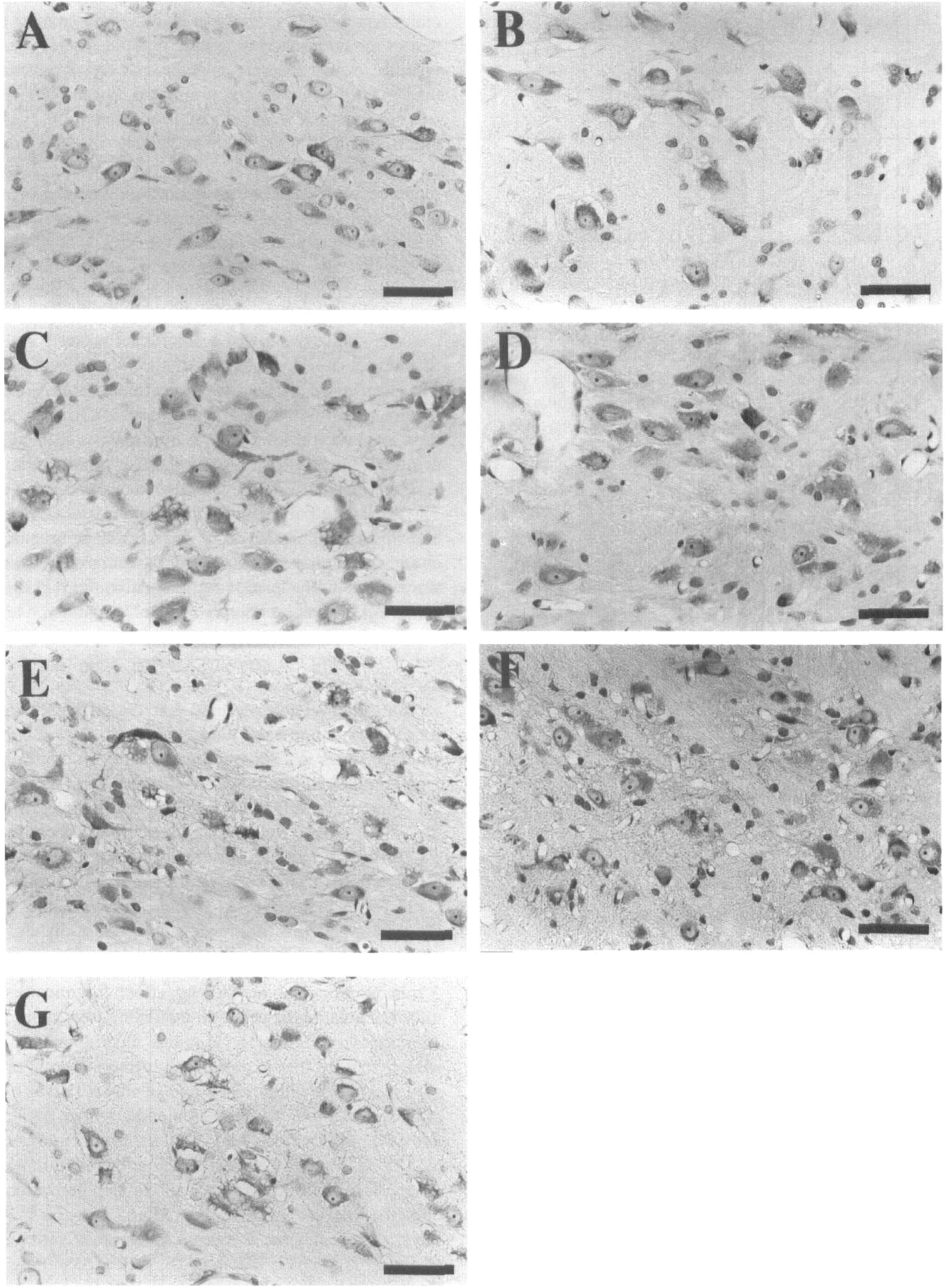
Microscopic changes in the thalamic reticular nucleus neurons in the early recirculation phase. Cresyl violet stain. (
Chronic neuronal damage
Some rats subjected to 5 min of cardiac arrest died of status epilepticus manifested as audiogenic seizures (Kawai et al., 1995b) within a few days. The mortality rates in the three groups were not significantly different.
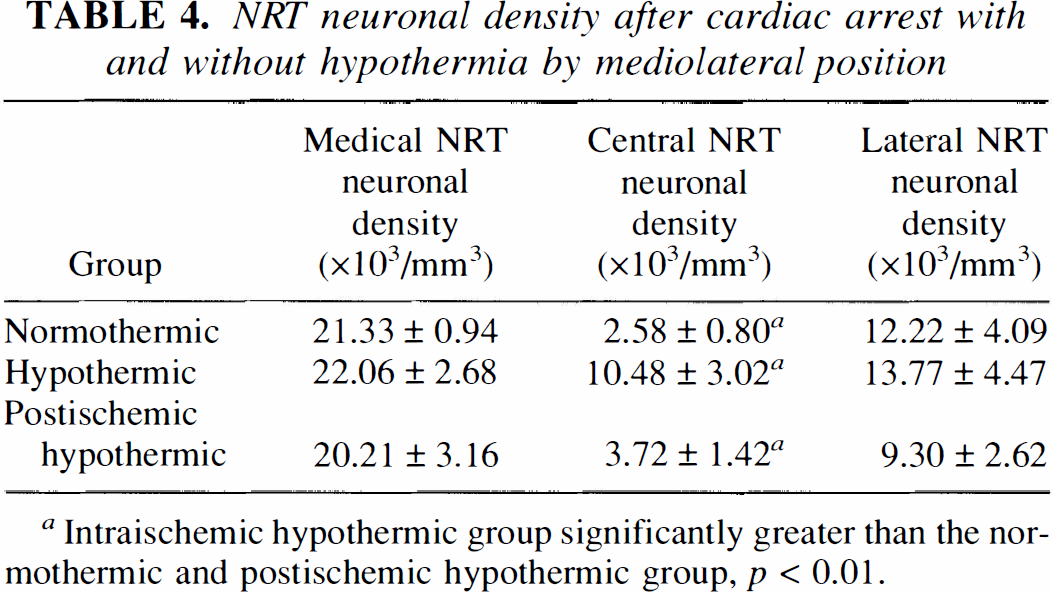
As previously reported, parvalbumin immunostaining clearly defined the limits of the NRT lesions (Kawai, et al., 1995a). The extent of NRT damage was confined to the middle portion of the nucleus in the rostrocaudal and dorsoventoral dimensions (Fig. 4). This pattern remained the same even when temperature modification was added. However, the lesion area in each section and the rostrocaudal extent of the lesion (i.e., number of sections in which the lesion was detected) in hypothermic rats were clearly smaller than in normothermic rats. The estimated volumes of the lesions were 0.224 ± 0.080, 0.088 ± 0.033, and 0.170 ± 0.088 mm3 in the normothermic, hypothermic, and postischemic hypothermic groups, respectively (Fig. 5). The lesion volume of the intraischemic hypothermic group was significantly smaller than that of the normothermic group. A trend whereby lesion volume in postischemic hypothermic rats was smaller than in normothermic rats was noted, but the difference was not statistically significant. Protection of the NRT by intraischemic hypothermia was characterized not only by the reduced volume of the lesion but by the presence of scattered surviving neurons in the lesion (Fig. 5). Virtually complete loss of the neuronal soma and neuropils were consistently recognized in the NRT lesions of normothermic rats, whereas dispersed neuronal somas with less a densely packed neuropils network remained in the NRT lesion of the hypothermic rats. Neuronal density analysis similarly showed a significant protective effect of intraischemic hypothermia but no significant protective effect of postischemic hypothermia (Table 4). In the mediolateral dimension of the central NRT where ischemic degeneration of NRT neurons was confined, the neuronal density of the intraischemic hypothermic rats was significantly greater than that of normothermic rats. The difference between normothermic rats and postischemic hypothermic rats was not significant. Neuronal density in the medial and lateral NRT, where most neurons were consistently spared after ischemia, did not significantly differ in the three groups.
NRT neuronal density after cardiac arrest with and without hypothermia by mediolateral position
Intraischemic hypothermic group significantly greater than the normothermic and postischemic hypothermic group, p < 0.01.
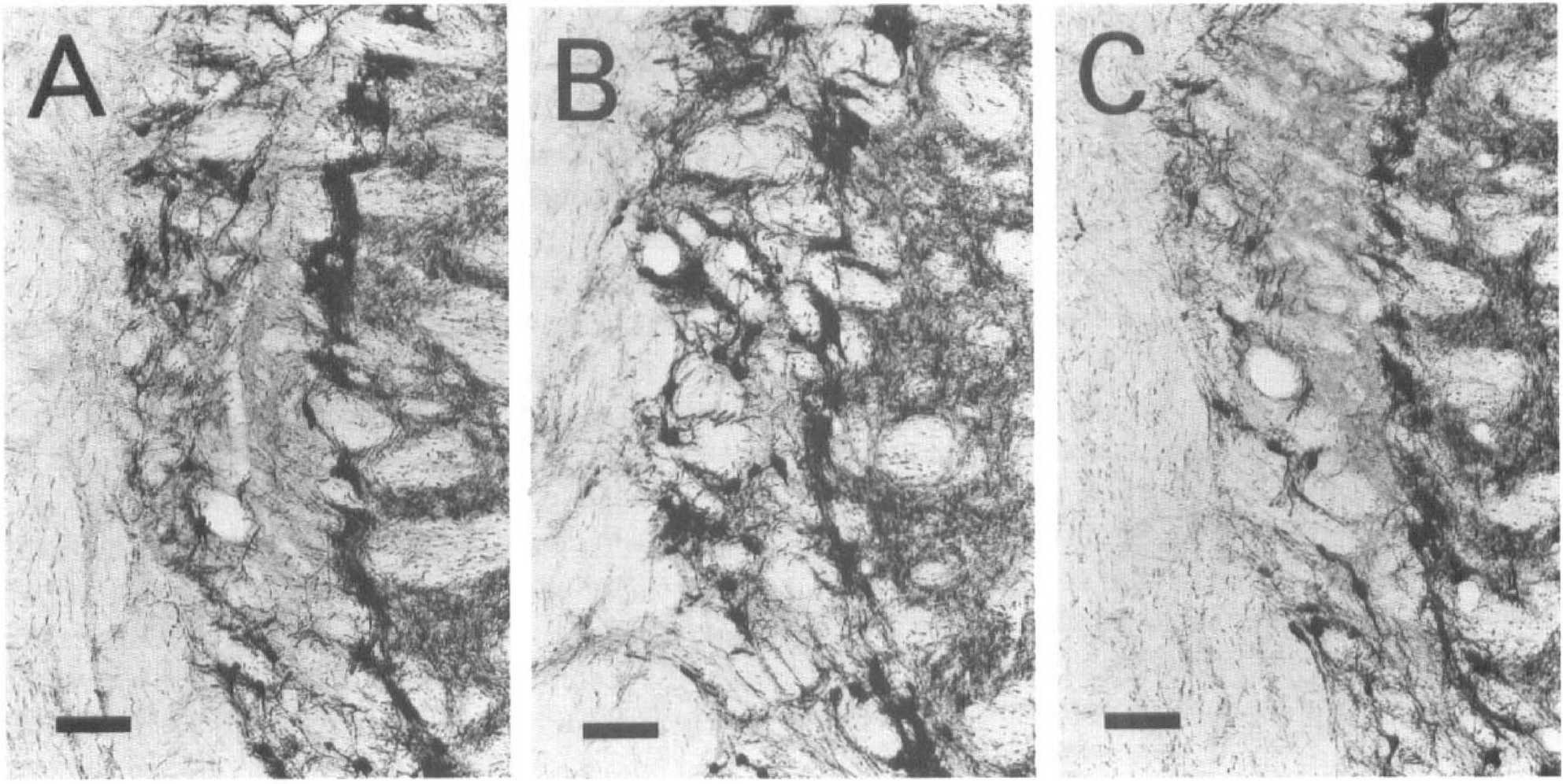
Middle segment of the NRT 14 days after cardiac arrest. Parvalbumin immunohistochemistry. Coronal sections 20-μm thick immediately posterior to the caudal end of the anterodorsal and anteroventral thalamic nuclei are shown. (
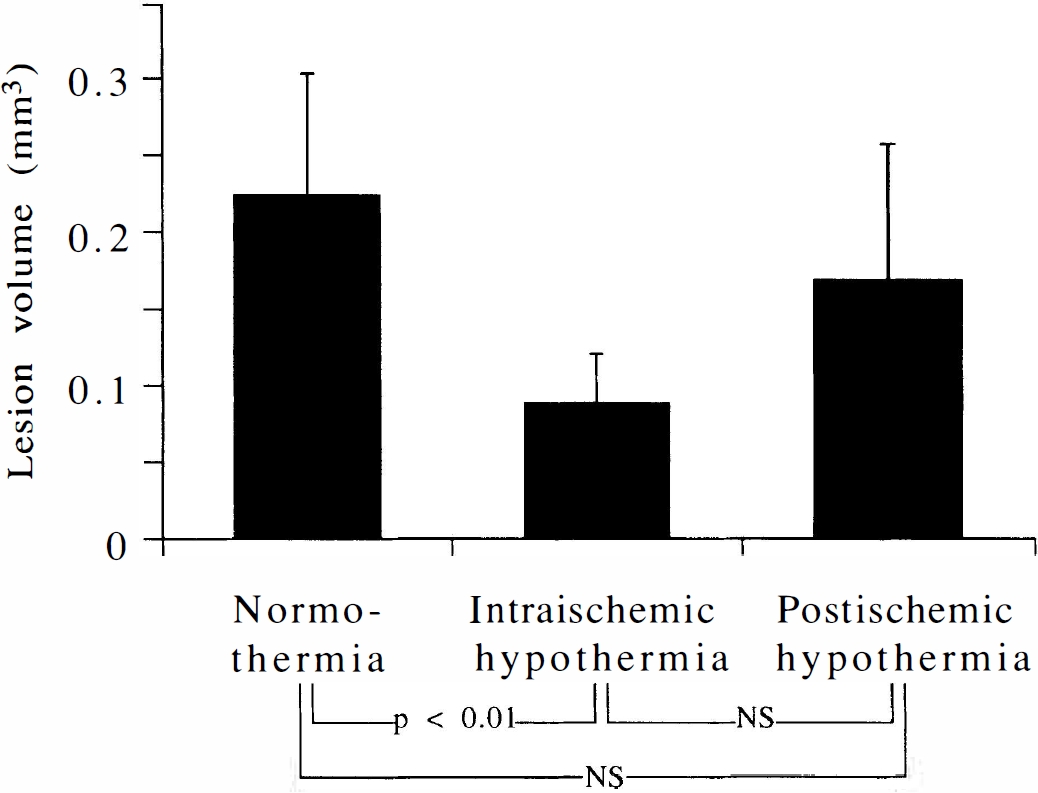
Bar graphs showing lesion volume in the thalamic reticular nucleus evaluated 14 days after cardiac arrest. The area of the lesion was measured in serial parvalbumine-stained sections, and the volume of the lesion was calculated as described in the text. Lesion volume in the intraischemic hypothermic group was significantly smaller than in the normothermic group, while the difference between the normothermic group and postischemic hypothermic group was not significant. NS, not significant.
DISCUSSION
The present study revealed that the difference in intraischemic temperature resulted in marked morphological differences in NRT neurons as early as after 15 min recirculation, which may explain the much less significant protective effect of postischemic hypothermia in this nucleus when evaluated 14 days after ischemia; and the protective effect of intraischemic hypothermia clearly shown in the early recirculation phase remained significant when evaluated 14 days after ischemia.
The temperature difference during ischemia did not affect the degree of initial halo formation within the first 5 min of recirculation, but did affect the transition to microvacuolation or neuronal darkening after more than 15 min of recirculation. Previous electrophysiological studies showed that hypothermia during ischemia or anoxia resulted in a decreased rate of initial rise in extracellular potassium concentration and a longer latency of anoxic depolarization (Katsura et al., 1992; Hiramatsu et al., 1993). However, when ischemic/anoxic duration is longer than 2–3 min, and once anoxic depolarization occurs, the increase in free fatty acid and decrease in ATP are similar in degree in spite of temperature differences (Busto et al., 1989a; Welsh et al., 1990). Assuming that the halo formation is morphological evidence that neurons have experienced catastrophic ionic movements across the cell membrane and water entry subsequent to anoxic depolarization, the similarity of the degree of halo formation observed in the NRT in normothermic and hypothermic rats in this study further supports the results of these previous biochemical studies, showing that temperature differences do not significantly affect the very early phase of ischemia when the duration of ischemia is long enough. Initial halo formation was observed in various neuronal populations, most of which were GABA-containing neurons, after transient global ischemia (Kawai et al., 1992a). GABA-containing neurons usually have a higher metabolic rate (Celio, 1986), which may explain their tendency toward ischemic halo formation, because the initial rise in the extracellular potassium concentration is proportional to the metabolic rate (Katsura et al., 1992). However, this halo formation was reversible in brain regions other than the NRT in rats whose rectal temperature was maintained at approximately 37°C (Kawai, et al., 1992a). Based on the present results showing similar degrees of halo formation at 5 min but significant differences in neuronal degeneration after more than 15 min, it is speculated that the temperature difference affected not the very early phase but the late phase of the transition from reversible halo formation to irreversible microvacuolation or dark neuron in which severe disturbances of the integrity of the cellular membrane and cytoskeleton may occur. It is interesting in this context that prolonged degradation of the cytoskeleton protein spectrin was observed in the cerebral cortex of rats whose brain temperature was maintained higher after middle cerebral artery occlusion (Tadashi Morimoto and Myron D. Ginsberg, personal communication).
The stability of the blood-brain barrier after ischemia is also affected by temperature differences. Horseradish peroxidase leakage was markedly decreased by hypothermia in the thalamus, including the NRT (Dietrich et al., 1990). In the present study, the reactive hypertension after cardiac arrest was less prominent in hypothermic rats. A previous study using the same animal model showed that the reactive hypertension phase coincided with reactive hyperemia in the brain (Kawai et al., 1992a), which is known to aggravate blood-brain barrier breakdown and brain injury (Kuroiwa et al., 1985; Ting et al., 1986; Seida et al., 1988). Although the cause-effect relationship of less-prominent reactive hypertension and lesser damage in the NRT is unknown, the increased stability of the blood-brain barrier after hypothermic ischemia may be another mechanism underlying the protective effect on early neuronal changes in the NRT.
When evaluated 14 days after cardiac arrest by volumetry of the lesion and by neuronal density analysis in the present study, vulnerable NRT neurons were found to have been partially but significantly rescued by intraischemic hypothermia. The results of previous studies were not conclusive in terms of the effect of hypothermia on ischemic neuronal damage in the NRT. In a cisternal infusion model of global cerebral ischemia in the rat, 5–10 min of normothermic (33–38°C, infusion fluid temperature) ischemia resulted in minimal to extensive neuronal loss in the NRT, whereas 5–10 min of hypothermic (22–25°C, infusion fluid temperature) ischemia resulted in no neuronal loss in the NRT. Thalamic reticular nucleus neurons were relatively less protected by hypothermia than hippocampal CA1 neurons (Ross and Duhaime, 1989). The protective effect of mild intraischemic hypothermia (35°C) on NRT damage was statistically significant in a study using 2-vessel occlusion model of 15-min forebrain ischemia in the rat (Minamisawa et al., 1990a) but not significant in another study using the same model of 5–15 min ischemia (Minamisawa et al., 1990b). However, ischemia in the thalamus produced by 2-vessel occlusion model, in contrast with that produced by cisternal infusion and cardiac arrest models, may be nonuniform and variable because of collateral circulation afforded by the highly anastomosed thalamic arterial network (Ross and Duhaime, 1989). Furthermore, in those studies degeneration of the NRT was evaluated only on a semiquantitative 4-grade scale and not in serial-sections through the entire rostrocaudal extent of the nucleus. As shown in the present and in previous studies (Ross and Duhaime, 1989; Kawai et al., 1992a and 1995a; Ross et al., 1995), degeneration of the NRT was confined to the central part of the nucleus in all three dimensions. A rostrocaudal shift in selecting the coronal sections in which histological evaluation is made may result in great variability in estimating degeneration in the NRT. Therefore, clearly identifying the extent of the nucleus and of the lesion in the nucleus by immunostaining for a marker protein of NRT neurons, such as parvalbumin, and tracing the damage through its entire rostrocaudal extent in serial sections seemed essential to evaluating the NRT damage (Kawai et al., 1995a). Furthermore, the neuronal density analysis recently reported by Ross et al. (1995) has enabled more direct and detailed quantitative evaluation of the NRT damage, especially in its mediolateral extent. In the present study, we used these two recently reported methods to evaluate NRT damage and showed conclusive findings concerning the protective effect of intraischemic hypothermia on ischemic NRT damage after global cerebral ischemia.
In human cardiac arrest cases, a brief period (≤5 min) of ischemia resulted in selective NRT degeneration without definite lesions in the cerebral cortex and in the thalamus. Whereas profound intraoperative hypothermia was insufficient to prevent the selective loss of NRT neurons, the extent and severity scores of NRT degeneration were greater in a patient who had sustained hypothermic brief ischemia than in three patients who had sustained hypothermic brief ischemia (Ross and Graham, 1993). These findings in patients with brief cardiac arrest seemed to be consistent with our results in the rat cardiac arrest model, showing a limited but significant protective effect of intraischemic hypothermia on the NRT damage after global cerebral ischemia. In contrast, a longer period of (≥10 min) cardiac arrest resulted in extensive cortical and thalamic neuronal loss with sparing of the NRT in human cardiac arrest patients, whereas in rat global ischemia models, longer duration (5–15 min) of ischemia resulted in severer NRT damage, and even longer ischemia (>15 min) seemed to cause “saturated” damage of the NRT unrelated to temperature modification (Ross and Duhaime, 1989; Minamisawa et al., 1990b). We used brief (5 min) cardiac arrest in our study in order to clearly show the protective effect of hypothermia and to avoid the effects of the complicated resuscitative procedures required after longer cardiac arrest. However, the discrepancy in NRT damage in the human and rat after longer global cerebral ischemia with or without temperature modification is another important issue that remains to be characterized.
Although the degenerative mechanisms operative in the ischemic loss of NRT neurons are unknown, it was suggested that an excitotoxic component mediated by AMPA (DL-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid)-type glutamate receptors is involved in the process (Ross and Duhaime, 1989; Ross and Graham, 1993; Ross et al., 1995). Neurons in the central part of the NRT in the mediolateral dimension, which are classified as large fusiform neurons associated with the ventrobasal thalamic nuclei (Spreafico et al., 1991), were partially rescued from ischemic cell loss after cardiac arrest by the AMPA antagonist NBQX (2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline), while neurons in the medial and lateral borders of the NRT, which are classified as small fusiform neurons associated with the ventrolateral thalamic nuclei, were not rescued by NBQX but by the partial μ-opiate-agonist buprenorphine (Ross et al., 1995; Sabol Jones and Ross, 1995). The hypothermic protection found in our study was localized in the central part of the NRT, which resembled the pattern found after NBQX treatment. Hypothermia may ameliorate the NRT damage by decreasing the release of glutamate from the presynaptic terminals of input fibers to NRT neurons, thus reducing the excitotoxic process via AMPA receptors in NRT neurons, because ischemic release of glutamate is known to be decreased by mild hypothermia in the striatum and the hippocampal CA1 region (Busto et al., 1989a; Mitani and Kataoka, 1991).
The protective effect of postischemic hypothermia was much less marked for NRT neurons. Whereas the protective effect of postischemic hypothermia has been controversial (Busto et al., 1989b; Welsh and Harris, 1991; Dietrich, et al., 1993), a recent report revealed that postischemic hypothermia exerted its protective effect when started early enough after ischemia and when it lasted long enough (Colbourne and Corbett, 1995). In the present study rats were cooled immediately after resuscitation and hypothermia was maintained 15 to 60 min after ischemia, whether longer postischemic hypothermia would unequivocally protect NRT neurons requires further study. However, the fact that many neurons in the NRT showed obvious degenerative changes 15 min after recirculation in normothermic rats and 30 min after recirculation even in postischemic hypothermic rats makes the effect of postischemic hypothermia questionable.
Induced hypothermia has recently been introduced in clinical settings not only for prevention (i.e., intraischemic hypothermia such as during cardiac surgery) but also for treatment of patients after head injury or stroke (i.e., postischemic hypothermia). Not only is it necessary to protect the hippocampus, which shows delayed neuronal death, but also as many structures in the brain as possible. Thus, more precise knowledge on the effects and limitations of hypothermia as a means of brain protection for extensive clinical application should be obtained. We believe that the results of the present study will add valuable information to the accumulating knowledge on the protective effect of hypothermia against cerebral ischemia.
Footnotes
Acknowledgments:
This study was supported by a Grant-in-aid for Scientific Research from the Ministry of Education, Science, and Culture of Japan, by a Research Grant for Cardiovascular Diseases from the Ministry of Health and Welfare of Japan, and by research grants from the Ono Pharmaceutical Company and the Eisai Pharmaceutical Company. We thank Ms. Noriko Kishino, Ms. Tomomi Iwasawa, and Ms. Kimi Tatebe for their technical assistance.