
Abstract
This study examined whether prolonged hypothermia induced 1 hour after resuscitation from asphyxial cardiac arrest would improve neurologic outcome and alter levels of stress-related proteins in rats. Rats were resuscitated from 8 minutes of asphyxia resulting in cardiac arrest. Brain temperature was regulated after resuscitation in three groups: normothermia (36.8°C × 24 hours), immediate hypothermia (33°C × 24 hours, beginning immediately after resuscitation), and delayed hypothermia (33°C × 24 hours, beginning 60 minutes after resuscitation). Mortality and neurobehavioral deficits were improved in immediate and delayed hypothermia rats relative to normothermia rats. Furthermore, both immediate and delayed hypothermia improved neuronal survival in the CA1 region of the hippocampus assessed at 14 days. In normothermia rats, the 70-kDa heat shock protein (Hsp70) and 40-kDa heat shock protein (Hsp40) were increased within 12 hours after resuscitation in the hippocampus. Delayed hypothermia attenuated the increase in Hsp70 levels in the hippocampus but did not affect Hsp70 induction in the cerebellum. Hippocampal expression of Hsp40 was not affected by hypothermia. These data indicate that prolonged hypothermia during later reperfusion improves neurologic outcome after experimental global ischemia and is associated with selective changes in the pattern of stress-induced protein expression.
Hypothermia during global cerebral ischemia reduces subsequent neuronal injury (Busto et al., 1987; Colbourne et al., 1993; Dietrich et al., 1993), perhaps by reducing oxidative damage (Baiping et al., 1994), maintaining mitochondrial function (Canevari et al., 1999), or reducing excitatory amino acid release (Busto et al., 1989) and metabolism (Busto et al., 1987). This intraischemic hypothermia is used clinically to minimize brain injury during cardiopulmonary bypass, aneurysm surgery, or other periods of predictable cerebral ischemia. Interestingly, reducing brain temperature during reperfusion after ischemia also reduces the development of neuronal injury (Busto et al., 1987; Kuboyama et al., 1993; Coimbra and Wieloch, 1994; Nurse and Corbett, 1994; Xiao et al., 1998). Postischemic hypothermia could prove useful for reducing brain injury after stroke, cardiac arrest, or other unpredictable ischemia. However, the mechanisms responsible for the beneficial effects of hypothermia during reperfusion are unknown.
The duration of hypothermia during reperfusion dramatically influences the efficacy of this intervention. Brief periods (1 to 3 hours) of hypothermia ameliorate neurologic injury only if initiated immediately after reperfusion (Kuboyama et al., 1993) and do not prevent the delayed development of neurologic injury (Dietrich et al., 1993). In contrast, prolonged hypothermia (5 hours or longer) is beneficial even if initiated several hours after reperfusion from forebrain ischemia (Coimbra and Wieloch, 1994; Coimbra et al., 1996), and this benefit appears to be permanent (Colbourne and Corbett, 1994, 1995; Coimbra et al., 1996).
The time window during which induced hypothermia is most efficacious for improving neurologic outcome provides clues as to the basis for the effects of hypothermia. For example, free radical generation, excitatory amino acid release, and metabolic failure occur primarily during early reperfusion (Rehncrona et al., 1979; Busto et al., 1989; Eleff et al., 1991; Katz et al., 1998), and are unlikely to be affected by hypothermia induced hours after reperfusion. In contrast, increased expression of stress-related proteins progresses over many hours after reperfusion (Gonzalez et al., 1989; Simon et al., 1991; Takemoto et al., 1995; Gillardon et al., 1996) and could be altered by delayed hypothermia. Stress-related proteins include heat shock proteins and immediate early gene products that can influence the subsequent repair, activation, or synthesis of other gene products. In addition to these mechanistic implications, clinical studies indicate that feasible induction of therapeutic hypothermia for survivors of cardiac arrest requires approximately 1 hour using conventional techniques (Bernard et al., 1997). Thus, immediate induction of hypothermia after reperfusion has less chance of clinical application for unpredictable ischemia than does delayed induction.
This study examined the effect of prolonged, mild hypothermia induced during reperfusion in a rat model of asphyxial cardiac arrest that reproduces many features of the global cerebral ischemia produced during human cardiac arrest (Katz et al., 1995). In contrast to arterial occlusion models (Perito et al., 1987), circulatory arrest produces uniform and complete ischemia that injures both forebrain and hindbrain regions (Radovsky et al., 1997). Previous studies have noted that different models of global cerebral ischemia exhibit different responses to therapeutic interventions (Vaagnes et al., 1997), and arterial occlusion models of cerebral ischemia may not predict the effect of an intervention on cardiac arrest. Mild hypothermia (32 to 34°C) previously has been found sufficient for improving recovery after circulatory arrest when induced immediately on reperfusion (Leonov et al., 1990; Weinrauch et al., 1992; Wass et al., 1995). Therefore, this study tested whether delayed induction of hypothermia would improve functional and histologic outcome using a cardiac arrest model. Furthermore, this study examined whether prolonged hypothermia during reperfusion altered the expression of stress-related proteins in distinct brain regions. The data suggest that hypothermia during reperfusion improves neurologic recovery and produces anatomically specific changes in the expression of particular stress-induced proteins.
MATERIALS AND METHODS
Animal protocols were approved by the University of Pittsburgh Animal Care and Use Committee. Male Sprague-Dawley rats (Harlan, Indianapolis, IN, U.S.A.) were housed two per cage with free access to food and water. On the day of the ischemic insult, the average weight of rats was 356 ± 29 g.
Experiment 1
The effect of hypothermia on neurologic recovery was examined in 28 rats. Global cerebral ischemia was induced in rats by 8 minutes of asphyxia resulting in reversible circulatory arrest. Rats were randomly assigned to one of three treatment groups: normothermia, immediate hypothermia, or delayed hypothermia. Rats in the normothermia group (N = 12) were maintained at their normal temperature (36.8°C) for 24 hours after resuscitation. Cooling was initiated immediately after resuscitation in the immediate hypothermia group (N = 8), reaching a brain temperature of 33°C in less than 10 minutes. Rats in the delayed hypothermia group (N = 8) were maintained at their normal temperature (36.8°C) for 45 minutes after resuscitation and were cooled to 33°C during the interval between 45 and 60 minutes. Hypothermia was maintained for 24 hours in both the immediate hypothermia and delayed hypothermia groups. After this period of hypothermia, rats were rewarmed to 36.8°C over 2 hours. Neurobehavioral testing was conducted daily, and brains were collected for histologic analysis on day 14 of survival.
Experiment 2
Separate rats were employed for biochemical and immunohistochemical studies. Global cerebral ischemia was induced using the identical asphyxial cardiac arrest as in experiment 1. Rats (N = 3 per group) were subjected either to normothermia (36.8°C continuously) or delayed hypothermia (33°C beginning 60 minutes after resuscitation) during reperfusion. Brains were collected for biochemical analysis after 6, 12, or 24 hours of reperfusion. Brains from other rats (N = 6) were collected for immunohistochemical analysis after 24 hours of reperfusion. Sham-operated rats subjected to anesthesia and femoral cut-downs without asphyxia were used as controls for biochemical and immunohistochemical studies.
Temperature measurements
At least 4 days before the asphyxial insult, rats were lightly anesthetized with 1.5% halothane in oxygen and prepared for intracranial temperature monitoring. A 4.5-mm × 20-gauge stainless-steel guide cannula was stereotactically placed into the frontoparietal cortex (posterior −2 mm, lateral +2 mm relative to bregma, and 2 mm below skull surface according to the Atlas of Paxinos and Watson, [1997]). Dental cement and skull screws (#080, Small Parts Inc., Miami Lakes, FL, U.S.A.) were used to secure the cannula and protective cap designed to protect and secure the temperature probe housing. Battery-operated wireless temperature probes (XH-FM-BP, MiniMitter, Sun River, OR, U.S.A.) were inserted on the day before ischemic insult. Brain probes were calibrated against a 0.1°-C graduated mercury thermometer (Fisher Scientific, Pittsburgh, PA, U.S.A.) after each battery change and were checked for accuracy between rats. The 6 mm × 25-gauge temperature probe extended 1.5 mm beyond the guide cannula into the cerebral cortex. Ultimate probe positions were confirmed by tracks visible on histologic sections to terminate in the superficial to middle layers of the cortex.
The temperature at the tip of the brain probe was transmitted to an FM receiver and recorded every 3 seconds by a PC-compatible computer using commercial software (Vital View, MiniMitter, Sun River, OR, U.S.A.). This arrangement allowed continuous temperature monitoring in the freely moving rat (Colbourne et al., 1996). Baseline recordings revealed diurnal and activity-related changes in brain temperature with a mean brain temperature of 36.8°C. Moreover, the temperature data were used on-line to thermostatically control a 100-W heating lamp and a cooling fan by means of software-driven relays. During active temperature control, the heating lamp and fan were placed approximately 30 cm away from the rat and were directed at the thorax. Rat cooling was facilitated by use of a cold water spray and by shaving of the back. This arrangement allowed for precise automated control of brain temperature. Frequent checks were made during these procedures to ensure accurate temperature control, and rats were considered out of protocol if the records demonstrated a 1-minute deviation of more than 1.0°C from the desired set-point.
Asphyxial cardiac arrest
Global cerebral ischemia was induced by asphyxial cardiac arrest as described previously (Katz et al., 1995, 1998). Rats were anesthetized using halothane, intubated with a 14-gauge intravenous catheter, and placed on a mechanical ventilator (Harvard Rodent Ventilator, Harvard Apparatus, South Natick, MA, U.S.A.). The left femoral vein and artery were exposed by means of an incision and cannulated with polyethylene catheters (PE-50 tubing, Harvard Apparatus, South Natick, MA, U.S.A.). The arterial catheter was connected to a pressure transducer for continuous arterial blood pressure recording and for arterial blood gas analysis (I-STAT, Sensor Devices Inc. Waukesha, WI, U.S.A.). Corrections for temperature were made to blood gas measurements in the hypothermia rats. In particular, pH and p
After reducing the fraction of inspired oxygen to 0.21 (room air) for 2 minutes, rats were chemically paralyzed with intravenous vecuronium (2 mg/kg). In order to minimize anesthetic effects, halothane was weaned and discontinued during this period. Unparalyzed rats do not regain consciousness during a similar interval, and profound hypoxemia ensues quickly after the onset of asphyxia (Katz et al., 1998). Asphyxia was induced by disconnecting the ventilator at the end of expiration for 8 minutes. Complete circulatory arrest was documented by a fall of central arterial blood pressure to equal central venous pressure. Circulatory arrest occurred within 200 seconds in all rats. After 8 minutes the ventilator was reconnected, and ventilation resumed with pure oxygen at a rate of 60 respirations/minute. Intravenous epinephrine (0.005 mg/kg) and bicarbonate (1.0 mEq/kg) were administered, and external chest compressions were performed at a rate of 200 compressions/minute. Rats without return of spontaneous circulation within 2 minutes were considered out of protocol and were excluded from analyses. Physiologic measures were recorded before the arrest (baseline), and at 10, 30, and 60 minutes after resuscitation.
After stabilization, arterial and venous catheters were removed and the femoral vessels ligated. Wounds were cleaned and sutured. After confirming adequate spontaneous respirations on room air, rats were extubated. All rats received ampicillin, 50 mg/kg subcutaneously, at the beginning of the surgical procedures and at 12-hour intervals during the first 24 hours of recovery. During the entire 14 days of recovery, rats were offered food and water by hand until adequate self-feeding was demonstrated. All rats requiring assistance with feeding were given subcutaneous injections of 5% dextrose in 0.45% saline (20 mL/kg) at 12-hour intervals.
Neurobehavioral measurements
Rats were evaluated daily using a standardized neurologic examination similar to the test described previously (Katz et al., 1995). This test measured sensorimotor function (grooming, forepaw grasp, limb movement, and sensation for each limb) and a global assessment of level of consciousness, respirations, cranial nerve function, axial body tone, righting, locomotion, and balance. A neurologic deficit score is assigned from 0 to 26, with a higher number corresponding to a more severe insult. The scoring system used was found to correlate well with the previously validated neurologic deficit score (Katz et al., 1995) but had less interrater variability (Spearman's r = 0.999, N = 88, P < 0.001). Rats were tested by an investigator (C.C.) who was blinded to the experimental condition of the rat. On the first day after resuscitation, rats were tested after the rewarming period.
On postrecovery days 8, 9, and 10, exploration, habituation, and locomotion were examined in rats with neurologic deficit scores of 5 or less using an open-field test. Rats with neurologic deficit scores greater than 5 were unable to ambulate in the test chamber and were not tested. For comparison, experimentally naïve control rats were also tested in the open field. The open field was a 100-cm × 100-cm × 30-cm Plexiglas box with black walls and a Plexiglas floor. Rats were habituated to the darkened (25-W black light) and sound-attenuated testing room for 60 minutes before being placed in the open field. Each rat was placed into one corner of the open field, and an observer blinded to the prior treatment recorded the activity over the next 20 minutes. Locomotor activity was quantified by dividing the open field into a 4 × 4 grid of 25-cm × 25-cm square sectors. Total crossings between sectors and total rearings per 5-minute epoch were recorded for each rat.
Histology
Halothane-anesthetized rats were transcardially perfused with 100 mL of 0.1 mol/L phosphate-buffered saline (PBS), followed by 100 mL of 4% paraformaldehyde in PBS. Brains were removed and placed in 4% paraformaldehyde in PBS at 4°C. Twenty-four hours before sectioning, brains were placed in 20% sucrose in PBS. Coronal sections (20-μm thick) were sliced on a freezing microtome, mounted on slides, and stained with hematoxylin and eosin. The number of pyramidal neurons in an 0.7-mm field was counted in the CA1 region of the anterior hippocampus (corresponding to −3.60 mm from bregma according to the Atlas of Paxinos and Watson, 1997). The number of Purkinje cells was counted in a 0.7-mm field in the second lobule of cerebellum (corresponding to −1.3 mm interaural). Two fields from two separate sections at the same level were examined by two separate observers and results averaged (total of 4 observations per brain). The CA1 region was selected because of its known vulnerability to ischemia, and because its laminar structure and characteristic cell morphology allow for quantifiable assessments of neuronal survival. Although other regions are injured after asphyxial cardiac arrest, these regions require more subjective neuropatho-logic assessments (Katz et al., 1995; Radovsky et al., 1997). Investigators were blinded to the treatment group while examining histology.
Immunoblotting
At 6, 12, or 24 hours after resuscitation from asphyxial cardiac arrest, rats were anesthetized with halothane and decapitated. Brains were quickly dissected into ice-cold PBS. After cooling for 60 seconds, the hippocampus, frontal cortex, cerebellum, and brainstem were dissected on a cold metal stage and then frozen at −70°C. Frozen tissue was solubilized by sonication for 5 seconds on ice in 0.5 mL of buffer (50 mmol/L Tris-HCl, 100 mmol/L NaCl, 5 mmol/L EDTA, pH 7.4) containing detergent (1.0% Nonidet P-40), protease inhibitors (100 mmol/L PMSF, 1 μg/mL aprotinin, 5 μg/mL leupeptin, 1 μg/mL pepstatin) and phosphatase inhibitors (1 mmol/L Na3VO4, 1 mmol/L NaF, 1 μg/mL microcystin). After centrifugation of this lysate at 12, 000 × g for 15 minutes, the supernatant was collected and stored at −70°C. Protein concentration was determined by the Bradford method using a Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA, U.S.A.).
Equal aliquots of protein (20 μg) were denatured in a sodium dodecyl sulfate sample buffer and separated electrophoretically on 10% polyacrylamide gels containing sodium dodecyl sulfate. Gels were then electrophoretically transferred to Immobilon-P membranes (Millipore Corp., Bedford, MA, U.S.A.). Immunolabeling of the resulting blots followed standard protocols at room temperature. In brief, membranes were incubated for 1 hour with 5% dry milk in PBS containing 0.1% Tween-20 before incubation for 2 hours with a primary antibody. Primary antibodies included a 1:2000 dilution of anti-Hsp70 (SPA-810, StressGene, Victoria, BC, Canada) and 1:10, 000 dilution of anit-Hsp40 (SPA-400, StressGene, Victoria, BC, Canada). Labeling with antiactin antibody (Boehringer Mannheim, Mannheim, Germany) was used to confirm equal loading and transfer of protein. A horseradish peroxidase-conjugated species-specific anti-IgG secondary antibody (Bio-Rad Laboratories, Hercules, CA, U.S.A.) was used in 1:10, 000 dilution to label the primary antibody, and the resulting complexes visualized by enhanced chemiluminescence using a commercial kit (Renaissance, New England Nuclear, Boston, MA, U.S.A.). The labeled membranes were exposed to X-ray film (X-OMAT, Kodak, Rochester, NY, U.S.A.) for 5 to 15 minutes. The amount of protein loaded and exposure time was adjusted to yield an optical density that corresponded to the linear range of protein concentration. The resulting images from duplicate experiments were scanned and quantified using the NIH Image software.
Immunohistochemistry
To confirm the anatomic localization of Hsp70 after the asphyxial cardiac arrest, rats were instrumented and treated as in the behavioral and biochemical study. At 24 hours after normothermia (N = 3) or delayed hypothermia (N = 3) resuscitation from asphyxial cardiac arrest, rats were deeply anesthetized with halothane. Each rat was transcardially perfused with 100 mL PBS followed by 100 mL of 4% paraformaldehyde in PBS. The brain of each rat was removed and postfixed in 4% paraformaldehyde in PBS at 4°C for 6 to 12 hours. Brains were then transferred to 20% sucrose in PBS at 4°C for 12 to 24 hours. Brains were cut on a freezing microtome into 20-μm sections. These sections were stored at −20°C in a cryopreser-vative solution (30% ethylene glycol, 30% sucrose, 1 % polyvinyl-pyrrolidone, 0.1 mol/L PBS, pH 7.2) before further processing.
Floating sections were immunostained using standard protocols. In brief, sections were washed at room temperature in PBS for 15 minutes, followed by 1 % horse serum (in 0.1 mol/L PBS, pH 7.4) for 30 minutes. Sections were then incubated in 1:1000 or 1:2000 dilutions of primary antibody (SPA-810, StressGene, Victoria, BC, Canada) at 4°C for 48 hours. After washing in PBS, immunolabeling was visualized using the Vectastain Elite ABC kit (Vector Laboratories Inc, Burlingame, CA, U.S.A.). Sections were incubated with the universal biotinylated secondary antibody (horse antimouse and antirabbit IgG). This biotinylated antibody was labeled with a streptavidin-HRP conjugate and the resulting complexes developed with diaminobenzamide (0.5 mg/mL in 0.1 mol/L PBS, pH 7.4) and H2O2 (0.03%) in PBS.
Statistics
Physiologic variables were compared by repeated-measures ANOVA, followed by post-hoc Tukey tests. Mortality between groups was compared using a survival curve analysis with a Mantel-Cox log rank test for differences in survival. Neurologic deficit scores were compared using nonparametric ANOVA (Kruskal-Wallis test) or the Mann-Whitney U-test. Open-field data were analyzed by repeated-measures ANOVA with epoch and days as within-subject factors and group as the between-subject factor. Histologic analysis was compared by univariate ANOVA. Significant ANOVA effects were examined by post-hoc Tukey tests. For biochemical measures, differences between normothermia and delayed hypothermia groups were examined by multiple factor ANOVA. In all cases, an alpha error rate < 0.05 was used as the criterion for significance.
RESULTS
Baseline physiologic parameters did not differ between groups
Baseline physiologic measurements did not differ between any of the treatment groups (Table 1). Average halothane time was 42.4 ± 7.0 minutes. At baseline, brain temperature was poorly correlated with tympanic (Pearson = −0.12, N = 27) and rectal (Pearson = 0.03, N = 23) temperature. Temperature at the different recording sites differed by as much as 1.2°C; this variability in temperature sites confirms previous studies (Busto et al., 1987; Colbourne et al., 1993). Spontaneous circulation was restored after 71 ± 10 seconds of chest compressions and mechanical ventilation. There was a significant effect of time on heart rate (F[3, 72] = 34.51, P < 0.001), mean arterial pressure (F[3, 72] = 79.43, P < 0.001), p
Physiologic variables
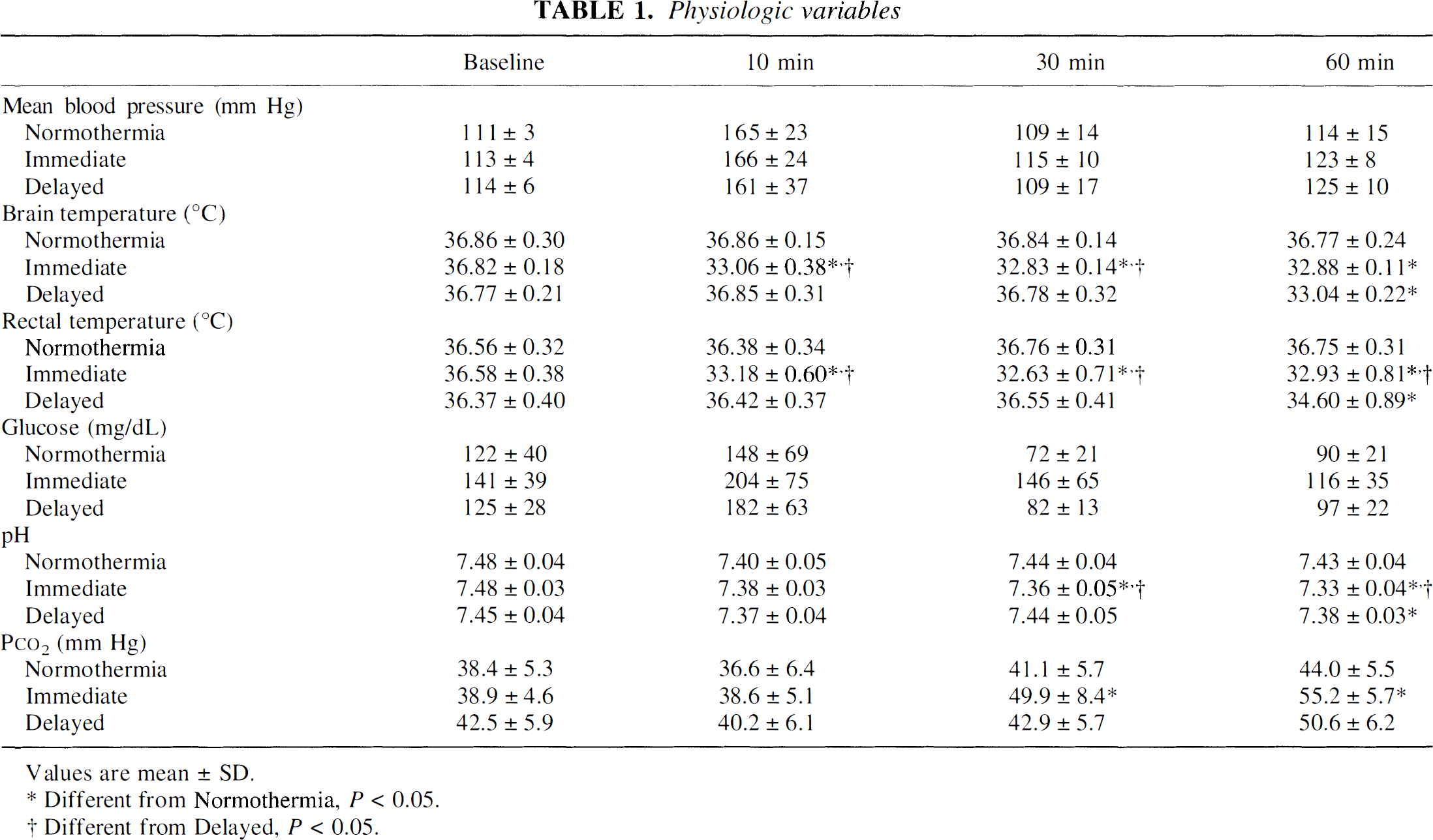
Values are mean ± SD.
Different from Normothermia, P< 0.05.
Different from Delayed, P < 0.05.
In rats destined for immunohistochemistry or biochemical analyses, physiologic variables were monitored for up to 12 hours of reperfusion with normothermia (N = 4) or delayed hypothermia (N = 4). Because of concern for infectious or thromboembolic complications associated with indwelling arterial catheters, this prolonged monitoring was not performed in rats destined for 14-day survival. Mean arterial pressure, heart rate, glucose, and blood gases did not vary between treatments during the first 12 hours after resuscitation (Table 2). In normothermia rats, hemodynamic variables and blood gas values did not change between 12 hours and 24 hours. Hypothermia rats resumed spontaneous activity during this same interval and would not tolerate invasive monitoring without potentially confounding sedation.
Prolonged monitoring of physiologic variables
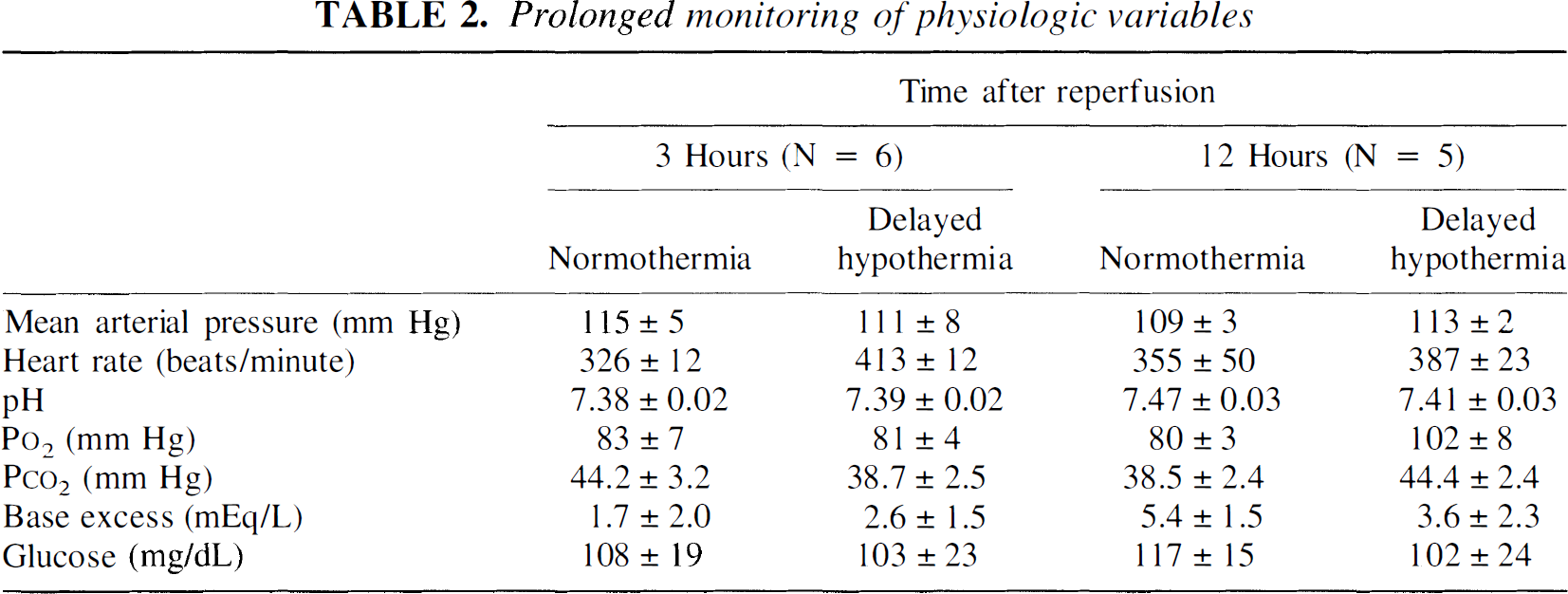
Different from Delayed, P < 0.05.
Experiment 1
Hypothermia reduced mortality after reperfusion. Mortality in the normothermia group was greater than in the hypothermia groups (Mantel-Cox log rank = 6.13, df = 2, P < 0.05). All hypothermia rats survived to day 14, whereas only 66% of normothermia rats survived to day 14 postinsult. Two deaths occurred during the first 24 hours, one on day 4 and another on day 7. Gross internal examination did not reveal an obvious cause for these deaths, and available physiologic data including weight loss did not distinguish these rats from surviving members of the normothermia group.
Neurologic recovery was improved by induced hypothermia. After cardiac arrest, all rats remained immobile, without righting reflex or response to stimuli for approximately 12 hours. Normothermia rats remained unable to ambulate, eat, or groom for 3 to 5 days. In contrast, all hypothermia rats exhibited some locomotion within the first 24 hours after resuscitation. Persistent deficits in all groups included spasticity of the limbs and loss of balance.
Confirming these observations, neurobehavioral scores for the normothermia group were worse than in the hypothermia groups on all 14 days of measurement (Fig. 1), using both the established (Katz et al., 1995) and modified neurologic deficit scores. Scores did not differ between delayed and immediate hypothermia groups (Fig. 1). There was a significant effect of day on neurologic deficit scores (Kruskal Wallis = 85.92, P < 0.001), reflecting that scores improved for all groups over time. Neurologic deficits persisted in the normothermia group during the 14 days, in contrast to the full recovery seen in hypothermia groups within 2 to 3 days.
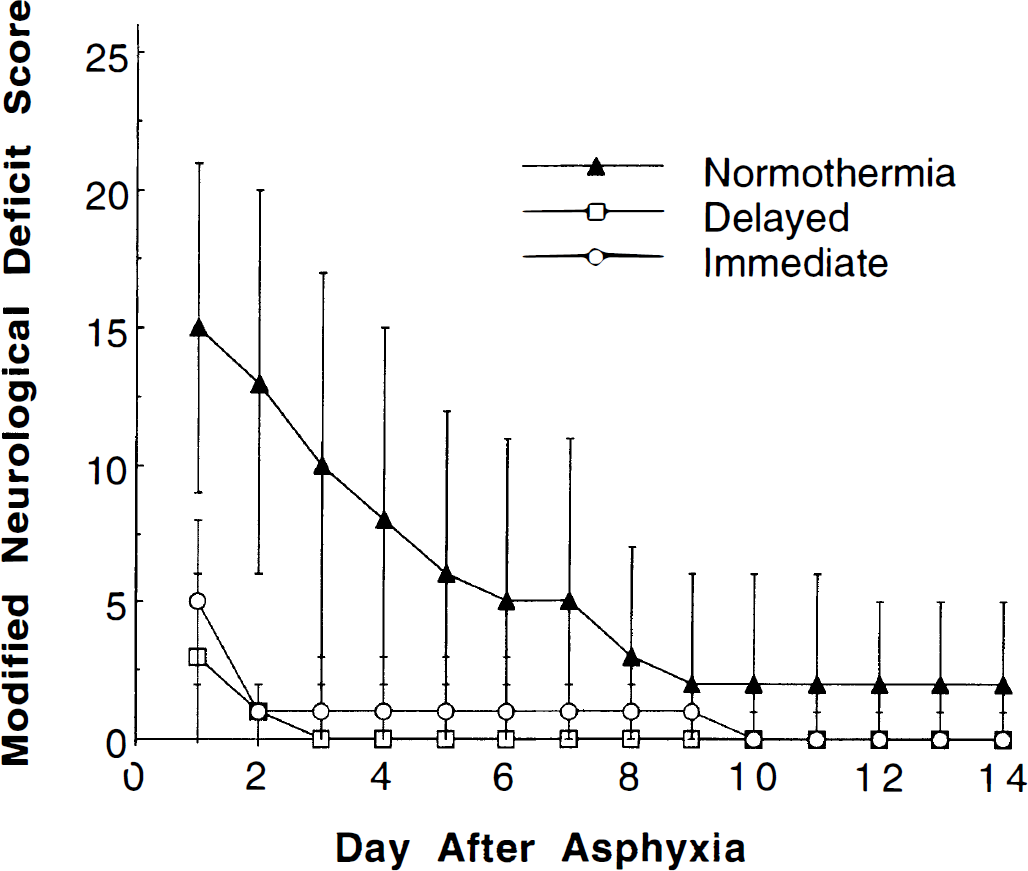
Modified neurologic deficit score for the normothermia, immediate hypothermia, and delayed hypothermia group (mean ± SD). Higher scores correspond to greater neurologic deficits. Both hypothermia groups were significantly improved compared to the normothermia group on all 14 days (Kruskal Wallis, P < 0.05). The immediate hypothermia group did not differ from the delayed hypothermia group.
Rats recovering from cardiac arrest were less active in an open-field test. Behavioral testing in the open field revealed a decrement in initial exploration and impaired habituation among rats recovering from asphyxia (Fig. 2). All rats subjected to asphyxial cardiac arrest and resuscitation exhibited less total activity than naïve rats. This decrement in total activity was reflected by a significant effect of group on sector entries (F[3, 16] = 5.49, P < 0.01). Habituation of rats to the open field was observed in all groups as a decrease in activity between the first and last epoch. Habituation was reflected by a significant effect of time on sector entries (F[3, 35] = 136.72, P < 0.001) and on rearings (F[3, 54] = 29.99, P < 0.001) (Fig. 2). Resuscitated rats also exhibited a smaller relative decrement in activity with time (group × time interaction, F[9, 48] = 3.64, P < 0.001), perhaps indicating less habituation to the test chamber. Interestingly, among asphyxiated rats, the immediate hypothermia group exhibited the greatest decrease in activity between the first and last epoch, suggesting that habituation in this group resembled the habituation in control rats. However, post-hoc comparisons could not distinguish the immediate hypothermia from the delayed hypothermia or normothermia groups. No differences between groups were observed for rearings. Furthermore, no effect of testing day was noted for sector entries or rearings, and data from all 3 days were combined for illustration (Fig. 2).
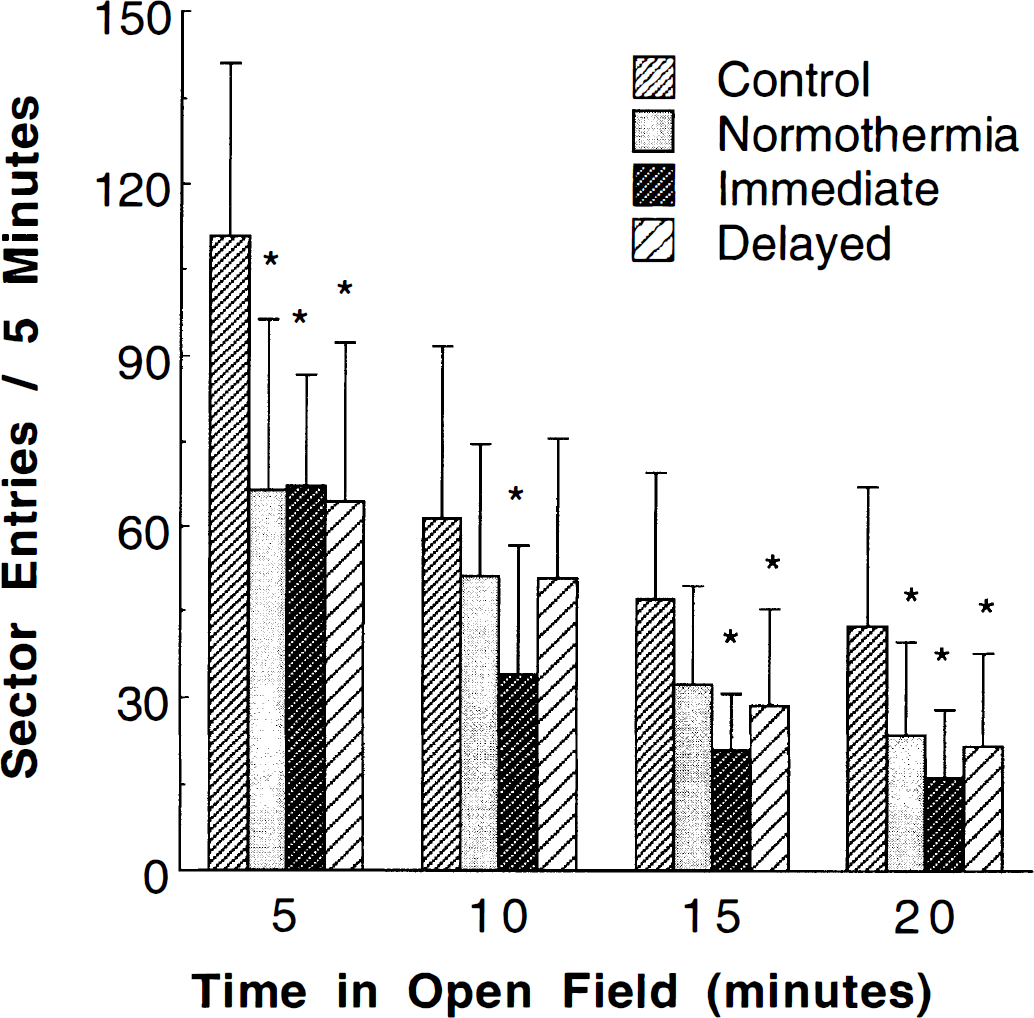
Mean number of sector entries per 5-minute epoch in the open-field experiment. All asphyxia groups showed decreased total activity compared to the control group (*, P < 0.05 compared to controls). Habituation to the testing chamber was reflected by a decrease in activity between the first and last epoch. The percentage decrease in activity between first and last epochs was less in the asphyxia groups than in the control group.
Delayed hypothermia improved histologic outcome in CA1 sector of hippocampus. Neuronal density in the hippocampus varied by treatment group (F[3, 102] = 11.59, P < 0.001). Total neuron number in the CA1 region of the hippocampus was decreased by 42% in the normothermia group relative to shams (normothermia = 40.0 ± 18 and sham = 68.8 ± 6.5). Immediate and delayed hypothermia rats showed only a 22% decrease in total cell number compared to shams (Fig. 3). There was no difference between hypothermia groups, and both hypothermia groups had significantly more neurons than the normothermia group. Histologic damage in the hippocampus was symmetrical after asphyxia and reperfusion. More prominent gliosis was evident in sections from normothermia rats (data not shown). In contrast to the hippocampus, no difference was found in the number of Purkinje cells of cerebellum between groups (neurons per 0.7-mm field: controls = 14.4 ± 2.1, normothermia = 16.8 ± 4.3, immediate hypothermia = 15.9 ± 3.4, and delayed hypothermia = 14.9 ± 3.2).
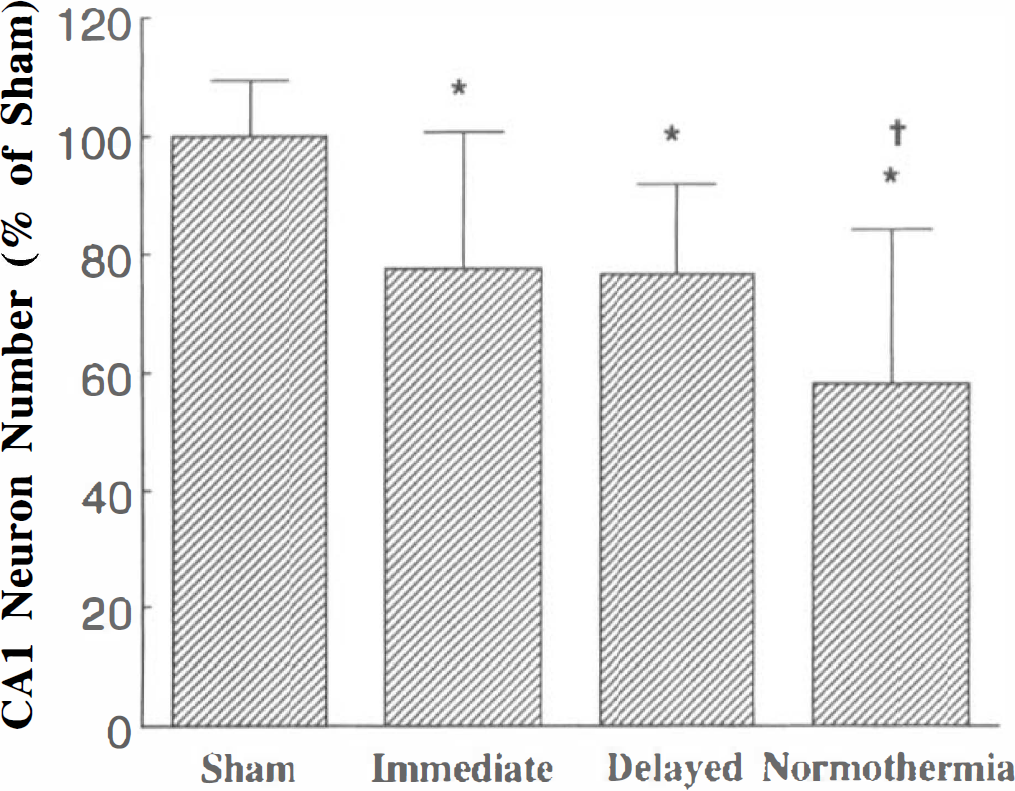
Neurons per high-powered field in the CA1 region of the hippocampus. Neuron number is expressed as percent of neuron number in shams. All asphyxia groups differed significantly from the sham group (*, P < 0.05). The normothermia group also differed from both hypothermia groups (§, P < 0.05). There was no difference between the hypothermia groups. Relative to shams, the normothermia group showed a 42% decrease in total cell number, compared to a 22% decrease in the hypothermia groups.
Experiment 2
Delayed hypothermia selectively decreased induction of Hsp70 in hippocampus. Delayed hypothermia selectively altered the cellular response to ischemia and reperfusion in the hippocampus. The time course of Hsp70 levels at 6, 12, and 24 hours after ischemia showed a significant effect of time (F(2, 17) = 38.4, P < 0.001) and group (F(1, 17) = 14.9, P < 0.002) between normothermia and delayed hypothermia rats. As expected, levels of stress-inducible Hsp70 are undetectable by immunoblot analysis in hippocampal or cerebellar extracts from control rats that did not undergo asphyxia. In contrast, Hsp70 was detected at 6 hours after asphyxia and markedly increased at 12 hours after asphyxia in normothermia rats (Fig. 4). Interestingly, in the hippocampus of delayed hypothermia rats at 12 hours, Hsp70 levels were reduced by 55% relative to normothermia rats (t = 5.39, df = 4, P < 0.05). This decrease was not seen in the cerebellum of delayed hypothermia rats. Reprobing of membranes for actin confirmed that there were no significant differences in the amounts of protein loaded and transferred between groups.

Immunoblot analysis of Hsp70 concentration in the hippocampus (A). Protein extracts were analyzed from 3 different rats in each treatment condition. Hsp70 was undetectable in shams but was robustly increased after asphyxia. Time course of Hsp70 levels in hippocampus 6, 12 and 24 hours after ischemia (B).
In contrast to Hsp70, which was expressed only after injury, Hsp40 was detected in control rats that were not subjected to any ischemia (Fig. 5). Relative to controls, hippocampal levels of Hsp40 were decreased at 6 hours and then increased at 12 hours after the asphyxial cardiac arrest, as indicated by a significant effect of time (F[2, 17] = 15.82, P < 0.001). Hsp40 protein levels in the normothermia and hypothermia groups did not differ. No significant induction of Hsp40 in cerebellum was detected (data not shown).
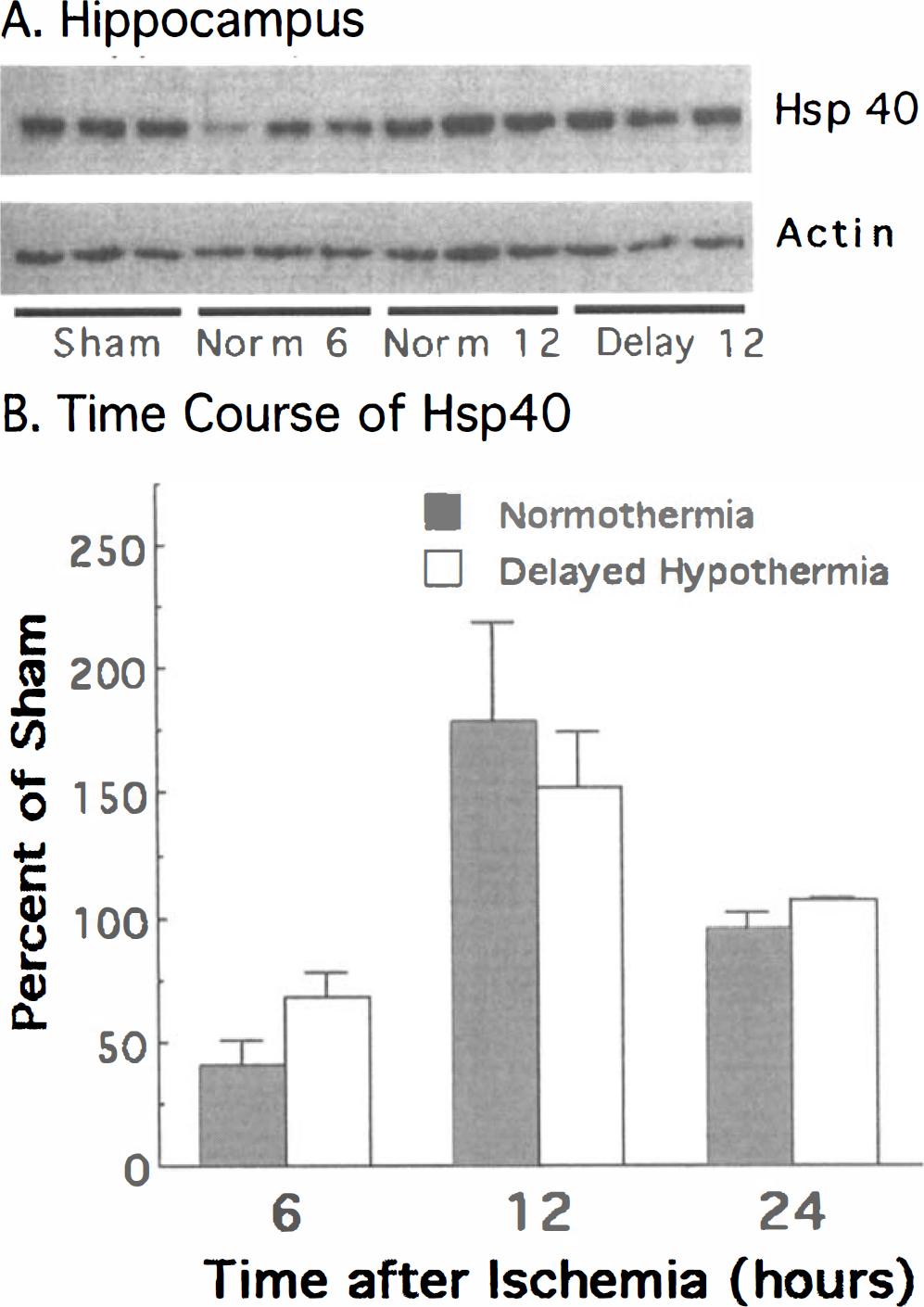
Immunoblot analysis of Hsp40 concentration in the hippocampus (A). Hsp40 is detected in shams, and Hsp40 levels decrease transiently at 6 hours, increase above sham levels at 12 hours after asphyxia, and then return to baseline at 24 hours (B). Groups are the same as in Fig. 4.
Hsp70 is localized to neurons in the hippocampus and cerebellum after cardiac arrest. Immunohistochemistry revealed the cellular localization of Hsp70 in the cerebellum and hippocampus of rats subjected to 8 minutes of asphyxia and 24 hours of normothermic and delayed hypothermic reperfusion. Hsp70 could be detected in the cytoplasm of almost every neuron of the CA1 sector of hippocampus (Fig. 6). Less intense staining was observed in the CA2 and CA3 sectors of the hippocampus. No difference in the localization of Hsp70 staining could be detected between normothermic versus delayed hypothermic reperfusion (Fig. 6B). A subset of neurons in the dentate gyrus also expressed Hsp70. In the cerebellum, Purkinje cells exhibited the most intense Hsp70 immunoreactivity, although some staining was evident throughout the molecular layer. No Hsp70 immunoreactivity was observed in glial cells after this duration of asphyxia. Hsp40 immunoreactivity was localized in the pyramidal cells of all hippocampal regions, the Purkinje cells, and molecular layer of cerebellum in both control rats and rats resuscitated from asphyxia (data not shown).
DISCUSSION
This study characterized the improvement in functional outcome after complete cerebral ischemia by delayed induction of prolonged hypothermia. Delayed and prolonged hypothermia has been reported to improve recovery after forebrain ischemia induced by arterial occlusion (Coimbra and Wieloch, 1994) but has not been examined using a cardiac arrest model, although the response to therapeutic interventions is known to vary depending on the methods of inducing cerebral ischemia (Vaagnes et al., 1997). In contrast to asphyxial cardiac arrest, arterial occlusion models of forebrain ischemia produce incomplete ischemia in the forebrain and spare much of the hindbrain (Pulsinelli et al., 1982). As a result, asphyxia results in more severe and global neurobehavioral deficits after a shorter total duration of ischemia. Interestingly, the histologic damage observed after asphyxial cardiac arrest is less pronounced than that observed in the vessel occlusion models. For example, neuronal density in the CA1 region of the hippocampus is reduced by 42% after asphyxia and normothermic reperfusion (Fig. 3), although this duration of asphyxia is associated with 33% mortality and severe behavioral injury (Fig. 1). In contrast, 10 or 20 minutes of carotid occlusion can produce a complete loss of CA1 neurons, without changes in climbing, ambulation, or other gross behaviors (Pulsinelli et al., 1982). The more severe behavioral changes observed after asphyxia probably results from the fact that neurons are damaged in multiple brain regions, many of which are spared in the vessel occlusion models (Radovsky et al., 1997).
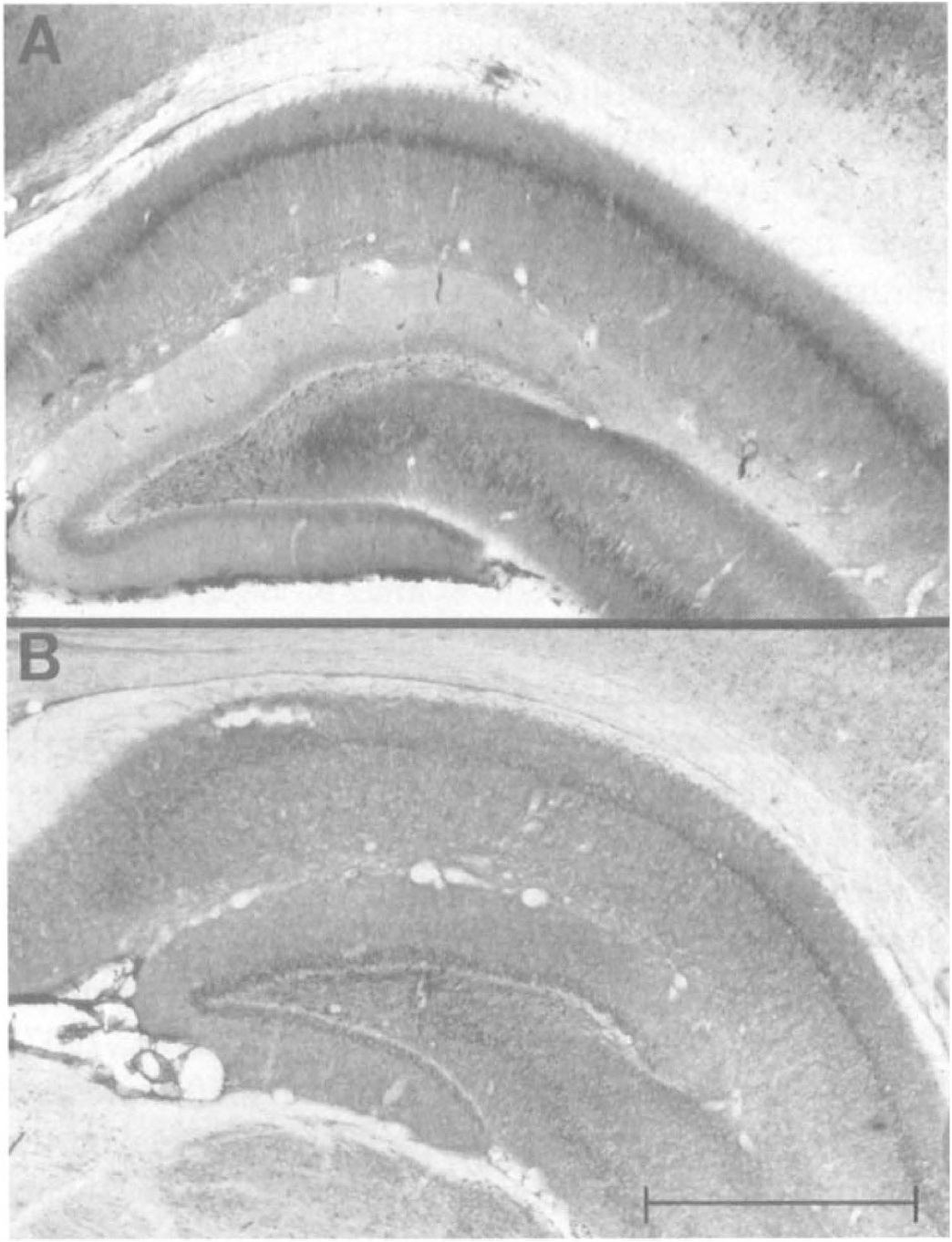
Photomicrographs of hippocampal region CA1 and dentate gyrus from normothermia (A) and delayed hypothermia (B) rats sacrificed 24 hours after an asphyxial cardiac arrest and immunostained for Hsp70. No Hsp70 was detected in control brains. The regional pattern of Hsp70 did not differ between temperature groups. In both regions, Hsp70 staining was localized to neurons. Almost every neuron in CA1 exhibited Hsp70 immunoreactivity. (Calibration bar = 1.0 mm).
Immediate and delayed induction of hypothermia were equally effective for improving functional recovery after asphyxia. The increased mortality of normothermia rats (33%) when compared to hypothermia rats (0%) probably reflects the severity of functional disability in this group. Extracerebral physiologic parameters measured during the first 12 hours (Table 2) as well as weight loss did not explain the increased mortality. Moreover, assessment of neurologic function using the neurobehavioral rating scales indicated that both hypothermia groups returned to normal within 2 to 3 days after reperfusion. In contrast, most surviving normothermia rats (5 of 8) exhibited gross neurologic deficits during the entire 2 weeks of recovery.
Induction of hypothermia decreased neuron loss in the CA1 region of the hippocampus. There was no difference between CA1 neuronal density in the delayed and immediate hypothermia groups (Fig. 3). However, normothermia rats had 42% fewer neurons in CA1 after 14 days of recovery. Although this single region is not responsible for all behavioral deficits encountered in resuscitated rats, it does serve as a quantifiable marker of structural brain damage. Interestingly, even the 22% decrement in CA1 neuron number observed in the hypothermia groups (Fig. 3) was associated with impaired exploratory activity and habituation in the open field (Fig. 2). The influence of hypothermia on neuronal degeneration in other regions exhibiting cellular stress is of great interest, but the hematoxylin and eosin staining used for this study provides for difficult quantification in less laminar and homogenous regions. Future quantification of damage in other regions may use specific degeneration stains (Schmued et al., 1997).
Hypothermia during reperfusion selectively alters expression of stress-related proteins. For example, the induced Hsp70 expression associated with neuronal stress in the hippocampus was attenuated by hypothermia during reperfusion (Fig. 4). This effect was selective because hypothermia did not inhibit the more modest induction of Hsp40 (Fig. 5), nor did it alter Hsp70 levels in cerebellum. Previous studies found that intraischemic hypothermia decreases heat shock protein message after global ischemia in swine (Shaver et al., 1995) and rat (Kawagoe et al., 1993), as well as after focal ischemia in rat (Chopp et al., 1992). Conversely, other studies demonstrate that increased postischemic temperature (≥39°C) augments neuronal injury as well as levels of hsp70 mRNA (Suga and Nowak, 1998; Kim et al., 1998). This study extended those previous reports by restricting hypothermia to reperfusion and by delaying the initiation of hypothermia until 60 minutes after reperfusion. Although not directly examined in this study, the decrease in Hsp70 levels observed for hypothermic rats may also result from decreased transcription of hsp70 message. Furthermore, the influence of temperature on Hsp70 has not been reported previously using a cardiac arrest model.
The basis for the decrease in hippocampal Hsp70 levels with delayed hypothermia is unknown. It is unlikely that the decrease in Hsp70 levels was a result of an overall decrease in protein synthesis, because induced expression of Hsp40 was not decreased (Fig. 5). Furthermore, Hsp70 levels were not decreased in the cerebellum. Previous work with temperature manipulations after cerebral ischemia induced by arterial occlusion suggests that Hsp70 expression is controlled at the level of mRNA synthesis (Chopp et al., 1992; Kawagoe et al., 1993; Shaver et al., 1995). Although increased levels of Hsp70 have been suggested to facilitate the recovery of neurons from ischemia (States et al., 1996; Plumier et al., 1997). The current data indicate that increased Hsp70 expression does not mediate the therapeutic effects of delayed hypothermia. However, these data do suggest that hypothermia during reperfusion can produce selective changes in induced protein expression and that these changes are anatomically specific.
The induction of Hsp70 illustrates the pattern of cell stress during reperfusion after asphyxial cardiac arrest. The CA1 sector of the hippocampus is the region of most intense and uniform Hsp70 labeling after 24 hours of reperfusion (Fig. 6). Hsp70 expression is correlated with cell injury in many models, including excitotoxicity (Gonzalez et al., 1989), global ischemia (Nowak 1985, 1991; Simon et al., 1991; Gaspary et al., 1995), and focal ischemia (States et al., 1996; Wagstaff et al., 1996). Thus, these data support the idea that the CA1 region is selectively vulnerable to neuronal degeneration after the ischemia of cardiac arrest and supports the examination of CA1 for quantification of histologic injury.
These data emphasize the importance of monitoring and controlling brain temperature during reperfusion after ischemia (Busto et al., 1987; Nurse and Corbett, 1994). Studies describing induction of hypothermia as a therapeutic maneuver for brain ischemia frequently examine the effect of cooling that is initiated during or immediately after the ischemic insult (Busto et al., 1987; Maier et al., 1998; Xiao et al., 1998). The influence of changes in temperature at some delay after reperfusion is less well known. Previous studies have reported that the delayed induction of mild hypothermia improves outcome after incomplete forebrain ischemia in gerbils (Nurse and Corbett, 1994) or in rats (Coimbra and Wieloch, 1994; Colbourne and Corbett, 1995). The importance of these delayed effects of cooling on outcome increases when using animal models that impair the normal behavioral control over temperature (Nurse and Corbett, 1996).
The present data did not delineate the maximum delay after asphyxia for which inducing hypothermia remained therapeutic. However, no significant differences in outcome variables were noted between the hypothermia groups, suggesting that initiation delays even longer than 60 minutes may be effective. Previous studies using cerebral artery occlusion models have found that induction of hypothermia as long as 6 hours after reperfusion can still reduce neuronal damage (Colbourne, and Corbett, 1994, 1995; Coimbra et al., 1996). In further support of this conclusion, three rats were not included in the present study because they unintentionally developed hypothermia at later time points (3, 4, and 7 hours) caused by a warming lamp failure. These rats exhibited very mild neurologic deficits and thus resembled hypothermia rats more than normothermia rats. The efficacy of delayed hypothermia after cardiac arrest is important for clinical applications, because therapeutic hypothermia requires approximately 1 hour for induction in patients resuscitated from cardiac arrest (Bernard et al., 1997). Taken together, these data suggest that hypothermia ameliorates some events leading to neuronal death that occur hours after reperfusion.
Therapeutic induction of hypothermia during reperfusion is unlikely to have any direct effect on free radical generation or excitatory amino acid release. Early reperfusion causes oxidative stress after asphyxia that is demonstrated by a depletion of endogenous antioxidants (Katz et al., 1998). However, the 50% depletion of reduced glutathione, ascorbate, and α-tocopherol observed after 10 minutes of reperfusion returns to normal by 120 minutes. Although hypothermia can decrease free radical activity, brief hypothermia during this period of oxidative stress does not alter ultimate neurologic outcome (Dietrich et al., 1993; Kuboyama et al., 1993). Similar to oxidative stress, increased intracellular calcium levels (Silver et al., 1990; Erecinska et al., 1992) and excitatory amino acid release (Busto et al., 1989) occur during early reperfusion and may serve to trigger events that mediate delayed neuronal death. However, the lack of lasting therapeutic benefit with brief hypothermia during early reperfusion argues that these mechanisms are not the targets of delayed and prolonged hypothermia. Finally, it is unlikely that hypothermia relieved active inhibition of protein synthesis commonly seen in response to ischemia (Nowak, 1985, 1991; DeGracia et al., 1996). In fact, expression of Hsp70 in the hippocampus was not impaired in the more severely injured normothermia group but was selectively decreased by delayed induction of hypothermia.
This study notes that hypothermia during reperfusion can selectively alter the expression of particular gene products. Recent studies have suggested that activation of caspases (Chen et al., 1998; Ni et al., 1998) or increased expression of apoptosis-promoting genes (Chen et al., 1996) may mediate neuronal death after transient ischemia. The present data suggest that hypothermia during reperfusion could affect these or other specific signaling and gene expression pathways. Further experiments will be required to determine the basis for this selectivity and the contribution of this altered expression to neuron survival.
In conclusion, the present study determined that mild hypothermia during reperfusion improves neurologic recovery after asphyxial cardiac arrest in rats. Prolonged mild hypothermia is equally effective when induced immediately after reperfusion or 1 hour after reperfusion. The improvement in behavioral and histologic outcome is associated with a regionally specific decrease in Hsp70 expression in the hippocampus. Taken together, these results suggest that induction of hypothermia after reperfusion may retard some of the delayed cellular events that mediate ischemic neuronal death. These data emphasize the importance of prolonged temperature monitoring and control during the recovery period in both mechanistic and outcome studies of experimental ischemia and support further explorations of induced hypothermia to improve recovery after clinical cerebral ischemia.
Footnotes
Acknowledgments
The authors thank Andrea J. Freel and Jeremy F. Monikowski for their assistance with histology.