
Abstract
We assessed the expression of several genes encoding pro-apoptotic cysteine proteases similar to interleukin-1β converting enzyme (ICE) and nematode Ced-3 in association with delayed neuronal death (DND) after transient forebrain ischemia in Mongolian gerbil. The levels of the two species of Nedd2 mRNA concomitantly increased about twofold in the whole forebrain at 3–6 h after 10-min ischemia and declined to the basal level by 24 h. In situ hybridization revealed that the Nedd2 gene was up-regulated in some neuronal populations in CA1 and CA3 regions of the hippocampus. In contrast, expression of ICE, CPP32/Yama/Apopain, and TX/ICErelII did not change within 48 h. These observations raise the possibility that up-regulation of Nedd2 in the vulnerable neurons may contribute to the proteolytic processes preceding the manifestation of apoptosis and/or necrosis after ischemic insult.
Keywords
In the developing mammalian nervous systems, more than half of all neurons are eliminated by apoptosis (Cowan et al., 1984; reviewed by Kerr et al., 1987). Genetic analyses have identified several genes essential for programmed cell death during the development of the nematode Caenorhabditis elegans (reviewed by Ellis et al., 1991). The programmed cell death regulated by a gene, ced-3, is suppressed by another gene, ced-9 (Hengartner and Horvitz, 1994). ced-3 encodes a cysteine protease similar to mammalian interleukin-1β converting enzyme (ICE) (Cerretti et al., 1992; Thornberry et al., 1992; Yuan et al., 1993), while ced-9 is a homologue of a mammalian proto-oncogene, bcl-2 (Tsujimoto et al., 1984). Overexpression of ICE induces apoptosis in cultured cells owing to its cysteine protease activity, which is countered by the expression of bcl-2 (Miura et al., 1993). However, no neurological abnormalities have been found in ICE-deficient mice (Kuida et al., 1995; Li et al., 1995).
From a cDNA library derived from mouse neuronal precursor cells, we isolated a gene, Nedd2, that is expressed in the ventricular zone of the neural tube and down-regulated during development (Kumar et al., 1992, 1994). Nedd2, also known as Ich-1 (Wang et al., 1994), encodes an ICE/Ced-3-like cysteine protease that can induce apoptosis in cultured cells, which is preventable by the expression of bcl-2 (Kumar et al., 1994). Expression of antisense Nedd2 mRNA delayed apoptosis of a hematopoietic cell line upon removal of cytokines, suggesting that Nedd2 is a component of the apoptotic pathway (Kumar, 1995a).
The Nedd2/Ich-1L precursor protein of 51 kDa (pro-Nedd2 or p51) encoded by Nedd2/Ich-1L mRNA is processed into two polypeptides of 19 kDa (p19) and 12 kDa (p12) (N. L. Harvey et al., unpublished), which probably assemble to form enzymatically active tetramer (p19/p12)2 as suggested for other ICE/Ced-3-like proteases (Thornberry et al., 1992; Fernandes-Alnemri et al., 1994, 1995; Wilson et al., 1994; Faucheu et al., 1995; Munday et al., 1995; Nicholson et al., 1995; Ramage et al., 1995; Tewari et al., 1995; Duan et al., 1996). The processing mechanisms of pro-Nedd2 and the substrates for active Nedd2 protease are still elusive (reviewed by Kumar and Lavin, 1996). A splicing variant, Nedd2
In contrast to the embryonic nervous systems, Nedd2 is markedly down-regulated in adult mouse brain (Kumar et al., 1994). It was, however, observed that a fraction of neurons in adult brain continue to express low but significant levels of Nedd2 mRNA. It is thus possible that under certain conditions, such as ischemia and epilepsy, Nedd2 is up-regulated, and it may result in triggering or aggravation of the following neuronal degeneration. We tested such a hypothesis in a paradigm of delayed neuronal death (DND) after transient forebrain ischemia in adult Mongolian gerbils (Kirino, 1982). This is a highly reproducible model of selective neuronal death, in which the hippocampal neurons in Ammon's horn degenerate in 48–72 h (Kirino, 1982), and eventually up to 95% of the CA1 pyramidal cells die (Bonnekoh et al., 1990). Morphological changes in most of the dying neurons suggest that the major cause of DND is necrosis (Deshpande et al., 1992). However, apoptosis is also implicated (reviewed by Charriaut-Marlangue et al., 1996), since the dying neurons show biochemical features characteristic of apoptosis, such as internucleosomal DNA fragmentation (MacManus et al., 1993; Okamoto et al., 1993; Sei et al., 1994; Nitatori et al., 1995) and interference with a nuclease inhibitor (Roberts-Lewis et al., 1993). Moreover, ischemia- and glutamate-induced neuronal death is suppressed by inhibitors of transcription and translation (Goto et al., 1990; Shigeno et al., 1990; Kure et al., 1991; Linnik et al., 1993), suggesting that newly expressed gene products are essential for the degenerative process, at least in part (reviewed by Schreiber and Baudry, 1995). In this article, we present some evidence suggesting that Nedd2 may be one of such genes.
MATERIALS AND METHODS
Operation of animals
We used male Mongolian gerbils (Meriones unguiculatus) weighing 60 ± 10 g. Under inhalation anesthesia with ether, the common carotid arteries were exposed bilaterally and occluded by aneurysmal clips for 10 min. During the procedure, the rectal temperature was maintained at 36–37°C by warming the animals with heat pads. Control animals underwent the same operation without clipping. After reperfusion, each animal was allowed free access to food and water. The forebrains were dissected at 6 h after sham operation and at 1, 3, 6, 12, 24, 48, and 72 h after reperfusion. The effectiveness of the ischemic operation was immunohistochemically confirmed by degradation of microtubule-associated protein-2–positive structures (Matesic and Lin, 1994) for randomly selected samples.
RNA extraction and Northern blot analysis
Total RNA was extracted from the whole forebrains by Isogen (Nippon Gene, Tokyo, Japan). Poly(A)+ RNA (5 μg) was purified by oligo-dT-cellulose chromatography, resolved on 1.2% agarose/2.2 M formaldehyde gels, and transferred to a nylon membrane. Random-primed, 32P-labeled mouse Nedd2 probes were hybridized at 65°C in a hybridization buffer, RapidHyb (Amersham, Arlington Heights, IL, U.S.A.), for 2.5 h. The final wash was performed at 60°C in 0.1 × standard sodium citrate containing 0.1% sodium dodecyl sulfate. After exposure on Imaging Plate, densitometry was done with a BAS2000 Image Analyzer (Fuji Photo Film, Tokyo, Japan). The same membranes were reprobed with mouse β-actin cDNA to assess the quality and the quantity of each RNA sample. The full insert excised from a mouse Nedd2 cDNA clone, MS N2.4 (Kumar et al., 1994), was used to generate the Nedd2 probe. The β-actin probe was prepared from mouse brain RNA by reverse transcription-coupled polymerase chain reaction (RT-PCR).
RT-PCR
Poly(A)+ RNA (1 μg) was reverse transcribed from oligo-dT primer in 33 μl reaction using First Strand Synthesis Kit (Pharmacia, Uppsala, Sweden). One-tenth of the reaction was amplified using 0.2 μM primers (see below) and 1.25 μCi [α-32P]dCTP in a standard PCR reaction mixture (50 μl) under the following conditions for 15 cycles: 0.5 min at 94°C, 1 min at 55°C (CPP32, TX) or 60°C (Nedd2, β-actin, ICE), and 1 min at 72°C. Each sample was resolved on 5% polyacrylamide gel and analyzed by densitometry as described. Control experiments were performed to determine the range of PCR cycles over which amplification efficiency remained constant and to demonstrate that the amount of PCR product was proportional to the amount of input RNA. The identity of each PCR product was confirmed by the size and by direct sequencing of the amplified cDNA eluted from the gel. The following pairs of oligonucleotides corresponding to the sequences within the coding regions were used as primers (the DNA database accession numbers and the nucleotide positions are shown in parentheses): mouse Nedd2 (D28492), CAGAATTTTGCACAGTTACCTGCACACC (769–796) and GCATAGCCACATATCATGTCTGAGCG (1084–1109); mouse β-actin (X03672), AGAAGAGCTATGAGCTGCCTGACG (790–813) and TACTTGCGCTCAGGAGGAGCAATG (1067–1090); mouse ICE (L28095), ACACGTCTTGCCCTCATTATCTGCA (527–551) and ATCTGGTGTTGAAGAGCAGAAAGC (1025–1048); human CPP32 (U13738), ATAAAATGGATTATCCTGAGATG (173–195) and TTTTATGACACGCCATGTCATCATC (598–622); human TX (Faucheu et al., 1995), GAGAATCTGACAGCCAGGGATATG (570–593) and TACTTCCTCTAGGTGGCAGCACCA (1044–1067).
In situ hybridization
The procedure was essentially the same as we described previously (Kumar et al., 1994). In brief, the forebrains were cryostat-sectioned coronally (10 μm thick). After fixation using 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4), permeabilization, acetylation, and dehydration, each section was incubated at 42°C for 12 h with the hybridization buffer containing digoxigenin (DIG)–labeled sense or antisense probe (50 ng/400 μl per slide), which was transcribed from MS N2.4 and alkali digested to 100–150 bases. After hybridization, the sections were washed twice with 2 × sodium, sodium phosphate, EDTA (SSPE) at room temperature for 20 min each, treated with 10 μg/ml of RNase A at 37°C for 15 min, then washed with 2 × SSPE at 42°C for 20 min twice and 0.2 × SSPE at 50°C for 20 min. Chromogenic reaction was done with alkaline phosphatase–conjugated anti-DIG goat IgG (Boehringer, Mannheim, Germany) at room temperature for 12 h following the manufacturer's protocol. In control hybridization experiments, the signal intensity detected by the antisense probe was proportional to the graded concentration of the unlabeled sense probe spotted and ultraviolet-cross-linked on a nitrocellulose membrane.
RESULTS
Temporal profile of Nedd2 expression in forebrain after ischemia
Expression of Nedd2 gene in the gerbil forebrain was analyzed first by Northern hybridization using mRNA derived from the animals at 6 h after sham operation or at 1, 3, 6, 12, 24, or 48 h after 10-min ischemia. Under stringent hybridization and washing conditions, radiolabeled mouse Nedd2 cDNA probe detected a single band of ∼3.5 kb, a size close to that of mouse Nedd2 transcripts (Fig. 1A). Densitometric analysis revealed that the expression of Nedd2, normalized against that of β-actin, was up-regulated about twofold in the whole forebrain around 3–6 h postischemia and decreased to subbasal levels by 24 h (Fig. 1B).
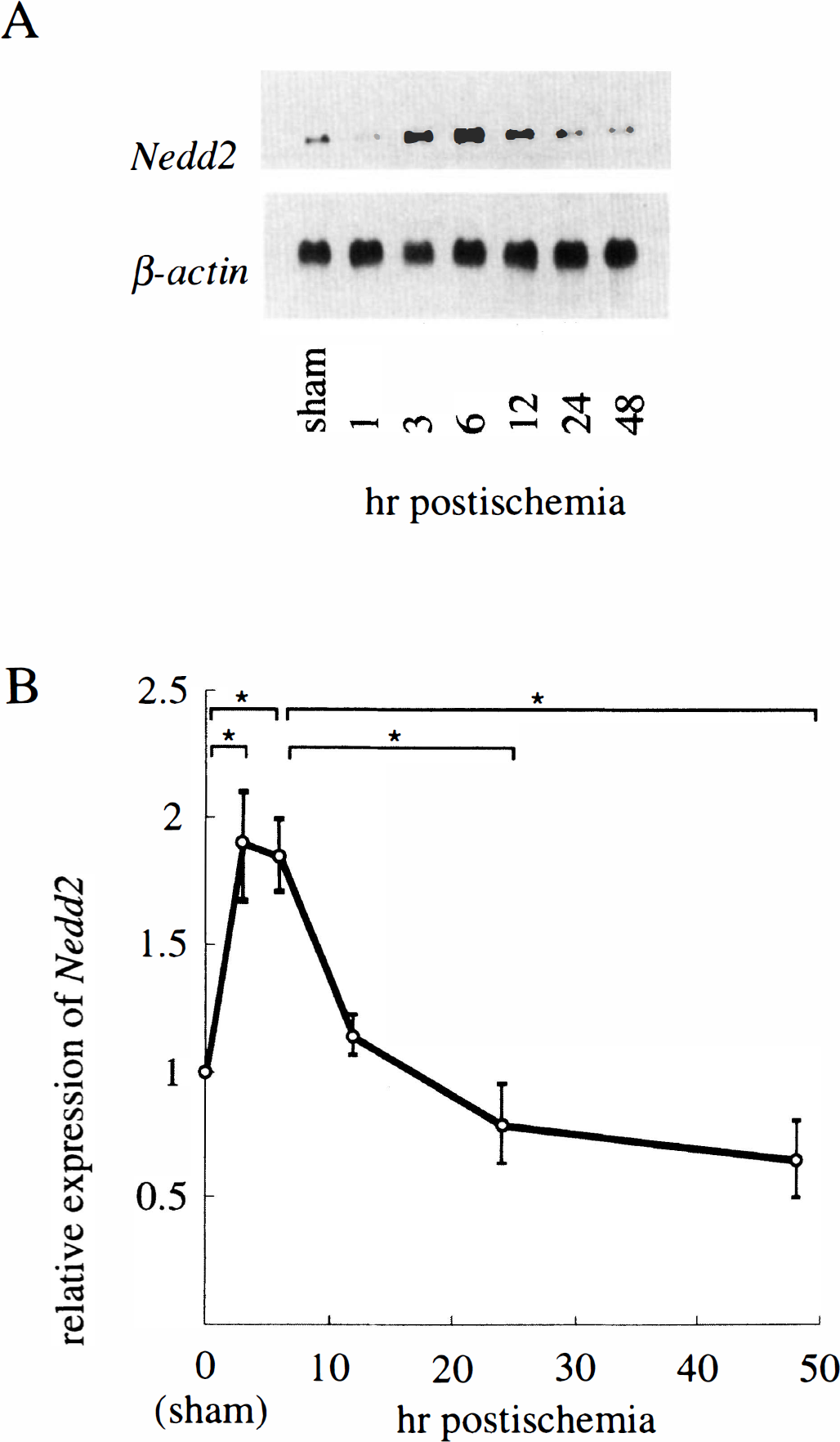
Northern blot analysis for Nedd2 expression in the gerbil forebrain after transient ischemia. (
Northern blot analysis failed to discriminate the two splicing variants derived from the Nedd2/Ich-1 gene, i.e., Nedd2/Ich-1L mRNA and Nedd2
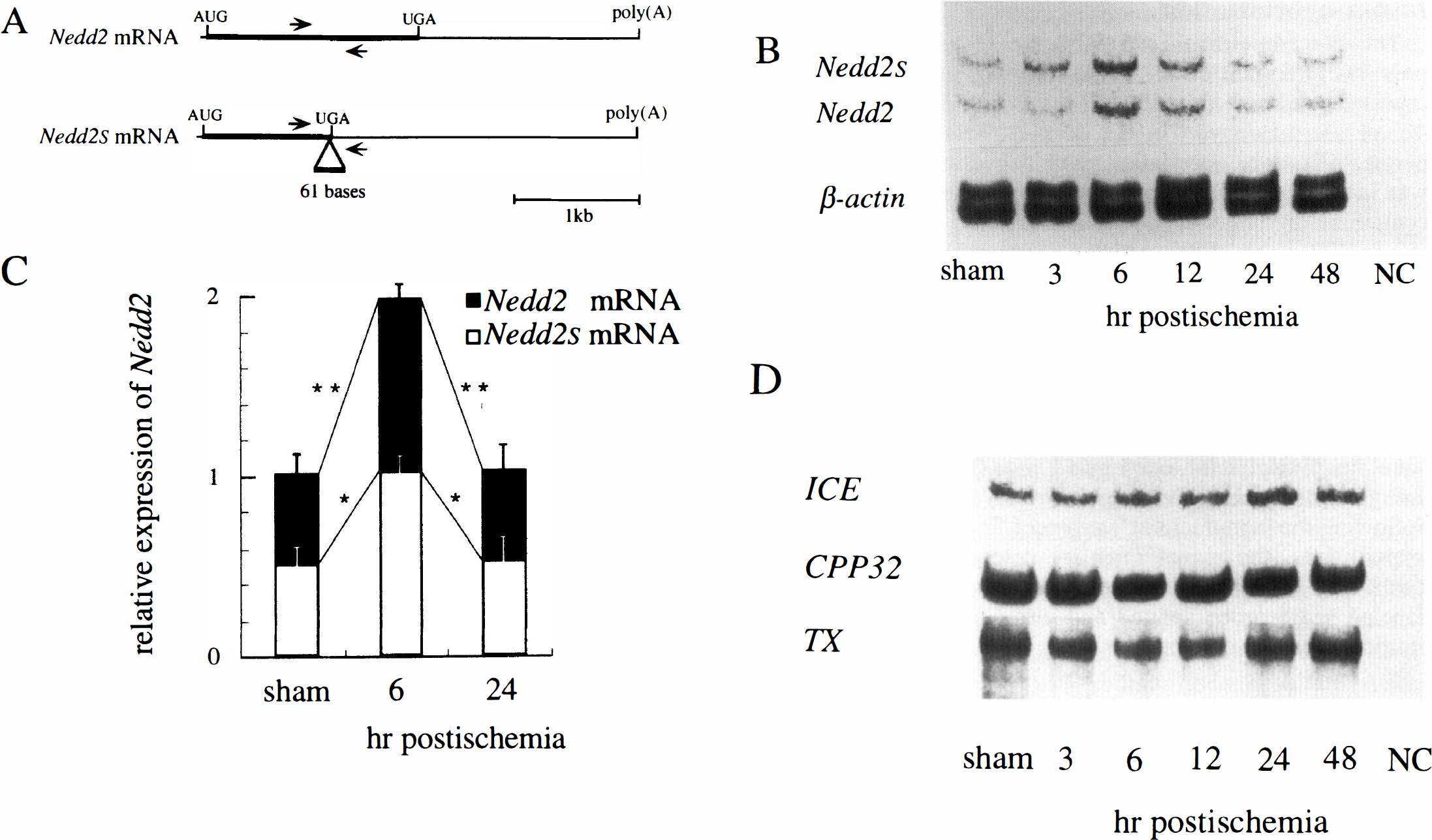
Reverse transcription–coupled polymerase chain reaction (RT-PCR) analysis of the splicing variants of Nedd2 and other interleukin-1β converting enzyme (ICE)–like protease genes. (
We also examined the expression of three other genes encoding ICE family proteases, i.e., ICE, CPP32/Yama/Apopain, and TX/ICErel
Spatial profile of Nedd2 expression in control and postischemic forebrains
To identify the cell population expressing Nedd2 in the control and the postischemic forebrains, we performed in situ hybridization using DIG-labeled RNA probes transcribed from the plasmid MS N2.4. Although this probe fails to discriminate the two splicing variants, we assume that, as the RT-PCR data suggest, the relative levels of Nedd2 and Nedd2s mRNAs stay roughly constant. Levels of the Nedd2 expression were variable from one neuronal population to another in the forebrain neurons of the sham-operated animals. In the hippocampus, the expression was minimal in the dentate gyrus (Fig. 3A), low in CA1 (Fig. 3E) and CA3 (Fig. 3G), and moderate to high in CA4 (Fig. 3C). These expression patterns were consistently observed in all the control animals (n = 4) and are similar to those observed in adult mouse brain (Kumar et al., 1994).
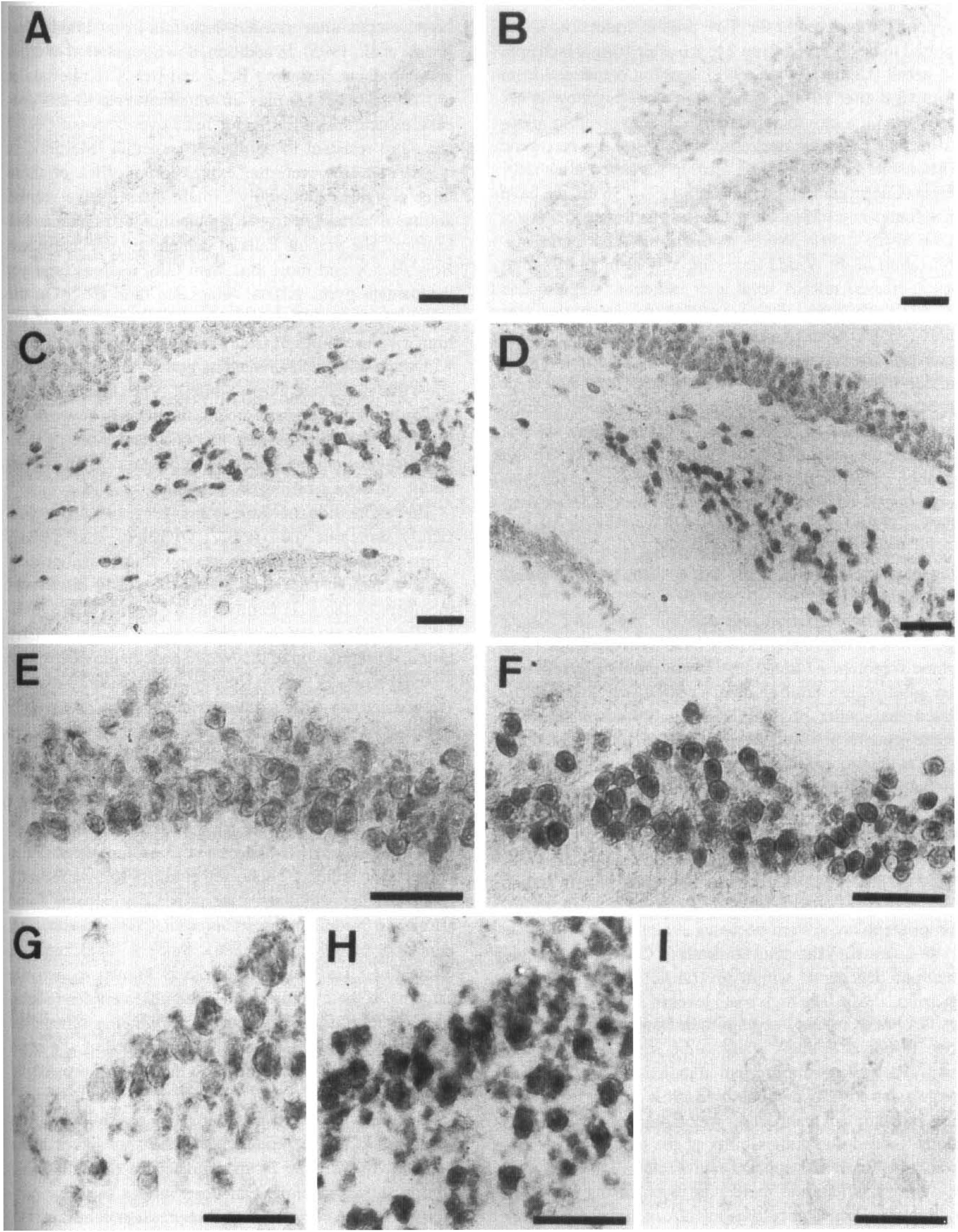
Spatial expression patterns of Nedd2 gene in the control and the postischemic forebrains. The sections from sham-operated animals (
In the forebrains of the animals dissected at 3 h after ischemia (n = 4), up-regulation of Nedd2 was prominent in CA1 (Fig. 3F) and CA3 (Fig. 3H) and moderate in the cerebral cortex (data not shown). Nedd2 expression increased in a large population of pyramidal cells in CA1 and CA3, but the intensity of the signals was variable among these neurons. The expression level in dentate gyrus and in CA4 did not change significantly (Fig. 3B and C). Essentially the same expression pattern was observed in the animals at 6 h postischemia (n = 4), but the pattern became indistinguishable from that in the control animals by 24 h postischemia (n = 4) (data not shown). These expression profiles are consistent with those of the Northern blot and RT-PCR analyses described. We therefore concluded that Nedd2 is up-regulated at 3–12 h postischemia in some specific neurons such as those in CA1, CA3, and the cerebral cortex, while its expression is rather constitutive in CA4 and stays low in dentate gyrus.
DISCUSSION
We have shown that the two transcripts from the gerbil Nedd2 gene were transiently and concomitantly up-regulated in specific neuronal subpopulations in the forebrain from 3 to 12 h after ischemia. This may be due to transactivation of the Nedd2 gene mediated by Fos, Jun, or other related transcription factors known to be induced immediately after ischemia (Uemura et al., 1991; reviewed by Nowak et al., 1993; Dragunow et al., 1994; Takemoto et al., 1995), since AP-1 elements (reviewed by Morgan and Curran, 1991) exist in the upstream region of the mouse Nedd2 gene (M. Kinoshita et al., unpublished). The time course of Nedd2 up-regulation also supports this hypothesis: Fos protein induction is reported to be observed from 2 to 8 h after 10-min ischemia in gerbil (Uemura et al., 1991), and Jun is induced from 5 to 48 h after 15-min ischemia in rat (Dragunow et al., 1994). p53, a key transcription factor involved in apoptosis that can mediate transactivation of the bax gene (Miyashita et al., 1994), is also up-regulated after ischemia (Chopp et al., 1992; Li et al., 1994). So far, we have not found any p53-binding element in the upstream region of the mouse Nedd2 gene. On the other hand, stabilization of the Nedd2 transcript may also contribute to the increased mRNA level after ischemia, since mouse Nedd2 mRNA contains two mRNA destabilization signals (AUUUA) in its relatively long 3′ untranslated region and therefore may be readily degraded in the cells under normal conditions. Nevertheless, the molecular mechanism of Nedd2 gene regulation in these specific neurons will be an important subject for further studies.
Since the ratio of the two Nedd2 transcripts did not change significantly after ischemia, the splicing machinery appears unaffected during this process. The molecular ratio of Nedd2 and Nedd2s proteins, however, has yet to be determined by, for instance, using specific antibodies that can discriminate the two species, which are currently unavailable.
In situ hybridization revealed that the gerbil Nedd2 gene was constitutively expressed to a detectable level in some forebrain neurons, just as we previously reported on adult mouse brain (Kumar et al., 1994). It is speculated that under physiological conditions, potentially toxic proteins like Nedd2 should be strictly regulated not only at the process of transcription but also posttranscriptionally. On the other hand, translation machinery is known to be disturbed after ischemia (Thilmann et al., 1986), and the levels of mRNA and protein tend to dissociate (reviewed in Sharp et al., 1993; Nowak et al., 1993). Thus, the time course of the Nedd2 protein and its activity after ischemia should be assessed when an appropriate assay system becomes available.
In contrast to the delayed death of CA1 neurons, CA4 neurons degenerate within several hours after ischemia (Kirino, 1982). The high basal level of Nedd2 expression in CA4 neurons may explain their immediate degenerative response or their ready-to-die state. More importantly, however, our observation indicates that the Nedd2 expression in CA4 neurons by itself is not sufficient for the induction of cell death under physiological conditions. Indeed, the vulnerability of the hippocampal neurons after ischemia appears multifactorially determined by energy failure and molecular mechanisms related to cell death. For instance, Fas mRNA is induced 6 h after ischemia in the gerbil forebrain (Matsuyama et al., 1994). Bax, which promotes cell death unless hererodimerized with Bcl-2 (Oltvai et al., 1993), is also up-regulated two- to threefold in the cerebral cortex and hippocampus after transient ischemia in rat brain (Krajewski et al., 1995). In addition, down-regulation of other gene products, including Bcl-2 and Bcl-X (Krajewski et al., 1995), may also play an important role in this process, as neurons of transgenic mice overexpressing bcl-2 are more resistant to permanent ischemia (Martinou et al., 1994). However, the expression profiles of these three proteins cannot fully explain the selective vulnerability of certain neuronal populations to ischemia. For example, the granule cells in dentate gyrus express less Bcl-2/Bcl-X and more Bax than CA3 neurons, and yet the dentate gyrus is less vulnerable than CA3. On the other hand, Nedd2 expression might account for the higher vulnerability of CA3 pyramidal cells as compared with the granule cells in dentate gyrus: Nedd2 expression stays low in dentate gyrus, while it is prominent in CA3. According to the current model, the balance between the levels of Bcl-2-like protein activities and those of ICE-like proteins may be a major determinant of the vulnerability of these neurons to ischemia.
The expression of three other genes encoding ICE family proteases, i.e., ICE, TX/ICErel
As a first step in determining the physiological relevance of ICE-like proteases in DND, we have shown that the Nedd2 gene is up-regulated in the vulnerable neurons after ischemia. Our observations raise the possibility that the elevated Nedd2 expression contributes to the pathophysiological events preceding the neuronal death, which should be assessed by using specific inhibitors or knockout mice. If the involvement of these proteases in DND is established, they will provide a promising therapeutic target to rescue neurons destined to die after ischemia.
Footnotes
Acknowledgment:
We thank Dr. D. B. Alexander for critical reading of the manuscript and Ms. A. Miyazaki for secretarial assistance.