
Abstract
Apoptosis is an active, gene-directed process of cell death in which early fragmentation of nuclear DNA precedes morphological changes in the nucleus and, later, in the cytoplasm. In ischemia, biochemical studies have detected oligonucleosomes of apoptosis whereas sequential morphological studies show changes consistent with necrosis rather than apoptosis. To resolve this apparent discrepancy, we subjected rats to 10 minutes of transient forebrain ischemia followed by 1 to 14 days of reperfusion. Parameters evaluated in the CA1 region of the hippocampus included morphology, in situ end labeling (ISEL) of fragmented DNA, and expression of p53. Neurons were indistinguishable from controls at postischemic day 1 but displayed cytoplasmic basophilia or focal condensations at day 2; some neurons were slightly swollen and a few appeared normal. In situ end labeling was absent. At days 3 and 5, approximately 40 to 60% of CA1 neurons had shrunken eosinophilic cytoplasm and pyknotic nuclei, but only half of these were ISEL. By day 14, many of the necrotic neurons had been removed by phagocytes; those remaining retained mild ISEL. Neither p53 protein nor mRNA were identified in control or postischemic brain by in situ hybridization with riboprobes or by northern blot analysis. These results show that DNA fragmentation occurs after the development of delayed neuronal death in CA1 neurons subjected to 10 minutes of global ischemia. They suggest that mechanisms other than apoptosis may mediate the irreversible changes in the CA1 neurons in this model.
The neuronal response to brain ischemia depends on the severity and duration of the ischemic insult as well as on intrinsic neuronal factors that impart selective vulnerability to specific brain regions (Scholz, 1953). Brief periods of global ischemia kill vulnerable neurons in the CA1 region of the hippocampus whereas longer intervals of vascular occlusion kill neurons in the cerebral cortex and corpus striatum as well (Ito et al., 1975; Pulsinelli et al., 1982a). Although seemingly undamaged, neurons in other brain regions such as the CA3 region of the hippocampus or the paramedian cerebral cortex undergo transient alterations as evidenced by reversible changes in their subcellular organelles (Petito and Pulsinelli, 1984aa, b ). The postischemic interval required for the apparent development of selective neuronal necrosis is inversely proportional to the severity or duration of the ischemic insult—the so-called maturation phenomena described by Ito et al. (Ito et al, 1975). This interval is especially prolonged in the CA1 neurons of the hippocampus (Ito et al., 1975; Pulsinelli et al, 1982a). The term delayed neuronal death has been applied to this process (Kirino, 1982).
During the past few years, the concept of cell death via apoptosis has become increasingly important. This mechanism of cell death is mediated by regulated pathways involving defined gene products rather than by direct injury to plasma membranes as occurs in necrosis (for reviews, see Wyllie et al., 1980; Kerr and Harmon, 1991; Majno and Joris, 1995). Apoptosis is the mode of cell death during embryogenesis or after physiological atrophy (Raff et al., 1993) but occurs in disease states as well. A number of studies suggest that focal or global cerebral ischemia also may cause neuronal apoptosis. The appearance of DNA ladders on gel electrophoresis of extracted nuclear DNA is characteristic of the oligonucleosomes in apoptotic cells. DNA ladders have been identified after ischemia of 12 or 20 minutes in the rat or 5 or 20 minutes in the gerbil (Heron et al., 1993; Ferrer et al., 1994; Sei et al., 1994; Charriaut-Marlangue et al., 1995;MacManus et al., 1995; Nitatori et al., 1995; Volpe et al., 1995). In addition, techniques that identify apoptotic oligonucleosomes in tissue sections label nuclei of postischemic neurons in the CA1 region (Ferrer et al., 1994; Sei et al., 1994; MacManus et al., 1995; Nitatori et al., 1995; Volpe et al., 1995). Similar findings have been reported in focal ischemia as well (Linnik et al., 1993; MacManus et al., 1994; Li et al., 1995).
A discrepancy exists between the above biochemical studies and virtually all morphologic studies that examine the sequential changes of cell death in global ischemia. As outlined by Deshpande et al. (1992), none show evidence of neuronal apoptosis. In an attempt to resolve the apparent discrepancy between morphological studies showing necrosis and biochemical studies demonstrating, or suggesting, apoptosis, we subjected rats to 10 minutes of transient forebrain ischemia followed by 1 to 14 days of reperfusion. Brains were examined for the presence of apoptosis by light microscopy, in situ detection of DNA oligonucleosomes, and increased transcriptional and translational activity of p53, one of several genes involved in apoptosis (Yonish-Rovach et al., 1991). Preliminary reports have been published (Petito et al., 1996, Torres-Munoz et al., 1996).
MATERIALS AND METHODS
Adult male Wistar rats, weighing between 200 and 250 g, were subjected to four-vessel occlusion as previously described (Pulsinelli and Brierley, 1979). Under inhalation anesthesia, the vertebral arteries were cauterized, loose ligatures were placed around the common carotid arteries, and a suture was placed under the posterior neck muscles. After an overnight fast, the common carotid arteries were occluded and the neck ligature tightened in the awake hand-restrained animal. Animals were accepted for the experiment if unconsciousness, pupillary dilatation, and loss of righting reflexes developed and persisted during ischemia and if seizures were absent. Body temperature was maintained at 37±0.5°C during ischemia and until the animal regained thermal homeostasis after cerebral recirculation. After 10 minutes, the clasps and neck suture were removed and the animals killed 1, 2, 3, 5, and 14 days later by perfusionfixation with 4% freshly prepared paraformaldehyde in 0.1 mol/L phosphate buffer. Brains were removed 2 hours later or overnight and stored in 0.1 mol/L phosphate buffer for 2 to 4 days before processing.
A coronal slice from the level of the mid-dorsal hippocampus, together with a segment of small intestine, were dehydrated, embedded in paraffin, and stained with hematoxylin-eosin in 5-μm sections. The term ischemic cell change (ICC) was used to describe dead neurons with cytoplasmic eosinophilia and nuclear pyknosis. Their number was evaluated in one 400× microscopic field of the CA1 region in the mid-dorsal hippocampus and expressed as a percentage of all neurons in that field.
In situ end-labeling (ISEL) of DNA was performed using terminal deoxynucleotidyl-transferase (Gavrieli et al., 1992; Gold et al., 1993) obtained in kit form (ApopTag kit; Oncor Laboratories, Gaithersburg, MD, U.S.A.). Sections were deparaffinized, digested, and incubated at 37°C with dATP, dCTP, dGTP, and digoxigenin (DIG)-labeled dUTP. After thorough washing, sections were incubated with anti-DIG conjugate (ApopTag kit) and hydrogen peroxide (H2O2) and 3,3′-diaminobenzidine. Pretreatment of sections with DNAase, and the presence of labeled nuclei in the apical villae of the small intestine in the experimental sections, served as a positive control for the enzymatic procedures; omission of the enzyme served as negative controls. The number of ISEL-labeled cells was expressed as a percentage of all neurons and of all neurons with ICC in one high power field (400×) of CA1 neurons in the mid-dorsal hippocampus. All attempts were made to use the same region of mid-dorsal hippocampus as was used for the hematoxylin-eosin evaluation.
Combined ISEL and immunohistochemistry for cell-specific markers was performed as previously described (Petito and Roberts, 1995). Antibodies used included those directed against glial fibrillary acidic protein for astrocytes (1:1000 dilution, Dako Corporation, Carpenteria, CA, U.S.A.); isolectin B4 for microglia (40 μg/mL, Sigma, St. Louis, MO, U.S.A.); transferrin for oligodendrocytes (Benkovic and Connor, 1993) (1:1000 dilution, Cappel Labs., Durham, NC, U.S.A.), and SMI 31 and 33 for phosphorylated and nonphosphorylated neurofilaments (1:2000 dilution, Sternberger Monoclonals, Inc., Baltimore, MD, U.S.A.). Chromagens included diaminobenzidine (brown), VIP (purple), and alkaline phosphatase-red. With glial fibrillary acidic protein, transferrin and SMI, ISEL (diaminobenzidine, brown) preceded immunohistochemistry (VIP, purple or alkaline phosphatase-red); with isolectin B4, ISEL (VIP, purple) followed immunohistochemistry (diaminobenzidine, brown).
Immunoreactivity for p53 antigen was examined on deparaffinized sections using overnight incubation with anti-p53 monoclonal antibodies (1:50 dilution, Dako Corporation) after 10-minute exposure to microwave radiation in citrate buffer. This was followed by sequential incubation with biotinylated secondary antibody, the avidin-biotin complex, and H2O2-diaminobenzidine. A p53-expressing glioblastoma multiforme and ovarian carcinoma were used as a positive control. Omission of the primary antibody served as a negative control.
In situ hybridization and Northern blot analysis for p53 used a digoxigenin-labeled cRNA-probe, prepared by transcription of the commercially available wt-p53 cDNA (Oncogene Science, Uniondale, NY, U.S.A.) according to the Boehringer Mannheim Dig-RNA labeling kit (SP6/T7) protocol. Frozen and formalin-fixed paraffin embedded ovarian carcinoma and a p53-secreting cell line were used as positive controls. RNA stability was confirmed by hybridization using digoxigenin labeled oligo-poly-T as probe. Negative controls were performed by using sense riboprobes for p53; omission of probe was used as background control. Deparaffinized sections of rat brain and intestine were rehydrated and pretreated with 0.2 mol/L Hcl for 20′ at room temperature; digested with proteinase K (10 μg/mL in 50 mmol/L Tris 20 mmol/L MgCl2 buffer) for 10′ at room temperature; and postfixed in 4% paraformaldehyde in phosphate buffered saline. The sections were washed, incubated in prehybridization solution at 42°C for 1 hour, drained, and incubated at 42°C for 16 hours in hybridization solution plus probe. The coverslips were removed and sections were washed in 2× SSC, 1× SSC, and 0.1× SCC, 10′ each, at room temperature. The sections then were treated with 0.3% Triton X-100 containing 2% sheep whole serum in 100 mmol/L Tris-Cl, pH 7.5, 150 mmol/L NaCl for 1 hour at room temperature, drained, and covered again with 0.1% Triton X-100 and 1% normal sheep serum in Tris buffer containing 1:500 dilution of alkaline phosphatase-red-anti DIG conjugate for 2 hours at room temperature. Sections were rinsed and covered with Tris-HCP buffer pH 9.5, 50 mmol/L MgCl for 5′ at room temperature followed by developing solution prepared according to supplier instructions (Boehringer DIG DNA labeling and detection kit). Slides were washed, counterstained with methyl green, and mounted with Brand ACCU mounting medium.
Northern blot analysis for p53 mRNA was performed on pooled RNA extracted from three dewaxed, rehydrated, and homogenized 20-μm paraffin-embedded sections (Quiagen RNeasy kit). GAPDH cDNA DIG labeled probe was used as a positive control. Extracted RNA was electrophoresed in 1% agarose gel containing 1 × 3-[N-morpholino] propane sulfonic acid buffer, pH 7.0; transferred by capillarity to a nylon membrane; and cross linked with 1600 μjoules/cm2 (UV light) (Sambrode et al., 1989). The RNA on nylon membrane was hybridized with the p53 DIG-labeled cRNA probe, using hybridization solution consisting of 50% Formamide 6× SSPE 5× Denhart's solution, 10% dextran sulfate, 0.5% SDS and 100 μg/mL herring sheared sperm DNA at 42°C for 16 hours. The membrane was washed twice in 2×, and 0.5× SSC containing 0.1% SDS, incubated in northern blocking solution in maleate buffer followed by a 2-hour incubation with 1:5,000 anti-DIG-alkaline phosphatase-red conjugate in northern blocking solution. The membrane was washed in maleate buffer for 15′ at room temperature and equilibrated in 100 mmol/L Tris-Cl pH 9.5, 100 mmol/L NaCl, and 50 mmol/L MgCl2 buffer. The membrane was immersed in freshly prepared solution of 45 μL of NTB plus 35 μL of X-phosphate solution in 10 mL of same equilibrating buffer. The membrane was left in the dark for 30 minutes. When the signal became visible, the membrane was washed with sterile water to prevent over-development.
Evaluation
All observations, including light microscopy, ISEL, and molecular biology were performed without knowledge of the experimental conditions. Three to four animals were used for each reperfusion period. Sham-operated rats (n = 4) served as controls. The animal surgery, including brain perfusion, was performed in the laboratory of Drs. Pulsinelli and Nowak and the morphological and molecular biology studies in the laboratory of Dr. Petito.
RESULTS
Nuclei of positive controls, intestinal epithelial cells, and all nuclei in sections pretreated with DNase were intensely labeled by the ISEL technique and were unstained when terminal deoxynucleotidyl-transferase was omitted. As described in more detail below, ISEL was confined to those neurons with ICC. It was absent in normal neurons, in neurons with cytoplasmic condensations and in approximately half of all neurons displaying cytoplasmic eosinophilia and nuclear pyknosis at day 3 and 5. Labeling intensity diminished over time, and was faint and confined to neuronal cytoplasm at day 14.
The light microscopic characteristics of the CA1 region of the hippocampus were similar to prior reports in this model (Pulsinelli et al. 1984). At one day after ischemia, mild nuclear swelling was apparent when compared with control animals (Fig. 1A and B). In situ end-labeling was absent. At 2 days after ischemia (Fig. 1C), many neurons displayed focal cytoplasmic condensations and increased basophilia of both cytoplasm and nucleus. However, nuclear changes characteristic of early apoptosis, including peripheral margination of chromatin crescents or chromatin clumps, were absent. In situ end-labeling was absent. Glial changes, which included mild hyperchromasia of perineuronal glial nuclei and slight enlargement of astrocyte nuclei, were first apparent at this time.
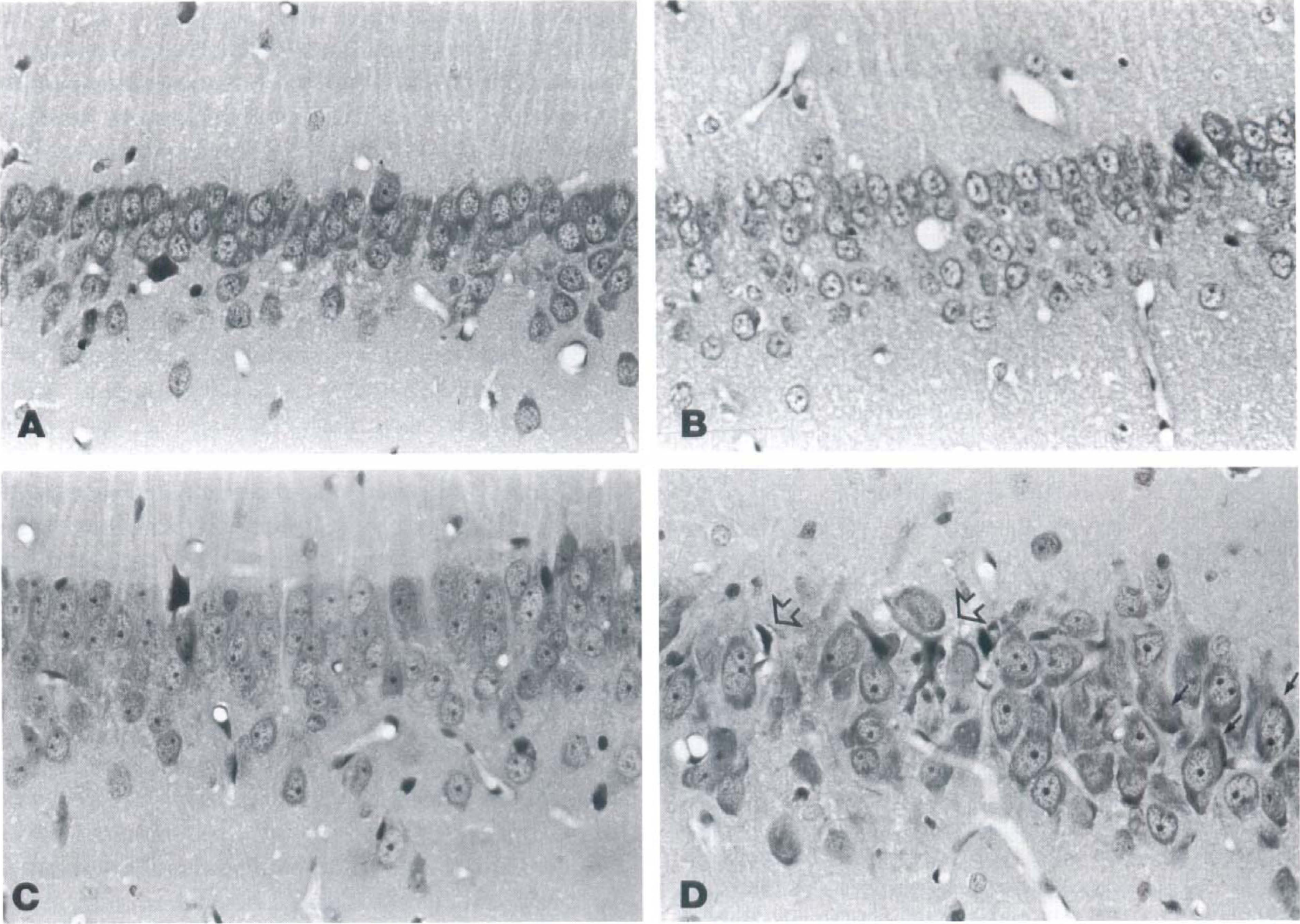
CA1 neurons in controls
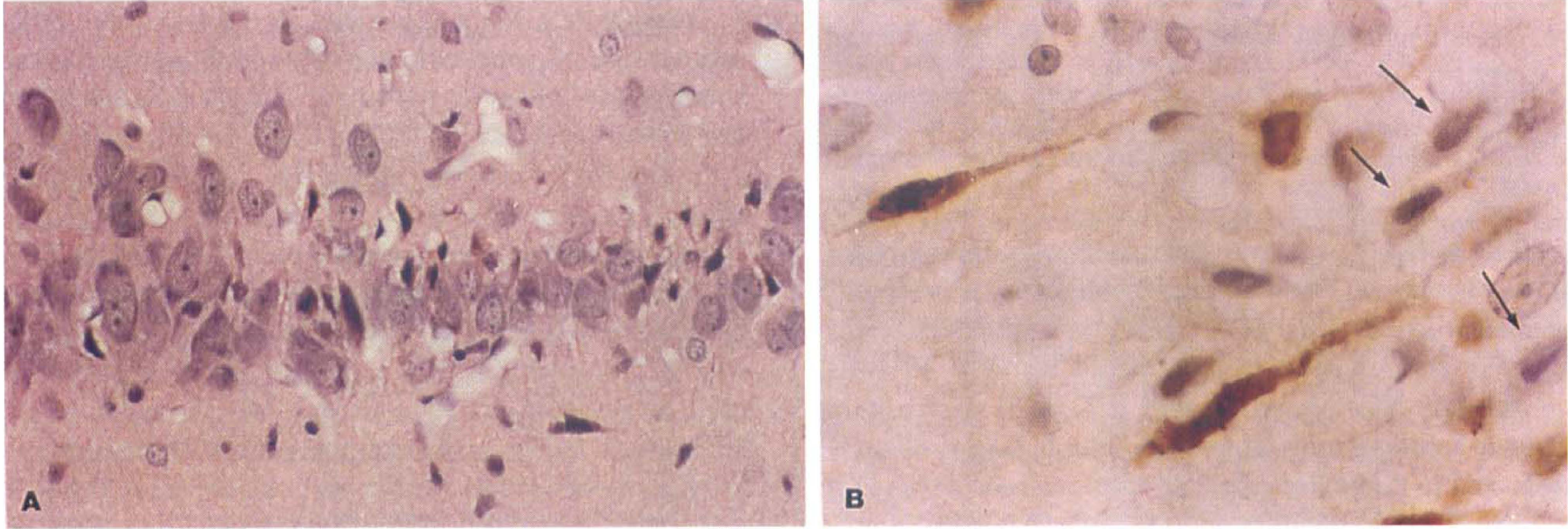
At 3 days after ischemia, the number of neurons with ICC averaged 66% (Fig. 1D). Two of these animals had moderate to severe neuronal necrosis that represented 76% and 98% of the total number of neurons. This was associated with microglial proliferation and phagocytosis that at times obscured the underlying ICC. In situ end-labeling was present in 50% and 100% of the neurons showing ICC in these two animals and remained absent in neurons without eosinophilic change. The third 3-day animal had milder damage with only 11% of neurons displaying ICC. In situ end-labeling was absent in both the neurons with ICC as well as in the neurons that appeared undamaged (Fig. 2A and B).
CA1 neurons, postischemic day 3.
At 5 days after ischemia, the number of ICC neurons averaged 28%. At this point, the neurons with ICC were more shrunken and often showed nuclear karyorrhexis, a process in which pyknotic nuclei in dead cells break up into multiple irregular fragments. Microglial proliferation and neuronal phagocytosis was prominent. At 14 days, the number of eosinophilic neurons had diminished, their neurons shrunken; nuclei had lost their hyperchromasia and cytoplasmic eosinophilia was decreased. Neuronal ISEL was present in most of the residual neurons with ICC (Fig. 2A and B).
These results are summarized in Table 1. The total number of CA1 neurons in one 400× field averaged 95 in controls, 104 at day 1, 107 at day 2, 167 at day 3, 88 at day 5, and 86 at day 14. The apparent increases at days 1 through 3 are related to the fact that neuronal shrinkage allowed more neurons to be included in the 400× field. The apparent decreases at days 5 and 14 reflect the fact that many of the dead neurons were obscured or removed by phagocytic cells. The average percentage of neurons with ICC was 66% at day 3, 37% at day 5, and 28% at day 14. Ischemic cell change and ISEL were absent at day 1 and 2. In situ end-labeling only labeled ICC neurons, although it was not always present in these; the ICC labeling incidence averaged 50% at days 3 and 5 and 100% at day 14.
Temporal relationship between the evolution of ischemic neuronal cell death and in situ end labeling of fragmented DNA in CA1 neurons
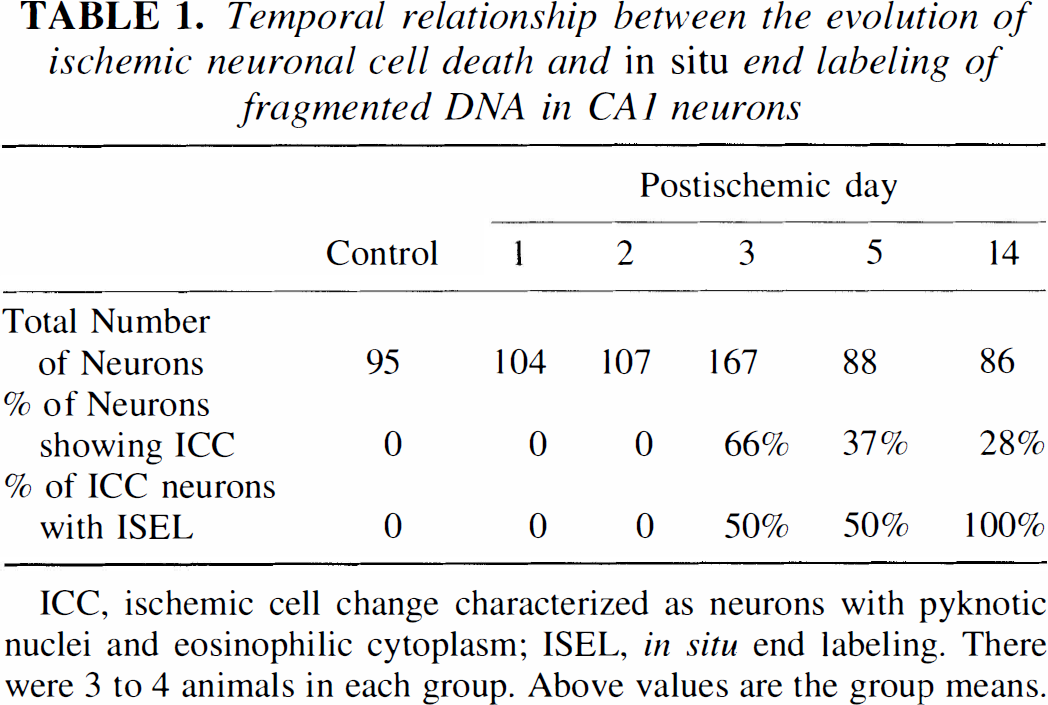
ICC, ischemic cell change characterized as neurons with pyknotic nuclei and eosinophilic cytoplasm; ISEL, in situ end labeling. There were 3 to 4 animals in each group. Above values are the group means.
As expected in this model (Pulsinelli et al. 1984), eosinophilic neuronal cell death developed in the distal CA4-6 region and represented 15% of these neurons at postischemic day 1 and 2, 19% at day 3, and 12% at day 5. Their cytoplasm was slightly labeled by ISEL at day 1 but no labeling was present at subsequent periods. Mild increases in astrocytic nuclei and numbers of activated microglia accompanied this necrosis between day 1 and 5. Neurons in the CA3 region were unchanged from controls.
Rare cells with one or more central hyperchromatic nuclear bodies were present in control and postischemic hippocampus and were ISEL-positive. Some colabeled with transferrin on postischemic days 1 through 3, suggesting an oligodendroglial origin as described in preliminary reports (Petito and Torres-Munoz, 1996; Petito et al., 1996).
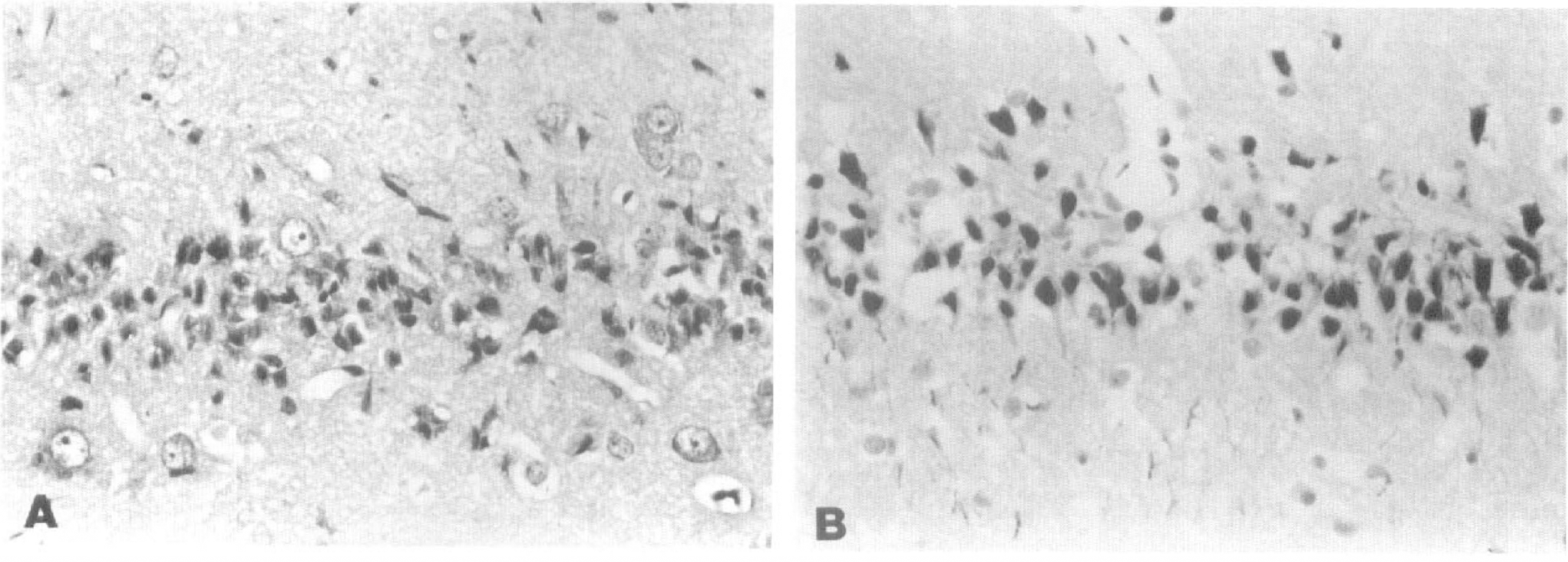
CA1 neurons, post-ischemic day 5. A. Severe neuronal necrosis is seen with hematoxylin-eosin stain. Residential neurons show mild nuclear swelling or cytoplasmic condensations. B. Most neurons display intense in situ end labeling (ISEL). Original magnification ×100.
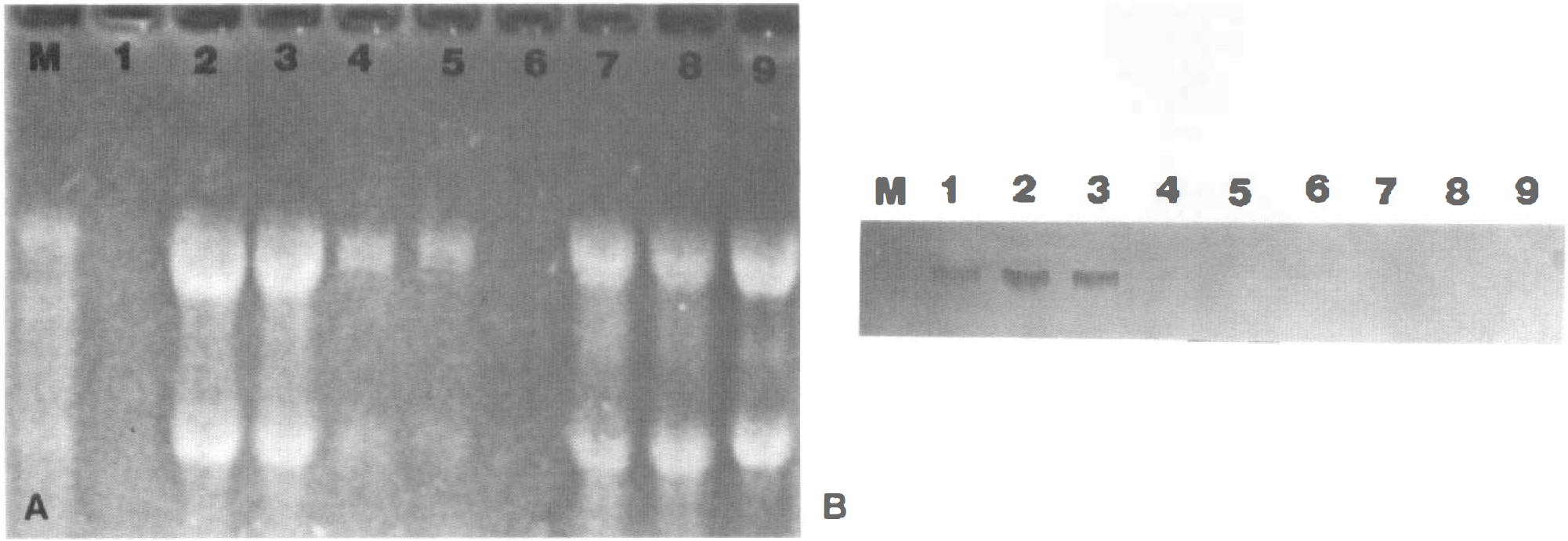
p53 protein and mRNA were detected in the positive controls but were absent in control and postischemic rat brain (data not shown). Similarly, northern blot analysis for p53 mRNA was positive in the p53 secreting Cos7 cell line and in the paraffin-embedded and fresh frozen ovarian carcinoma, but p53 mRNA was not detected in control or post-ischemic rat brain (Fig. 4).
Review of prior material
Plastic-embedded sections of CA1 neurons subjected to 30 minutes of forebrain ischemia (Petito and Pulsinelli, 1984b) showed mild cytoplasmic swelling at 24 hours followed by cytoplasmic and nuclear shrinkage between 36 and 48 hours. Peripheral accumulations of nuclear chromatin or the single or multiple round dense chromatin masses in neurons with intact cytoplasm were absent. Paraffin-embedded sections from rats exposed to 2 or 10 minutes of forebrain ischemia (Petito et al., 1990; Petito and Halaby, 1993) also revealed mild cytoplasmic dilation that preceded the appearance of eosinophilic neurons. Morphological evidence of neuronal apoptosis was absent.
DISCUSSION
Specific morphological criteria identify apoptosis in tissue sections (Kerr, 1965; Wyllie et al., 1980; Kerr and Harmon, 1991; Majno and Joris, 1995). Initially, focal accumulations of dense chromatin masses develop at the nuclear periphery which leads to progressive nuclear shrinkage with or without fragmentation into multiple round chromatin masses. The nuclear changes are initiated by DNA fragmentation into 180 bp oligonucleosomal fragments as a result of an activation of a Ca++Mg++ endonuclease (Wyllie et al., 1984; Arends et al., 1990). Once the nuclear alterations have progressed, the cell body shrinks and may form cytoplasmic buds, in which are incorporated the chromatin masses. The final appearance of the apoptotic cell can be twofold: a single body with a shrunken cytoplasm, and a solitary round, pyknotic nucleus, or multiple small apoptotic bodies formed from breakup of the cytoplasmic buds. During this process, the plasma membrane retains its structural and functional integrity (Daryznkiewicz et al, 1992). Apoptosis can take place in the absence of protein or mRNA synthesis, may be preceded by abnormalities in mitochondrial function, and may develop in anucleate cells in the absence of DNA fragmentation (Schwartzman and Cidlowski, 1993; Hockenbery, 1995; Kane, 1995; Kroemer et al., 1995;). Its control is based not only on nuclear susceptibility but also on specific cytoplasmic regulators (Polunovsky et al., 1996). Necrosis differs from apoptosis in the early stages because loss of plasma membrane integrity is one of the initial changes and leads to cell swelling which may or may not be transient. Although DNA breaks and nuclear pyknosis develop in necrosis, they appear after membrane damage and cytoplasmic changes and are not accompanied by peripheral chromatin condensations (Arends, et al. 1990; Gold et al. 1994).
DNA fragmentation can be detected in tissue sections by ISEL (Gavrieli et al., 1992; Wijsman et al., 1993). Either terminal deoxynucleotidyl-transferase or DNA polymerase I add labeled nucleotide tails to the 3′-OH ends of the cleaved DNA strands. Both methods bind nucleotides to the random nicks and breaks that occur in DNA of necrotic cells as well as to the oligonucleosomes of apoptotic cells (Gold et al., 1994). As a result, ISEL positivity alone does not distinguish between necrosis and apoptosis, a finding noted by a number of studies (Gold et al. 1993, Wijsman et al. 1993; Ansari et al, 1993; Mundle and Raza, 1995). If the cytoplasm already displays irreversible changes of cell death such as eosinophilic necrosis, ISEL-positivity cannot distinguish between these two processes. According to the current definition of apoptosis, ISEL identifies an otherwise normal-appearing cell as apoptosis only if the membrane integrity of the cell is unaltered. For example, both apoptosis and necrosis develop in experimental myocardial infarct produced by coronary artery occlusion (Kajstura et al. 1996). DNA strand breaks, indicated by nuclear ISEL, developed with both processes, but plasma membrane permeability was confined to, and used to identify, necrotic rather than apoptotic myocytes.
Sequential light and electron microscopic studies of CA1 neurons subjected to global ischemia clearly show that the early chromatin changes of apoptosis are absent and that the cytoplasmic changes precede nuclear changes (Kirino, 1982; Kirino et al., 1984; Petito and Pulsinelli, 1984b; Deshpande et al., 1992; Nitatori et al., 1994). In our prior study of hippocampal neurons exposed to 30 minutes of forebrain ischemia (Petito and Pulsinelli, 1984b), transient swelling of mitochondria, Golgi cisterns, and endoplasmic reticulum appeared at 3 minutes; disaggregation of polyribosomes and loss of Golgi cisterns at 15, 30, and 120 minutes; and cytoplasmic swelling between 24 and 36 hours. Nuclear changes of apoptosis were absent. Other studies examining the cerebral cortex or striatum in global ischemia also show that cytoplasmic changes precede nuclear changes and that the early nuclear changes of apoptotic cells are not present (Brown and Brierley, 1972; Dodson et al., 1977; Ekström et al., 1982; Petito and Pulsinelli, 1984a; Dietrich et al., 1991).
The results of the present study suggests that ISEL in postischemic neurons results from nucleotide binding to DNA of necrotic cells rather than to oligonucleosomes of apoptotic cells. The nuclear changes associated with early apoptosis, such as chromatin condensations or peripheral chromatin aggregation, were absent. Rather, karyorrhexis was observed in the already dead neurons at postischemic day 5. Karyorrhexis is a relatively late process of necrosis, and one that required 2 days to develop in the present study. It is distinguished from apoptotic nuclear fragmentation into round chromatin masses by the fact that it develops in an already dead cell and by the fact its fragments are small and irregular (Majno and Joris, 1995). Second, ISEL was never present in prenecrotic neurons at days 1−3. If the CA1 death had occurred by apoptosis, ISEL-positivity should have been observed in neurons with normal appearing cell bodies. Lastly, ISEL appeared to be a late event, following rather than preceding cytoplasmic eosinophilia and shrinkage. It was absent in many neurons displaying early cytoplasmic changes associated with necrosis. The labeling intensity in the ICC neurons decreased and by 15 days after ischemia, it was often weak and confined to neuronal cytoplasm, a localization representing a presumed leak of broken DNA strands from the nucleus into the cytoplasm of the dead cell (Russell, 1983; Rink, 1995).
If morphology and ISEL do not identify neuronal apoptosis after global ischemia, what then accounts for the oligonucleosomes that clearly have been detected by the presence of DNA ladders on gel electrophoresis after global ischemia in rodents. Several factors may explain this apparent discrepancy.
First, some of the oligonucleosomal ladders detected after ischemic insults could result from apoptosis of glial cells or of neurons outside the hippocampus. In a preliminary study, we showed that there are actually a large number of glial cells that undergo apoptosis after 10 minutes of global ischemia (Petito et al., 1996). They were most numerous in brain regions such as the cerebral cortex where neuronal cell death is absent or minimal. Glial and microglial apoptosis occur in other conditions of brain injury (Kane, 1995; Petito and Roberts, 1995; Lin et al., 1996) and their presence in ischemia is therefore not unexpected.
Second, DNA ladders follow rather than precede cell death in type II programmed cell death (Schwartz et al, 1993; Zakeri et al., 1993; Lockshin and Zakeri, 1996), and similarities to that in postischemic CA1 neurons suggests that this cell death mechanism could occur after transient ischemia as well. Type II programmed cell death, or autophagy, is a second type of cell death during embryogenesis that is in addition to the more familiar apoptosis or Type I programmed cell death. Cytoplasmic abnormalities initiate autophagy, with increases in lysosomes and loss of rough endoplasmic reticulum that precede eventual cell collapse. Chromatin condensation and nuclear pyknosis are late events and are accompanied by DNA fragmentation into oligonucleosomes that appear as DNA ladders on gel electrophoresis. Type II programmed cell death has been described in models of hepatic toxicity (Fukuda et al., 1993) and also takes place in neurons or oligodendrocytes deprived of growth factors or treated with corticosteroids (Schwartz et al., 1993). Similarities between postischemic neuronal death and Type II programmed cell death include cytoplasmic alterations that develop before nuclear changes; ISEL positivity that occurs with or follows the appearance of eosinophilic necrosis; and DNA fragmentation into oligonucleosomes after cellular evidence of necrosis. In addition, accumulation of lysosomes and proteases precede cell death in hippocampal neurons after global ischemia (Nitatori et al., 1995) whereas inhibition of lysosomal proteases protects against delayed neuronal death produced by global ischemia (Lee et al, 1991; Hoffmann et al, 1992; Robert-Lewis et al, 1993; Palm et al, 1995).
Lastly, the current definitions of apoptosis and necrosis may be too restrictive, excluding other mechanisms, such as transitional forms between apoptosis and necrosis (MacManus et al, 1994; Porter-Callilau et al, 1995). These authors hypothesize that the orderly sequence of nucleic and cytoplasmic events that occurs in apoptosis is interrupted after DNA fragmentation into the oligonucleosomes seen as DNA ladders, and evolution into necrotic cell changes rather than into apoptotic cellular changes develops.
The exploration of potential genes that are directly involved in ischemic neuronal death is in its infancy. There is evidence of transcriptional and translational activity of early-immediate genes including c-fos, c-jun, and heat shock protein (Kogure and Kato, 1993; Nowak, et al., 1993; Estus et al., 1994; Akin et al., 1996). Expression of c-fos and c-jun have been associated with apoptosis in other model systems (Barrett and Preston, 1994), although available evidence indicates such responses are more likely to be involved in neuroprotective mechanisms after ischemia (Nowak et al, 1992; Sommer et al, 1995; Abe and Nowack, 1996). Members of the interleukin-converting enzyme gene family and p53 are common apoptosis-driving genes (Yonish-Rovach et al, 1991; Miura et al., 1993; Steller 1994; Hale et al, 1994; Sakhi et al., 1994; Deshmukh et al, 1996) that have been associated with focal ischemia (Crumrine et al, 1994; Li et al, 1994). Their role in global ischemia is less clear as shown by the absence of p53 protein and mRNA in the present study and of interleukin-converting enzyme in an earlier study by Bhat et al (1996). However, their absence does not exclude apoptosis because their participation is not necessarily required for apoptosis to take place (Hale et al, 1995; Vasilakos et al, 1995; Sadoul et al, 1996). Bax, another apoptosis-driving gene, has been identified by immunoreactivity in necrotic neurons after global ischemia produced by transient cardiac arrest (Krajewski et al, 1995).
The present study does not rule out other conditions in which ischemia could produce neuronal apoptosis. For example, apoptosis may be responsible for the late delayed, ongoing neuronal cell death that occurs weeks after the ischemic interval. Such changes have been described in the cerebral cortex, substantia nigra, and basal ganglia after focal or global ischemia (Tamara et al., 1990; Hara et al., 1993; Saji et al., 1994; Dietrich et al., 1995). If this late delayed neuronal death is caused by apoptosis, it is likely to be caused by indirect factors such as loss of trophic factors that are known to induce neuronal apoptosis (Barde, 1989; Deckwerth and Johnson, 1993), rather than as the direct result of the ischemic insult itself.
The severity of ischemia could be another variable determining whether postischemic neurons die by apoptosis or necrosis. Precedence for this is found in a number of conditions, such as hyperthermia (Harmon et al., 1990) and ultraviolet irradiation (Fukuda et al., 1993), in which a given insult causes apoptosis if mild but necrosis if severe. Indeed, Kerr's original observation of apoptosis was in a model of hepatic ischemia which produced necrosis as well as apoptosis in liver cells (Kerr, 1965). In the penumbral zone of cerebral infarcts, regional cerebral blood flow declines to only 20% of control values (Nagasawa and Kogure, 1989), an insult that is relatively mild when compared with the CBF reduction to 3% of control values during global ischemia (Pulsinelli et al., 1982b). Thus, neuronal apoptosis might be expected at the periphery of infarcts, and has been described in some studies (Li et al., 1995).