
Abstract
Previous studies have shown that overexpression of bcl-2 in transgenic mice or by viral vectors protects the brain against cerebral ischemia. However, it is not known whether bcl-2, which is endogenously expressed in response to ischemia, exerts a protective effect. To address this question, the authors blocked the endogenous expression of bcl-2 after ischemia using antisense oligodeoxynucleotides (ODN). Antisense, sense, scrambled ODN, or vehicles were infused in the lateral ventricle of the rat for 24 hours after 30 minutes of temporary middle cerebral artery occlusion. Twenty-four hours later the brains were removed and bcl-2 protein expression was assayed by Western blot. Antisense ODN, but not sense or scrambled ODN treatment, significantly inhibited bcl-2 protein expression after ischemia. Bcl-2 protein expression was also studied 24 hours after 60 minutes of temporary middle cerebral artery occlusion in vehicle and antisense ODN-treated rats. After 60 minutes of ischemia and vehicle treatment, bcl-2 was expressed in many neurons in the ventral cortical mantle and the medial striatum. After antisense ODN treatment there were few neurons in this region expressing bcl-2, instead most neurons TUNEL labeled. Treatment with the antisense ODN, but not sense ODN, increased infarction volume as determined by cresyl violet staining 72 hours after ischemia compared with vehicle controls. These results suggested that endogenously expressed bcl-2 promoted survival in ischemic neurons and was not simply an epiphenomenon in neurons already destined to live or die.
Keywords
Bcl-2 is an oncogene that is overexpressed in b-cell lymphoma and a variety of other cancers (Tsujimoto et al., 1985). Bcl-2 is homologous to ced-9, a gene that suppresses the key committed step in the programmed cell death of neurons during the development of C. elegans (Hengartner and Horvitz, 1994). Overexpression of bcl-2 can inhibit apoptosis of neurons and other cells induced by a wide variety of stimuli. Thus, bcl-2 is a key gene in suppressing programmed cell death in the mammalian nervous system (Garcia et al., 1992).
There is substantial evidence that programmed cell death may occur after transient cerebral ischemia and that dysregulation of bcl-2 and related genes exacerbates ischemic neuronal injury. Protein synthesis inhibitors increase survival of neurons after both global and focal ischemia (Goto et al., 1990; Hossmann, 1993; Linnik et al., 1993). Fragmentation of DNA, one of the biochemical hallmarks of programmed cell death, also occurs after ischemia (Linnik et al., 1993). Expression of bcl-2 and bcl-x-l, a homologue of bcl-2 that also suppresses programmed cell death, is induced in neurons that survive after ischemia (Chen et al., 1995; Dixon et al., 1997; Gillardon et al., 1996; Hara et al., 1996; Niwa et al., 1997). These data suggest that bcl-2 and related genes could regulate neuronal death after ischemia.
Studies in which bcl-2 has been overexpressed support the hypothesis that bcl-2 promotes survival of ischemic neurons. Martinou et al. (1994) constructed a transgenic mouse with human bcl-2 driven by a neuron-specific enolase promotor. Transient middle cerebral artery occlusion produced significantly smaller infarctions in the transgenic mouse compared with wild-type controls. Overexpression of bcl-2 using viral vectors has also been shown to protect the brain against ischemic injury. Linnick et al. overexpressed bcl-2 with an amplicon-driven HSV vector in rat cortex and found a reduction in infarction size in animals transfected with the bcl-2 vector compared with control vectors (Linnik et al., 1995). Lawrence et al. (1996) found that transfection of bcl-2 protected striatal neurons against ischemia. These studies show that overexpression of bcl-2 in neurons destined to die after ischemia can prevent ischemic neuronal death. Such studies do not address whether endogenous expression of bcl-2 in neurons that survive after ischemia promotes survival of these neurons or whether it is simply an epiphenomenon in neurons already destined to survive. To address this question, the authors used bcl-2 antisense treatment to block expression of bcl-2 induced by ischemia and determined the effect of this treatment and controls upon infarction volume.
METHODS
Animal methods
All procedures were performed with the approval of the Institutional Animal Use and Care Committee at the University of Pittsburgh. The technique of Longa et al. (1989) was used to induce temporary focal ischemia in male Sprague Dawley rats (280 to 310 g). The rats were anesthetized with 4% isoflurane, intubated, then mechanically ventilated with 1.5% isoflurane (70% N20, 30% O2). Temporalis muscle temperature was maintained at 37.5°C ± 0.2°C with a thermostatically controlled heating lamp. A groin incision was made and one femoral artery cannulated for continuous blood pressure monitoring and for measurement of arterial blood gasses and blood glucose concentrations.
The bifurcation of the common carotid artery was exposed and the external artery ligated. The internal carotid artery was isolated. The external branch of the internal carotid artery was ligated close to its origin and a 3–0 monofilament nylon suture was inserted into the internal carotid artery lumen through the stump of the external carotid artery and advanced 20 to 22 mm past the carotid artery bifurcation. The suture was maintained in this position for either 30 or 60 minutes and then withdrawn to allow reperfusion of ischemic brain. The external carotid was then ligated and the wound closed. Sham-operated animals underwent identical surgical procedures but did not have the suture inserted into the artery.
Quantification of infarct volume
The animals were killed 72 hours after 60 minutes of ischemia with an overdose of chloral hydrate. The rats were perfused pericardially with 100 mL heparinized saline and 400 mL of 4% paraformaldehyde in 0.1 N phosphate buffer pH 7.4. The brains were then removed, frozen in −20°C 2-methylbutane, and coated with embedding matrix. They were then cut on the cryostat in 20-μm-thick sections. The sections were stained with cresyl violet and infarction volume was calculated using the previously described method (Swanson et al., 1990). The sections were obtained every 2 mm from the anterior caudate level to posterior hippocampus.
Western blotting
The rats were overdosed with chloral hydrate (600mg/kg IP) 24 hours after preconditioning and brains rapidly removed. Brains were placed on dry ice and dissected. The brains were first divided in the saggital plane and then the hippocampus and diencephalons removed. The remaining hemisphere was then bisected and the occipital half discarded. Finally, the striatum was dissected and discarded, leaving the fronto-parietal cortex, the portion of cortex in which the authors previously found expression of bcl-2 after 60 minutes ischemia (Chen et al., 1995b). This tissue was homogenized and lysed in 0.1 mol/L Tris-Cl, 0.001 mol/L EDTA, 1 μg/mL aprotinin, and 100 μg/mL phenylmethyl sulfonyl fluoride. The lysate was centrifuged at 14,000 g for 30 minutes, and boiled at 100°C in sodium dodecyl sulfate loading buffer (100 mmol/L Tris-HCl, 200 mmol/L dithiourea, 4% sodium dodecyl sulfate, 0.2% bromophenol blue, and 20% glycerol) for 6 minutes. Fifty μg of the resultant protein samples, as determined by spectrophotometry, (A280), were then electrophoresed on a 12% sodium dodecyl sulfate polyacrylamide gel.
Western blots were performed using the previously described technique (Chen et al., 1996) and transferred to polyvinyl difluoride membranes. The membranes were incubated with primary antibody to bcl-2 (Dayco, Carpinteria, CA, U.S.A.) at a dilution of 1:500 at 4°C overnight. This was followed by three washings in buffer (0.1% Tween 20, 0.5% bovine serum albumin, 1% nonfat dry milk in IX phosphate buffer), and then by incubation at room temperature for 60 minutes with secondary antibody. Secondary antibody was detected with chemiluminescent substrate (NEN) and CSPB (25 mm). The blot was exposed to Kodak X-OMAT film (Rochester, NY, U.S.A.). The density of bands was determined by the MCID image system (St. Catherines, Ontario, Canada). Each gel consisted of two lanes each for control, cerebrospinal fluid (CSF), vehicle, antisense, and scramble ODN-treated samples. Data were expressed as the ratio of the optical density of the appropriate band in each lane to the mean optical density of the control samples on that gel.
Infusion of ODN
A burr hole was drilled through the skull and a cannula inserted in the right lateral ventricle 30 minutes before ischemia. The brain infusion cannula (Alzet, Palo Alto, CA, U.S.A.) was placed stereotactically at 0.8 mm posterior to the bregma, 1.5 mm lateral to the midline, and 3.8 mm ventral to the dura. The cannula was then fixed to the skull with dental cement and anchored to a screw placed 5 mm away. CSF, bcl-2 antisense oligonucleotides, or bcl-2 sense oligonucleotides were continuously infused for 72 hours by an osmotic minipump (model 1003D; 1 μL/h flow rate; Alzet). Bcl-2 antisense phosphodiester oligonucleotide used for these experiments straddled the predicted translational initiation side of the rat bcl-2 mRNA. The antisense oligonucleotide was based on prior development of the human antisense oligonucleotide to bcl-2 (Kitada et al., 1993). The rat bcl-2 sequence (Sato, 1994) was used to target the same region as the human bcl-2 antisense. The antisense sequence used was 5′- TGTTCTCCCGGCTTGCGCCAT −3′. The sense ODN sequence was 5′- ATGGCGCAAGCCGGGAGAACA −3′. The scrambled sequence was 5′- GTGATCCCCTGCTCTTGCCGT −3′. In additional studies, the antisense ODN was end-labeled with fluorescein isothiocyanide and infused, using identical parameters, into the brains of 2 rats for 72 hours after 60 minutes of ischemia.
Immunocytochemistry and TUNEL
Rats were anesthetized with an overdose of chloral hydrate and then perfused pericardially with 100 mL heparinized saline followed by 500 mL of 2% paraformaldehyde. The brains were removed, fixed in 2% paraformaldehyde for 30 minutes, rinsed in phosphate buffer solution, then placed in 30% sucrose at 4°C before freezing in −30°C methylbutane. The brains were cut in 5-μm-sections using a cryostat, mounted, and kept frozen until staining. The slides were washed three times with PBS, then washed three times in PBS with 0.5% bovine serum albumen and 0.5% glycerine (buffer A).
Specific activity was blocked with 5% goat serum in buffer A. Sections were incubated at room temperature at 1 hour in a 1:100 dilution of the mouse monoclonal bcl-2 antibody (Dayco). Sections were then washed three times in buffer A and incubated for 1 hour in 1:3000 dilution of goat anti-mouse Cy3.18 immunoconjugate (Jackson Immunochemicals, West Grove, PA, U.S.A.). Sections were washed six times (5 minutes per wash) in buffer A, mounted in gelvatol, and coverslipped for light microscopy. To label cell nuclei, bis benzimide (similar to Hoechst 33258; Sigma, St. Louis, MO, U.S.A.) diluted in sterile water was applied to some sections for 30 seconds before coverslipping. Sections were examined using a light microscope equipped for epifluorescent illumination. Images were collected using an integrating 3 chip color video camera (700times600 pixels) equipped with a color frame grabber board. All images were collected while integrating at four frames per second.
For double and triple label experiments, TUNEL was performed on additional sections using modification of the previously reported technique (Gavrieli et al., 1992). Sections were washed twice in PBS, incubated in cold methanol for 30 minutes, then washed with PBS and incubated at 37°C for 1 hour in buffer containing terminal deoxynucleotidyl transferase and uridyldiphosphate (Boeringer Mannheim, Indianapolis, IN, U.S.A.). Sections were then processed for bcl-2 immunocytochemistry as described above. In some sections nuclear morphology was assessed using bis benzamide. Bis benzamide was diluted in sterile water and applied to some sections for 30 seconds before coverslipping. Sections were then examined by fluorescence microscopy. Digital images were collected using wavelengths of 550/565, 490/455, and 346/460 for bcl-2, TUNEL, and bis benzamide, respectively. Alternate sections were processed omitting the primary antibody to assess for nonspecific binding of secondary antibody. The bcl-2 antibody used for Western analysis and immunocytochemistry was raised to a peptide corresponding to amino acids 41 through 54 of human bcl-2. The authors have shown previously that this antibody reacts with the corresponding rat peptide (Clark et al., 1997).
Statistical analysis
All values expressed are mean ± SD. Differences between means were determined by one-way analysis of variance followed by Fisher's multiple comparison test. P < 0.05 was regarded as significant.
RESULTS
Treatment with antisense ODN, but not sense or scrambled control ODN, decreases expression of bcl-2 protein in response to sublethal ischemia
Figure 1 illustrates representative Western blots of brain tissue homogenates obtained 72 hours after 30 minutes of temporary focal ischemia in control rats or rats subjected to ischemia and perfused with either antisense, sense, or scrambled ODNs, or vehicle (artificial CSF). Each gel consisted of a pair of lanes from each experimental group. Densitometry was performed and the ratio of the experimental groups to the mean of the two control lanes was obtained. Changes in bcl-2 band density are shown in Fig. 2. Ischemia induced a significant increase in bcl-2 expression in CSF sense and scramble ODN-treated rats compared with controls. However, the antisense treated rats had no significant change compared with control. In contrast to bcl-2 there was significant basal expression of bcl-x long. After ischemia there was an increase in bcl-x long expression in all groups, but there was no effect of antisense treatment on bcl-x-long. To establish if ODN treatment had any effect on constitutively expressed proteins, the authors determined the effect of ODN treatment on the 73 kDa constitutive heat shock protein (hsc73) (Sorger and Pelham, 1987). hsc73 was constitutively expressed in control brain, but there was no change in expression induced by ischemia. Furthermore, there was no difference in hsc73 expression in the antisense treated group compared with other groups. These results suggested the bcl-2 antisense ODN selectively inhibited translation of bcl-2.
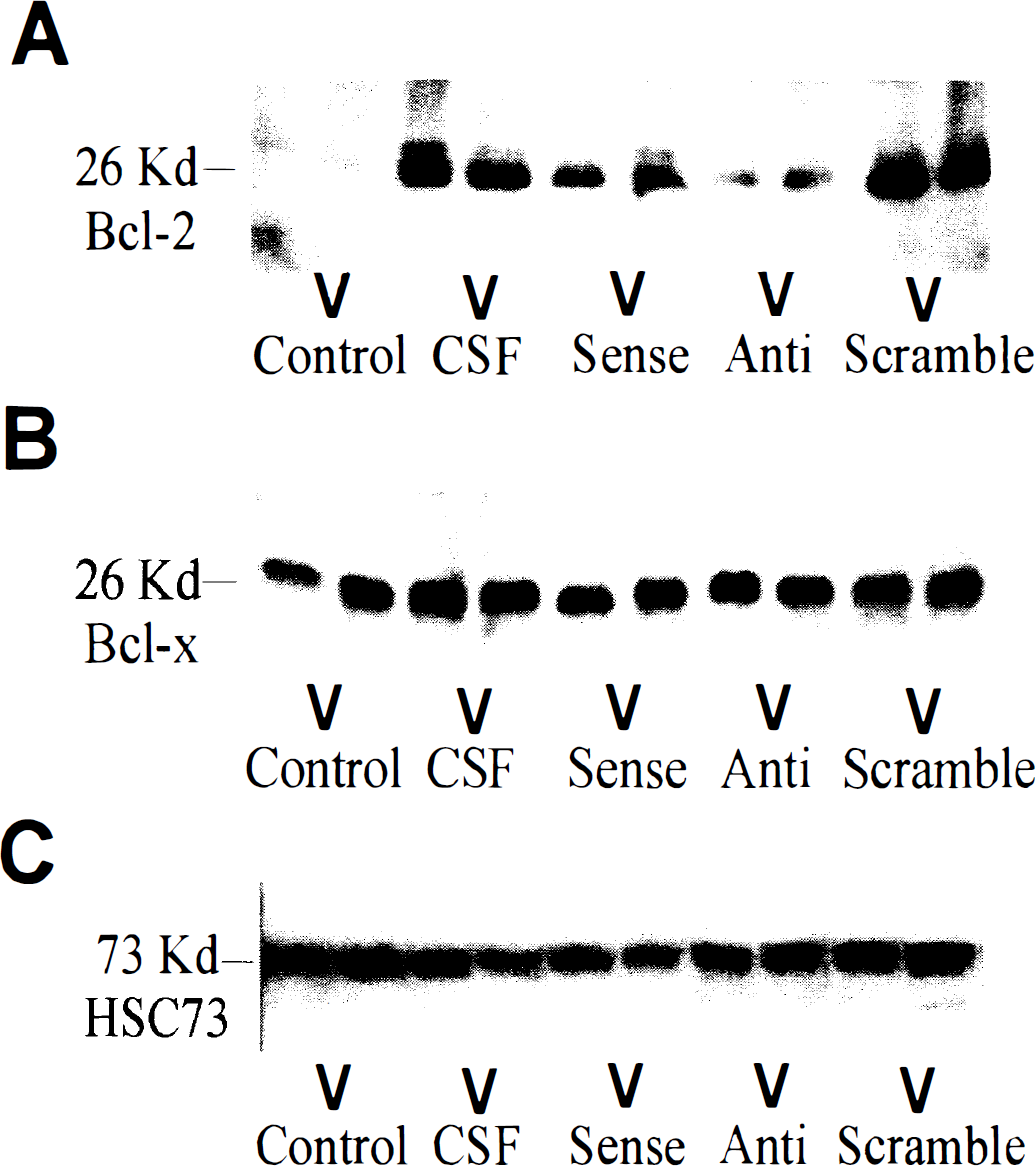
Representative Western blots of brain tissue homogenates obtained 24 hours after 30 minutes of temporary focal ischemia in rats subjected to ischemia and perfused with either antisense (Anti), sense (Sense), or scrambled (Scrambled) ODNs, or vehicle (CSF) or sham-operated rats not subjected to ischemia (Control). Pairs of samples were electrophoresed in adjacent lanes for each experimental condition.
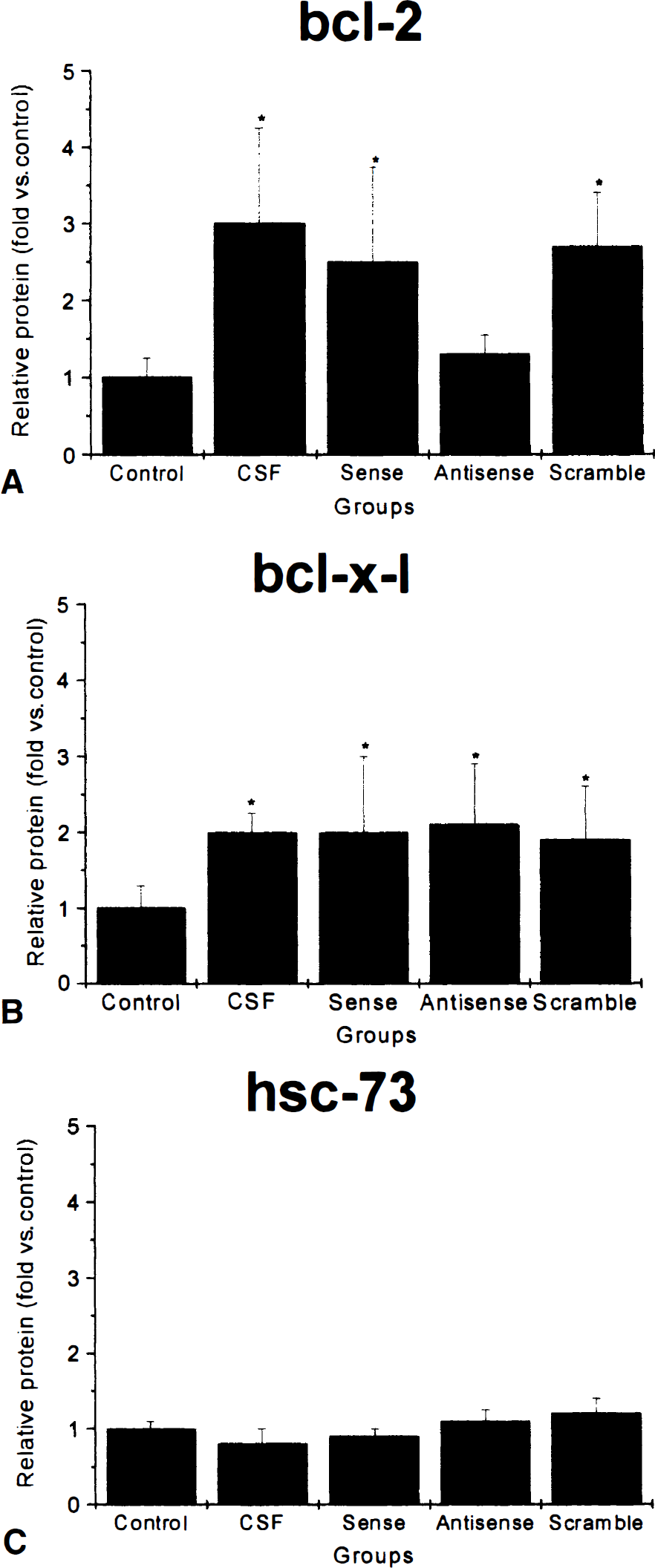
Relative changes in protein expression from 6 lanes for each experimental condition. Control, brain tissue removed from naïve rats. CSF, vehicle (mock CSF) infused for 72 hours after temporary middle cerebral artery occlusion. Sense, vehicle + sense ODNs infused for 72 hours after temporary middle cerebral artery occlusion. Anti, vehicle + antisense ODNs infused for 72 hours after temporary middle cerebral artery occlusion. Scramble, vehicle + scrambled ODNs infused for 72 hours after temporary middle cerebral artery occlusion.
The effect of antisense oligonucleotide treatment on bcl-2 expression was also studied using immunocytochemistry (Fig. 3). Sixty minutes of temporary focal ischemia was induced in rats. Brains were removed for Western blot analysis of bcl-2 protein expression 72 hours later. In control animals not subjected to ischemia there was increased expression of bcl-2 in neurons in the cortical mantle and medial striatum. These areas are adjacent to the border of the infarction and are ischemic in this model yet survive. By contrast, the rats that were treated with antisense ODN had expression of bcl-2 limited to only the most medial aspects of the striatum and the cortical mantle. Brains of two rats perfused with fluorescein isothiocyanide labeled antisense ODN were removed 72 hours after ischemia, frozen, and sectioned on a cryostat. Faint fluorescence was observable in many neurons in the cortical mantel ipsilateral to the middle cerebral artery occlusion, but not on the contralateral cortex (data not shown).
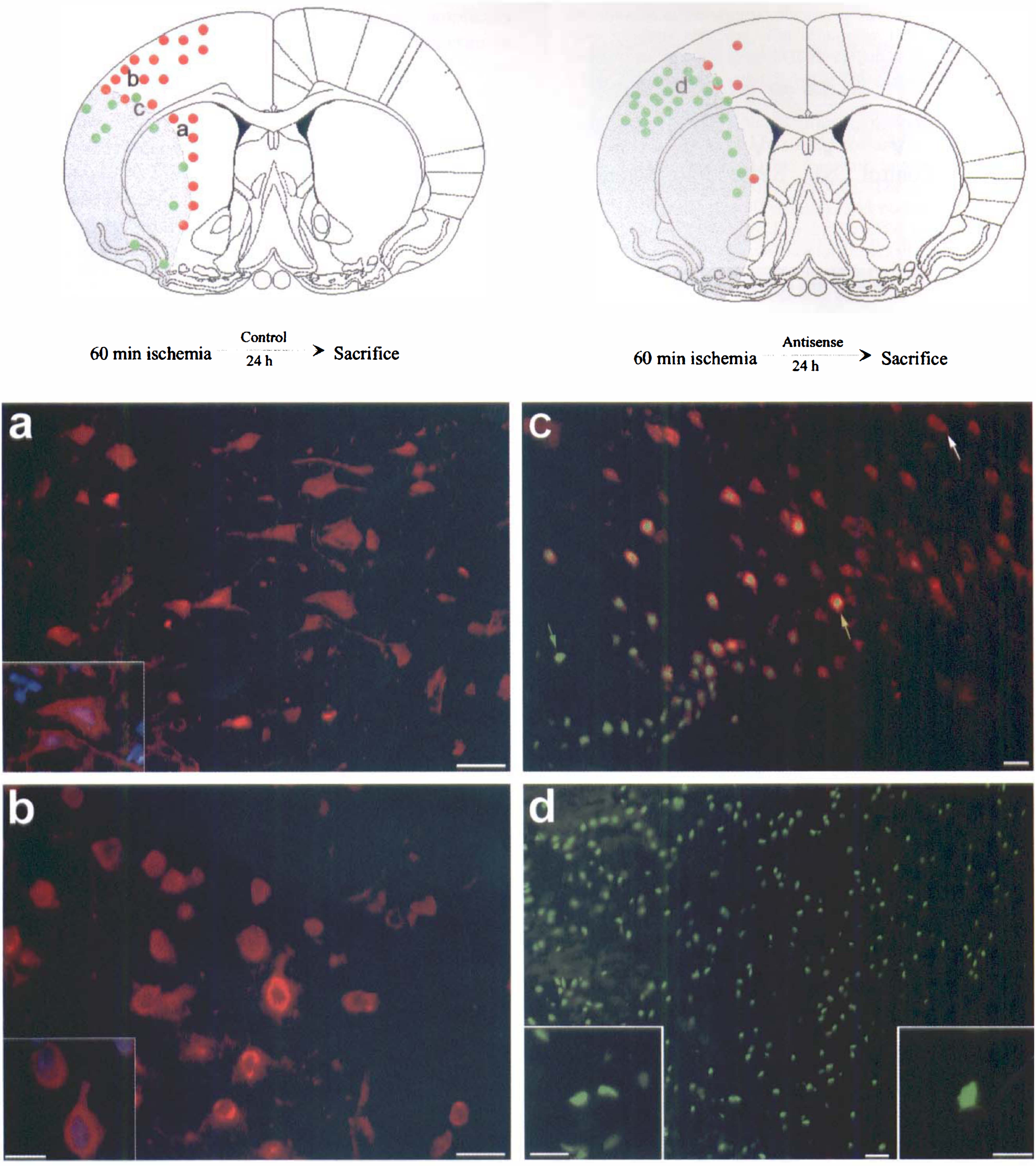
Immunocytochemical studies of bcl-2 expression after temporary focal ischemia. Schematic drawings illustrate distribution of bcl-2 immunoactivity (red dots) and TUNEL staining (green dots). The schematic illustration of the coronal section of the rat brain (left) indicates the distribution of bcl-2 immunoreactivity and TUNEL staining in a typical brain removed 24 hours after 60 minutes of ischemia; (right) illustrates the typical distribution of bcl-2 immunoreactivity and TUNEL staining in the brain subjected to 60 minutes of ischemia in which antisense oligonucleotides added to bcl-2 were infused for 24 hours postischemia; then, the rat was killed and the brain was removed for immunocytochemical analysis. The gray shaded area indicates the region that is infarcted on adjacent cresyl violet stained sections (not shown).
Treatment with bcl-2 antisense ODNs exacerbate ischemic neuronal death
The effect of antisense treatment on infarction volume was determined by infusion of either antisense or sense ODN, or vehicle (CSF) into the lateral ventricle for 72 hours after 60 minutes of temporary focal ischemia. Treatment with sense ODN resulted in no significant change in infarction volume from CSF vehicle controls; however, animals treated with bcl-2 antisense had a significantly larger infarction area at several sections and a larger infarction volume (Fig. 4). Furthermore, there was an apparent increase in the distribution of TUNEL staining neurons in the cortical mantel (Fig. 3D). Many of the neurons had TUNEL staining restricted to the nucleus and shrinkage of the nucleus, which are morphological characteristics that suggest apoptosis. That region in control brain contained many neurons that expressed bcl-2, but bcl-2 expression in this region was suppressed by antisense treatment.
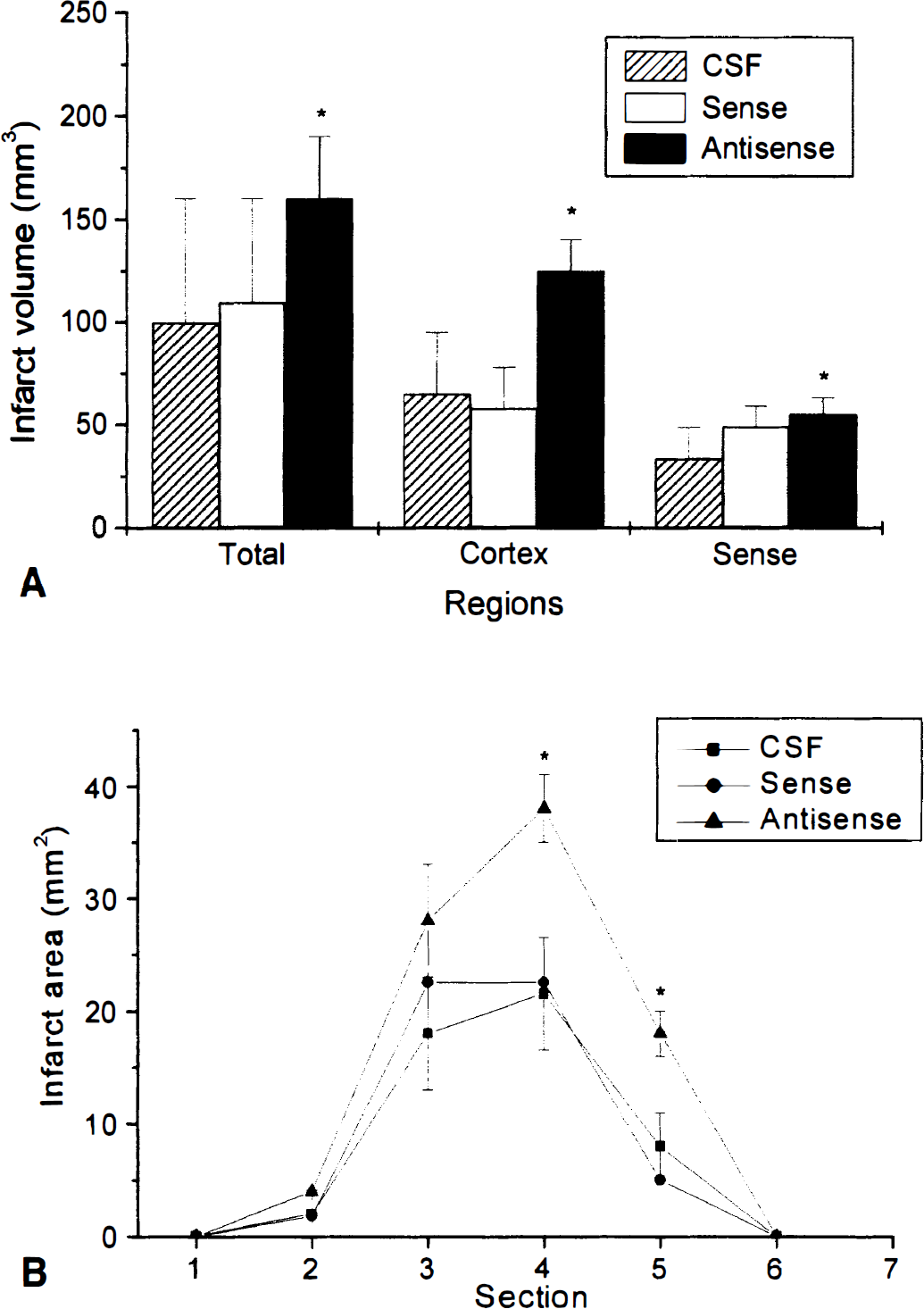
Effect of oligonucleotide treatment on infarction volume. Rats were subjected to 60 minutes of ischemia and antisense oligonucleotides or vehicle were infused for 24 hours through an intraventricular osmotic infusion pump.
DISCUSSION
The major finding of this study was that inhibition of bcl-2 expression in ischemic neurons by antisense treatment exacerbated neuronal injury and increased infarction size. A growing body of evidence supports the hypothesis that alterations in bcl-2 family genes occur after ischemia and exacerbate injury. Furthermore, overexpression of antiapoptotic members of the family, such as bcl-2, can protect neurons against ischemia. The current study showed that endogenously expressed bcl-2 might play an important role in determining the survival of ischemic neurons.
Several previous studies have examined expression of bcl-2 and related genes after cerebral ischemia. Expression of bcl-2 is barely detectable in normal nonischemic rodent brain, but expression is induced after ischemia in neurons that are ischemic but survive. Expression of bcl-2 is suppressed in severely ischemic neurons that are destined to die (Chen et al., 1995; Chen et al., 1997; Gugan et al., 1998; Krajewski et al, 1995). Expression of bax, a proapoptotic member of the bcl-2 family, is increased in these dying neurons (Chen et al, 1996; Gillardon et al., 1996; Hara et al, 1996; Krajewski et al., 1995), which is consistent with the hypothesis that the ratio of proapoptotic to antiapoptotic members of the bcl-2 family determines the fate of ischemic neurons. The current finding that bcl-2 expression was induced in neurons that do not TUNEL label was also consistent with this hypothesis.
There may be several mechanisms by which bcl-2 exerts its protective effect on ischemic neurons. Bcl-2 plays a key role in regulating initiation of programmed cell death by preventing egress of cytochrome C into cytoplasm. Cytochrome C, in turn, complexes with apaf-1 activating caspase 9, an important triggering event in apoptosis. Caspase 9 activates other caspases, including caspase 3, which then executes many of the biochemical changes that characterize programmed cell death. Thus bcl-2 regulates the key committed step in the programmed cell death cascade. Bcl-2 may also play a role in stabilizing the mitochondria and maintaining its membrane potential, thus preventing the generation of free radicals (Kane et al., 1993). Therefore, bcl-2 has antioxidant effects that may be important in necrotic and apoptotic cell death (Kane et al., 1995). Finally, bcl-2 also regulates calcium fluxes into the mitochondria (Murphy et al., 1996). An important event after ischemia is the influx of calcium into the mitochondria (Simon et al., 1984) that may uncouple mitochondrial respiration and otherwise disrupt mitochondrial function (Shears and Bronk, 1980). Thus, the expression of bcl-2 in neurons that are stressed may be an important event that ensures neuronal survival.
There were several limitations to the current study. As with any pharmacologic treatment, antisense oligonucleotides can have effects other than their desired action. Oligodeoxynucleotides can bind with membrane glycoproteins and ODN metabolic products such as nucleotides, and may interact with targets other than the desired sequence (Wagner, 1994). Nonetheless, the ODN did enter ischemic cortical neurons and had a sequence-specific effect on protein synthesis and infarct volume reduction. Furthermore, there was no effect of the antisense treatment on bcl-x-l expression or on expression of the constitutive heat shock protein hsc73. Thus, it appeared that the bcl-2 antisense ODN treatment had a selective effect on bcl-2 expression.
The current data shows that inhibition of bcl-2 expression by treatment with bcl-2 antisense after ischemia exacerbates neuronal death. These data support the hypothesis that endogenous expression of bcl-2 after ischemia is an important determinant of neuronal survival.
Footnotes
Acknowledgments
The authors thank Jingyu Luan and Jennifer Sinclair Sergi for technical support, and Pat Strickler for secretarial support.