
Abstract
We tested the hypothesis that treatment of transient focal cerebral ischemia in rat with antibodies directed against adhesion molecules reduces apoptosis. Rats (n = 31) were subjected to 2 h of middle cerebral artery (MCA) occlusion induced by intraluminal insertion of a nylon monofilament into the internal carotid artery. Upon reperfusion, animals were treated with monoclonal antibodies directed against intercellular adhesion molecule (ICAM)-1) (n = 8) or integrin CD11b/CD18 (n = 10), or administered IgG1 as a control (n = 13). At 48 h after ischemia, animals were killed and the brains analyzed for ischemic cell damage, using hematoxylin and eosin (H/E); apoptosis, using the terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick end labeling (TUNEL) method; and inflammatory cells, using immunohistochemistry with an anti-myeloperoxidase (MPO) antibody. Data revealed a significant reduction in the volume of infarction (p < 0.01) and a decline in the absolute (p < 0.001), and normalized (to the ischemic area, p < 0.05) numbers of apoptotic cells in both animals treated with anti-ICAM-1 and anti-CD11b antibodies compared to control animals. The numbers of immunoreactive MPO cells were also reduced in the treatment groups compared to those in the control group (p < 0.05). These data suggest that treatment with anti-adhesion molecule antibodies selectively reduce apoptosis, and that a contributing factor to the beneficial effect of antibody treatment for reducing ischemic cell damage may be a reduction in numbers of apoptotic cells.
Apoptosis is present in rat brain after onset of middle cerebral artery (MCA) occlusion and may contribute to ischemic tissue damage (Linnick et al., 1993; Charriaut-Marlangue et al., 1995; Li et al., 1995a-d). Cells exhibiting apoptotic morphology undergo a process of cell death that fundamentally differs from that of necrosis. Apoptosis is orchestrated by gene and/or protein expression, in contrast to necrosis in which the initiating and promoting events are primarily associated with disruption of ionic and metabolic homeostasis (Kerr et al., 1972; Wyllie et al., 1980; Ellis et al., 1991; Bursch et al., 1992; Vaux 1993; Vaux et al., 1994; Steller (1995).
Antibodies against adhesion molecules (Clark et al., 1991a,1991b; Bowes et al., 1993; Chen et al., 1994; Chopp et al., 1994; Jiang et al., 1994; Zhang et al., 1994, 1995), as well as antibiotics (Bowes et al., 1993), and proteins (Moyle et al., 1994; Jiang et al., 1995) have been used to reduce inflammatory cell response and cell damage after experimental cerebral ischemia, inflammatory cells presumably promote ischemic cell damage by microvascular occlusion, thereby prolonging or intensifying the ischemic event, and by cytotoxic means, in which neutrophils release free radicals and proteases (Engler et al., 1983; Braide et al., 1984; Harlan 1985; Hallenbeck et al., 1986; Schmid-Schönbein and Engler, 1986; Schmid-Schönbein 1987; Granger 1988; Mullane et al., 1988; Lucchesi et al., 1989; Weiss 1989). Although the volume of cerebral infarction is reduced after administration of agents designed to reduce neutrophil adherence to endothelium and subsequent migration into the ischemic tissue, inflammatory cells may exacerbate ischemic cell damage by other means, such as by inducing apoptosis.
In the present study, we tested the effects of administration of two anti-adhesion molecule antibodies, anti-CD11b and anti-intercellular adhesion molecule (ICAM)-1, on the numbers of apoptotic cells in rats that have been subjected to transient MCA occlusion. We selected these two antibodies because they both show efficacy in reducing ischemic cell damage in the rat even when administered hours after the onset of focal cerebral ischemia (Chopp et al., 1994; Zhang RL et al., 1995; Zhang ZG et al., in press). However, these antibodies differ in that the anti-CD11b acts against a β2 integrin molecule that is primarily expressed within the leukocyte, while the anti-ICAM-1 antibody acts against a glycoprotein that is primarily expressed on the surface of the endothelial cell. Although ICAM-1 and β2 integrins induce apoptosis in T cells (Damle et al., 1993), to our knowledge, there have been no studies on the role of postischemic inflammatory cells and adhesion molecules as mediators of apoptosis.
METHODS
Male Wistar rats (weighing 270–300 g, n = 31) were used in the experiments. Transient MCA occlusion was induced by advancing a 4–0 surgical nylon monofilament into the internal carotid artery (ICA) to block the origin of the MCA (Zea Longa et al., 1989; Chen et al., 1992). Briefly, animals were anesthetized with 3.5% halothane, and anesthesia was maintained with 1.0% halothane in 70% N2O and 30% O2 (vol/vol) using a face mask. Rectal temperature was maintained at 37°C throughout the surgical procedure using a feedback-regulated water heating system. The right femoral artery and vein were cannulated for measuring blood gases (pH, Po2, Pco2) before ischemia and for drug administration, respectively. A length of 18.5–19.0 mm 4–0 surgical nylon filament with its tip rounded by heating near a flame, was advanced from the external carotid artery into the lumen of the ICA until it blocked the origin of the MCA. At 2 h after MCA occlusion, animals were reanesthetized with halothane and reperfusion was performed by withdrawal of the filament until the tip cleared the ICA lumen.
Animals were divided into three groups. Anti-ICAM-1 group (n = 8) animals were subjected to MCA occlusion and an anti-rat ICAM-1 antibody was infused (i.v.) over a 3-min interval at a dose of 1 mg/kg upon reperfusion and 0.5 mg/kg at 22 h of reperfusion. Anti-CD11b group (n = 10) animals were subjected to MCA occlusion and anti-CD11b antibody was infused (i.v.) over a 3-min interval at a dose of 2 mg/kg upon reperfusion and 1.0 mg/kg at 22 h of reperfusion. Control group (n = 13) animals were subjected to MCA occlusion and an isotype-matched control antibody (mouse IgG1, Sigma Chemicals, St. Louis, MO, U.S.A.) was administered upon reperfusion and at 22 h of reperfusion, using the same volume dose as the experimental groups. The treatment antibodies have endotoxin levels of < 1.0 eu/mg.
The numbers of apoptotic cells within ischemic rat brain subjected to 2 h of MCA occlusion are maximum at 48 h after onset of ischemia in rats (Li et al., 1995a). Although it is preferable to kill animals at times >48 h when infarct volumes are to be measured, the primary hypothesis to be tested was that antibodies against adhesion molecules reduce numbers of apoptotic cells in ischemic tissue; therefore, 48 hour was selected as the most efficacious time for counting of apoptotic cells.
Antibody to rat ICAM-1, designated 1A29, was developed according to details provided in a recent publication (Tamatani and Miyasaka 1993) and was obtained from Drs. Anthony Manning and Donald Anderson at the Upjohn Company (Kalamazoo, MI, U.S.A.). The anti-CD11b antibody (clone 1B6c, F(ab′)2) was obtained from Repligen Corporation (Cambridge, MA, U.S.A.), and is a mouse anti-rat antibody (Mulligan et al., 1992, 1993; Zhang ZG et al., in press).
Animals were anesthetized (i.m.) with ketamine (44 mg/kg/body wt) and xylazine (13 mg/kg/body wt) at 46 h of reperfusion. Rats were transcardially perfused with heparinized saline and 10% buffered formalin, and brains were removed. Each brain was cut into seven 2 mm-thick coronal blocks (A-G), using a rat brain matrix. Brain tissue was processed and embedded, and 5 μm-thick paraffin sections were cut from each block and stained with hematoxylin and eosin (H/E) for histopathological evaluation. In addition, adjacent 5 μm-thick paraffin sections were obtained from block D and a molecular biological-histochemical system (ApopTag kit, Oncor, Gaithersburg, MD, U.S.A.) for specific staining of DNA fragmentation and apoptotic bodies, the terminal deoxynucleotidyl (TdT)-mediated dUTP biotin nick end labeling (TUNEL) stain (Gavrieli et al., 1992), and immunohistochemistry of myeloperoxidase (MPO) for neutrophil detection (Daugherty et al., 1994) were employed.
The area of infarction was measured using a Global Lab Image analysis program (Data Translation, Malboro, MA, U.S.A.). Each H/E section was evaluated at ×2.5 magnification. The area of infarction and the area of the ipsilateral hemisphere (mm2) were calculated by tracing the areas on the computer screen. To reduce errors associated with the processing of tissue for histological analysis, the area of infarction in each section was presented as the percentage of the infarct to the ipsilateral hemispheric area; the infarct volume was also presented as the percentage of infarct to the ipsilateral hemisphere.
Cells exhibiting DNA fragmentation were counted in both the ipsilateral and contralateral hemispheres using a light microscope (×200–300 magnification). Cells containing apoptotic bodies are referred to as apoptotic cells (Gavrieli et al., 1992; Wijsman et al., 1993; Wood et al., 1993; Li et al., 1995a). The TUNEL method is based on the specific binding of TdT to the 3′-OH ends of DNA and the ensuing synthesis of a polydeoxynucleotide polymer. Briefly, after deparaffinizing the brain sections, digesting the protein in specimens with proteinase K, then quenching endogenous peroxidase activity with 2% H2O2 in phosphate-buffered saline (PBS), slides were placed in equilibration buffer and then in TdT enzyme, followed by stop/wash buffer. After two drops of anti-digoxigenin-peroxidase was applied to the slides, peroxidase was detected with diaminobenzidine. Negative controls were performed using distilled water to replace TdT Enzyme. The labeling targets of the TUNEL staining were the new 3′-OH DNA ends generated by DNA fragmentation; these were typically localized in morphologically identifiable nuclei and apoptotic bodies. The direct immuno-peroxidase detection of digoxigenin-labeled DNA in thin sections of fixed tissue allowed analysis of scattered cells exhibiting DNA fragmentation at anatomical locations after MCA occlusion. In contrast, normal nuclei, which had relatively insignificant numbers of DNA 3′-OH ends, did not stain with the TUNEL method. Cells exhibiting necrotic morphologies contained, in some instances, stain-able concentrations of DNA ends; however, staining in necrotic cells appeared more diffuse than in apoptotic cells. Coronal sections stained with the TUNEL method were also counterstained with hematoxylin.
Numbers of neutrophils within brain were counted using immunohistochemical staining for MPO (Bradley et al., 1982; Daugherty et al., 1994). MPO is a marker for inflammatory cells, primarily neutrophils (Barone et al., 1991). Sections (5 UJTI thick) adjacent to those used for counting apoptotic cells were obtained and stained with a polyclonal rabbit anti-human MPO antibody (Dako no. A398, Copenhagen, Denmark). All immunoreactive cells were counted within the ipsilateral hemisphere using a light microscope (magnification ×132).
Welch's two-sample t-tests were used to compare the anti-ICAM-1 and anti-CD11b groups to the vehicle group with respect to lesion area, ratio of lesion area/ipsilateral hemisphere area, number of apoptotic cells, and ratio of the number of apoptotic cells to the lesion area. A Bonferroni-adjusted α-level of 0.025 (0.05/2) was used to interpret results of each test. P-values of ≦0.025 were considered to be statistically significant, while p-values of 0.025–0.05 were considered to be “marginally” significant. All measurements were performed blindly (by J.P., N.J., R.L.Z., Y.L.). All data are presented as mean value + SD.
RESULTS
Blood gases were within normal ranges for all animals and did not differ between groups (Table 1). Animals treated with anti-ICAM-1 and anti-CD11b antibodies exhibited percent cerebral infarct volumes of 18.0 ± 11.2 (n = 8) and 24.3 ± 11.0 (n = 10), respectively, that were significantly smaller (p = 0.01) than the percent cerebral infarct volumes obtained in control animals (36.5 ± 7.7; n = 13). These data are consistent with those of our previous reports (Chen et al., 1994; Jiang et al., 1994; Zhang et al., 1994).
Blood gases (mean ± SD) in the three groups
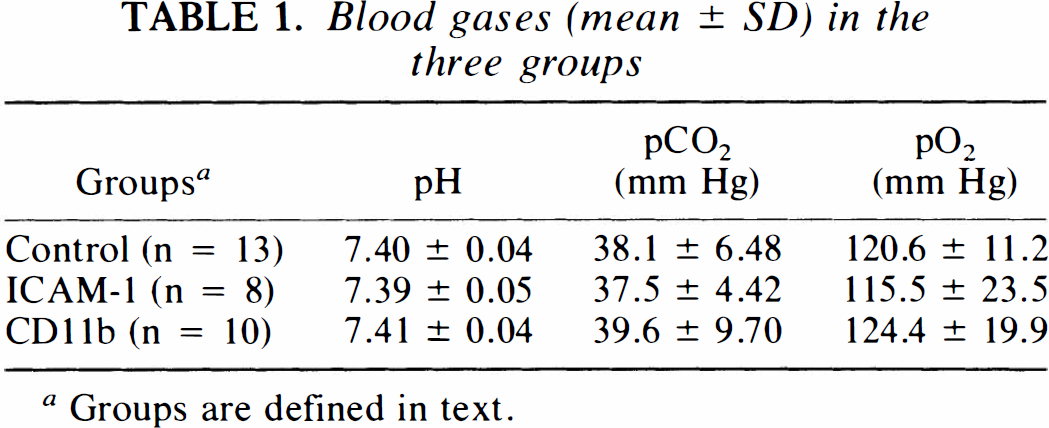
Groups are defined in text.
Figure 1 shows a representative coronal section of striatal tissue from a control animal in which apoptotic and inflammatory cells are readily apparent within the ischemic region. As we have previously reported in control ischemic animals (Li et al., 1995a–Li et al., 1995c), the apoptotic cells are primarily concentrated along the inner boundary of the zone of infarction.
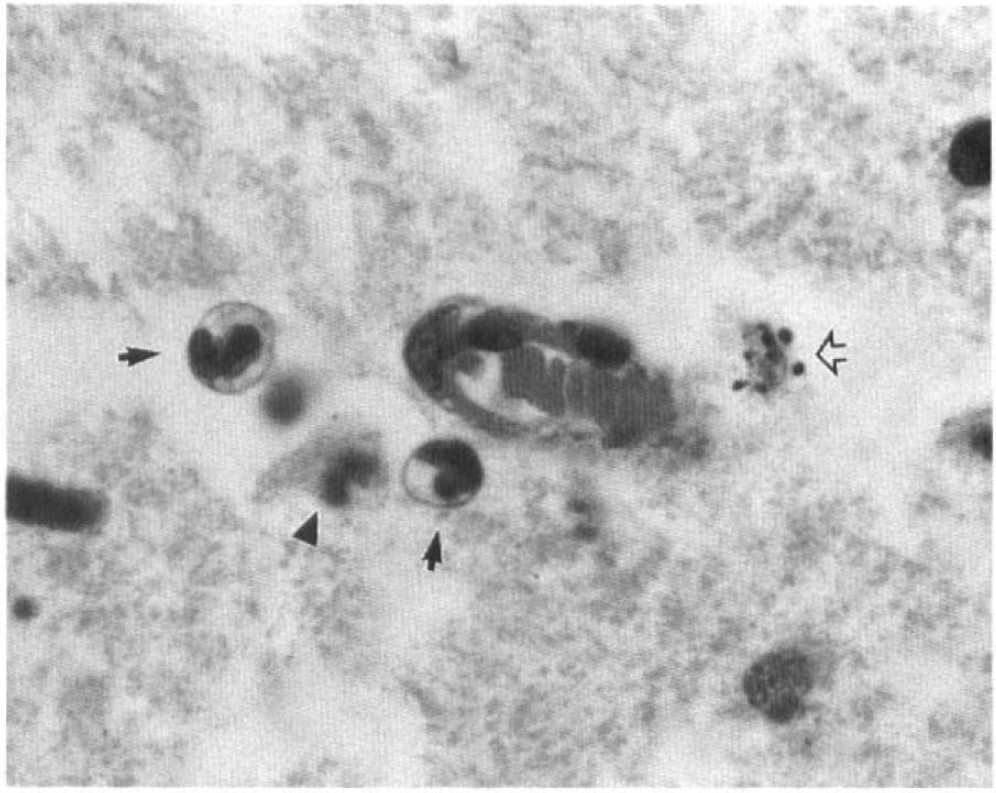
Coronal sections (5 μm thick) stained with hematoxylin and eosin from striatal tissue of an animal subjected to 2h of MCA occlusion and sacrificed 48 h after onset of reperfusion. ↑, neutrophil; ⇑, apoptotic cell; ▾, necrotic neuron original magnification ×660.
The difference in the mean number of apoptotic cells was found to be significantly different between vehicle (186.3 ± 43.4; n = 13) and anti-ICAM-1 (54.8 ± 57.0; n = 8) (p < 0.001), and vehicle and anti-CD11b (79.3 ± 57.1; n = 10) (p < 0.001). For the normalized numbers of apoptotic cells, the vehicle group (7.6 ± 2.6; n = 13) was found to be significantly different from the anti-ICAM-1 (4.2 ± 3.2; n = 8) (p = 0.023), and the vehicle group was marginally significantly different from the anti-CD11b (4.9 ± 3.1; n = 10) treatment group (p = 0.040).
The number and ischemic area normalized numbers of MPO immunoreactive cells were significantly (p < 0.01) reduced within the treated groups compared to the control group: control group (596 ± 331; 23.6 ± 11.6 mm−2; n = 5); anti-ICAM-1 group (166 ± 144; 13.8 ± 8.3 mm−2; n = 6); and anti-CD11b group (170 ± 142; 16.1 ± 10.5 mm−5; n = 5). No difference was detected in the absolute and normalized numbers of MPO immunoreactive cells between the anti-ICAM-1 and anti-CD11b groups. Numbers of immunoreactive MPO and apoptotic cells within adjacent sections of tissue were highly correlated (r = 0.78; p < 0.001). Figure 2 illustrates the relative distribution of apoptotic and immunoreactive MPO cells within a coronal section. Immunoreactive MPO cells exhibited a similar spatial pattern of distribution as the apoptotic cells.
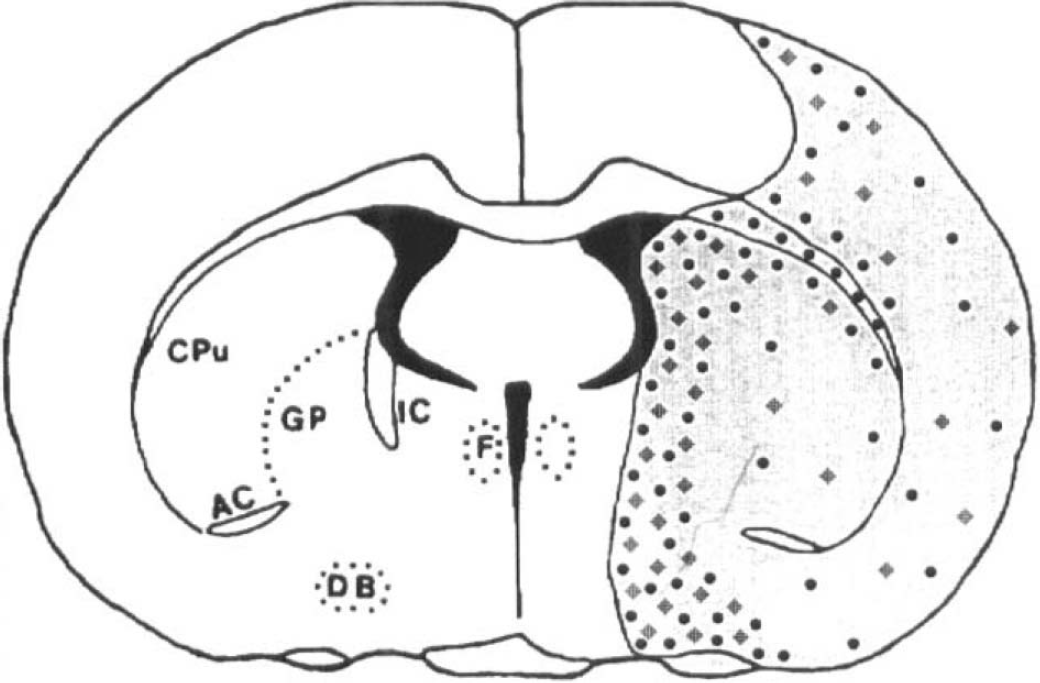
Schematic illustration of the spatial distribution of apoptotic: (⋄) and MPO (•) immunoreactive cells within a coronal section (D) of cerebral tissue. The gray shading corresponds to neuronal necrosis.
DISCUSSION
Our data indicate that administration of antibodies designed to reduce leukocyte adhesion and subsequent migration into ischemic tissue also reduces the numbers of apoptotic cells. There is a significant attenuation of apoptotic cells (absolute and normalized numbers) after treatment designed to reduce postischemic inflammatory response. Thus, the therapeutic benefit of administration of antibodies against adhesion molecules not only is associated with improvement of cerebral blood flow and reduction of cytotoxicity but also includes a reduction in the number of cells undergoing apoptosis.
There are several possible explanations for the reduction of apoptotic cells after treatment with anti-adhesion molecule antibodies. Reduction of apoptosis can be attributed to a reduction in numbers of inflammatory cells and/or to a direct effect of the antibodies on parenchymal cells. Inflammatory cells can induce apoptosis (Squier et al., 1995). Cytotoxic T lymphocytes undergo apoptosis and induce apoptosis in target cells (Duke et al., 1983; Ucker, 1987; Sellins and Cohen 1991; Nagata and Golstein, 1995). Although neutrophils undergo apoptosis (Squier et al., 1995), to date, there are no reports that neutrophils directly induce apoptosis in target cells. However, it is possible that the reduction in numbers of neutrophils within ischemic tissue indirectly causes reduction of apoptosis. This hypothesis is consistent with the robust correlation of immunoreactive MPO and apoptotic cells as well as the similar spatial distribution of immunoreactive MPO and apoptotic cells. Neutrophils are a potent source of oxygen-free radicals, and free radicals induce apoptosis (Hockenbery et al., 1991, 1993). Adherent neutrophils can release oxidizing radicals into the neural cell cytoplasm. Proteases, likewise, induce apoptosis (Sarin et al., 1993; Miura et al., 1994; Thompson, 1995), and leukocytes are a profuse source of proteases (Henson and Johnston, 1987; Weiss, 1989; Hansen, 1995). Expression of matrix-degrading proteases in normal tissues causes apoptosis (Talhouk et al., 1992). Interleukin 1β-converting enzyme (ICE) is a protease that facilitates apoptosis in mammalian cell lines (Los et al., 1995), and inflammatory cells express proteases, such as granzyme β, with similar apoptotic activity (Sleath et al., 1990; Howard et al., 1991; Odake et al., 1991; Shi et al., 1992; Caputo et al., 1994; Heusel et al., 1994).
Reduction of apoptotic cells compared to vehicle-treated animals was more pronounced with anti-ICAM-1 antibody treatment (p = 0.023) than with anti-CD11b antibody treatment (p = 0.040). However, the number of neutrophils within the ischemic tissue was nearly identical for the anti-ICAM-1 and anti-CD11b-treated groups; if apoptosis is caused by a direct attack of neutrophils on the parenchymal cells, then we expect the numbers of apoptotic cells to closely reflect the numbers of neutrophils within the tissue. It is, therefore, unclear why blocking the ICAM-1 sites shows a trend (although not statistically significant) towards being more effective in reducing apoptosis than blocking the CD11b sites. A possible explanation for the more potent anti-ICAM-1 reduction of apoptosis is that cell adhesion molecules (CAMs) directly affect neurons. During development of the nervous system, CAMs promote neuronal migration and axonal growth; yet, at other times, CAMs inhibit these events by maintaining stable adhesion between cells (Doherty and Walsh, 1994). Cytokines released in the course of inflammation induce expression of ICAM-1 on neurons, allowing them to be targeted by neutrophils (Birdsall, 1991). Adherence of neutrophils to cytokine-stimulated neural cells is mediated primarily by ICAM-1 and lymphocyte function antigen-I (LFA-1) interactions, and 70–90% of the binding can be blocked by monoclonal antibodies to either ligand (Birdsall, 1991). A direct role for adhesion molecule mediated-apoptosis was recently demonstrated in which T-cell receptor-directed engagement of integrins by their ligands ICAM-1 or VCAM-1 induced apoptosis of T cells (Damle et al., 1993).
Although the mechanism by which the anti-ICAM-1 antibody inhibits apoptosis is not known, it may be related to a coupling of ICAM-1, IL-1β, and ICE. The mammalian gene, ICE, encodes a cysteine protease involved in the processing of IL-1β. ICE is required for the conversion of pro IL-1β into active IL-1β. ICE induces apoptosis and activates IL-1β, which, in turn, induces ICAM-1 (Springer, 1994). Conversely, whether blocking ICAM-1 inhibits IL-1β and ICE, and, subsequently, reduces apoptosis is not known. An additional pathway relating adhesion molecules and apoptosis is related to the observation that tumor suppressor genes encode cell adhesion molecules (Hedrick et al., 1993). Tumor suppressor genes, e.g. p53, also induce apoptosis (Clarke et al., 1993). Thus, apoptosis may be facilitated by adhesion molecules, and, therefore, blocking adhesion molecules may reduce apoptosis, as demonstrated in the present study.
We have employed an immunohistochemical method using an anti-MPO antibody to measure inflammatory cells within the ischemic brain. This method has recently been applied to studies of human atherosclerotic lesions (Daugherty et al., 1994), and we adapted this method to the study of brain. MPO is a gold standard measurement for the presence of neutrophils (Barone et al., 1991). Although monocytes also possess MPO activity, MPO in monocytes accounts for <1% of the MPO recovered from inflammatory skin lesions (Bradley et al., 1982). We cannot exclude the possibility that other cells, such as monocytes and macrophages, may be included in the total MPO positive cell count.
The cellular target for the anti-adhesion molecule antibodies is not restricted to neutrophils, but may also include other exogenous inflammatory cells, such as monocytes and T lymphocytes. The antibodies employed in the present study are effective on different leukocytes. The CD11b receptor is most prominently expressed on neutrophils and monocytes (Kishimoto et al., 1989). In contrast, the ICAM-1 is a ligand for multiple β2 integrins, including those such as LFA-1, that are expressed on lymphocytes (Rothlein et al., 1986; Simmons et al., 1988; Staunton et al., 1988). ICAM-1 is also expressed in primary cultures of rat astrocytes (Shrikant et al., 1994). Although the focus of our study was apoptosis and MPO immunoreactive cells and although we did not measure the temporal profile of lymphocytes in ischemic tissue in the present study, T lymphocytes were present. Figure 3 shows a T lymphocyte within the ischemic tissue at 48 h after onset of reperfusion. It is, thus, possible, and further study is required to demonstrate, that the ability of the anti-adhesion molecule antibodies to reduce apoptosis may be related to the effect of antibodies, particularly ICAM-1, which is a ligand for LFA-1, on T lymphocytes or other inflammatory cells.
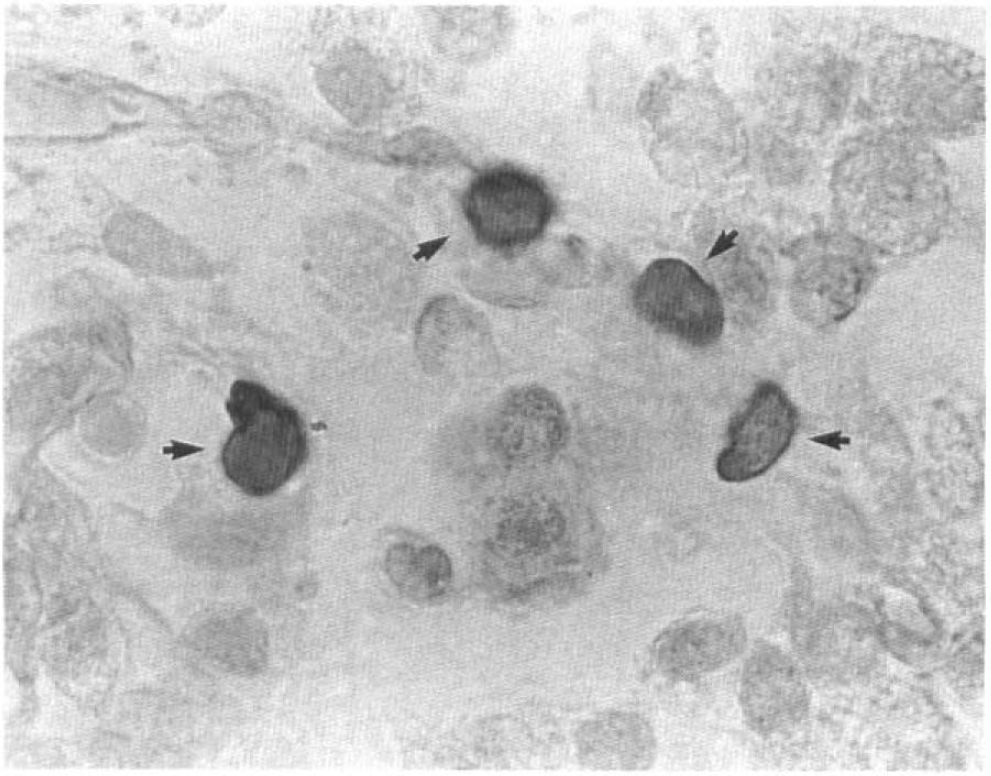
Coronal sections (5 μm thick), stained with hematoxylin and eosin, obtained from ischemic brain from a control animal. T-lymphocyte (↑) is present within the ischemic tissue. Original magnification ×660.
In summary, we have demonstrated that antibodies directed against CD11b and ICAM-1 adhesion molecules suppress apoptosis in ischemic brain. This observation raises the intriguing possibility that inflammatory cells and/or adhesion molecules and receptors promote apoptosis, and that antibodies against adhesion molecules suppress apoptosis in ischemic brain.
Footnotes
Acknowledgment:
The authors are grateful to Dr. Zhang G. Zheng for helpful discussions and technical assistance and Denice Janus for secretarial support. This work was supported in part from NINDS grants PO1 NS23393, RO1 NS33627, and RO1 NS34184.