
Abstract
Matrix metalloproteinase-9 (MMP-9) activity increases in the brain during the first day after focal ischemia and might be involved in the pathogenesis of tissue damage. We previously showed MMP-9 in the extracellular space of brain parenchyma along with neutrophil recruitment after ischemia. In the present study, we tested whether neutrophils were a direct source of enhanced MMP-9 in the ischemic brain. Neutrophil infiltration was prevented either by injecting an antibody against ICAM-1, which abrogates neutrophil adhesion to the endothelial vessel wall, or by inducing neutropenia. One-hour intraluminal middle cerebral artery occlusion with reperfusion was induced, and studies were performed at 24 hours. Circulating neutrophils expressed 95-kDa MMP-9 and dimers, and infiltrated neutrophils stained positive for MMP-9. The expression of MMP-9 (mainly 95-kDa proform and dimers and, to a lesser extent, 88-kDa form) increased in brain after ischemia/reperfusion. Treatments preventing neutrophil infiltration failed to preclude the ischemia-induced increase in 88-kDa MMP-9 form and gelatinase activity in neurons and blood vessels. However, these treatments prevented the major increase in 95-kDa MMP-9 form and dimers. We conclude that neutrophil infiltration highly contributes to enhanced MMP-9 in the ischemic brain by releasing MMP-9 proform, which might participate in the tissular inflammatory reaction.
Matrix metalloproteinases (MMPs) degrade the extracellular matrix and are involved in several brain diseases (Lukes et al., 1999; Mun-Bryce and Rosenberg, 1998; Yong et al., 1998). MMPs degrade components of the basal lamina, and several lines of evidence support their involvement in blood-brain barrier breakdown after ischemia (Asahi et al., 2000, 2001; Fujimura et al., 1999; Gasche et al., 1999; Heo et al., 1999; Rosenberg et al., 1998). MMP-9 becomes activated in brain tissue during the first day after cerebral ischemia/reperfusion (Asahi et al., 2000; Planas et al., 2000, 2001; Rosenberg et al., 1998, 2001) and also after permanent ischemia (Romanic et al., 1998). A correlation between MMP-9 activity and hemorrhagic transformation of the ischemic lesion has been reported (Heo et al., 1999; Lapchak et al., 2000; Sumii et al., 2002). Several strategies engineered to prevent activation of MMP-9 after ischemia, such as pharmacologic inhibition (Asahi et al., 2000; Romanic et al., 1998) or the availability of MMP-9 knockout animals (Asahi et al., 2000, 2001), are protective against brain infarct. The cellular source of MMP-9 is not fully known, as immunohistochemical studies have shown different results depending upon the specific antibodies used (Asahi et al., 2001; Planas et al., 2001, 2002; Romanic et al., 1998). Neutrophils express MMP-9 (Planas et al., 2002; Romanic et al., 1998), but whether the large increase of MMP-9 activity that is observed in brain after ischemia/reperfusion is caused by neutrophil recruitment remains to be proved. We previously showed that MMP-9 is released to the extracellular space of the ischemic brain concomitantly with neutrophil tissue recruitment (Planas et al., 2002). Neutrophil adhesion to the endothelial surface before infiltration into brain parenchyma is mediated by ICAM-1, among other adhesion molecules. In the present study, we tested whether neutrophils contribute to enhanced MMP-9 in focal cerebral ischemia/reperfusion by either causing neutropenia (Batteur-Parmentier et al., 2000) or by blocking ICAM-1 with a mAb (1A29) (Chopp et al., 1996; Furuya et al., 2001; Zhang et al., 1994, 1995).
METHODS
Animals and surgery for middle cerebral artery occlusion
Focal ischemia was produced in male Sprague-Dawley rats (280 to 320 g body weight) (Iffa-Credo, Lyon, France) by 1-hour intraluminal occlusion of the middle cerebral artery (MCA) with reperfusion, as reported previously (Justicia et al., 2001; Soriano et al., 1997). Rats were anesthetized (halothane) and intubated through the trachea for controlled ventilation. Mean arterial blood pressure (MABP) was monitored, and body temperature was maintained at 37.5°C during MCA occlusion. At 24 hours, rats were anesthetized with halothane and killed to obtain brain samples. For biochemical studies, rats were perfused through the heart with saline to remove blood from the cerebral vessels, and the region corresponding to the territory of the MCA ipsilateral and contralateral to the occlusion was dissected out and frozen at −80°C. For immunohistochemistry, rats were perfused with 4% paraformaldehyde, fixed overnight with the same fixative, and either embedded in paraffin and cut in 5-μm sections or cut in 50-μm sections with a vibratome. For the study of cellular gelatinase activity by in situ zymography, the brain was immediately frozen in isopentane at −60°C and sliced in 20-μm sections in a cryostat. Animal work was conducted in compliance with the Spanish legislation and in accordance with the Directives of the European Union.
Drug treatments
A mouse IgG1 against rat ICAM-1 (1A29) and a control nonbinding murine IgG1 against human (Ma et al., 1994) but not rat (Panes et al., 1995; Sans et al., 1999) P-selectin (P-23) were scaled up and purified by protein A/G chromatography at Pharmacia Upjohn Laboratories (Kalamazoo, MI, U.S.A.). ICAM-1 (n = 12) or P-23 (n = 12) antibodies (2 mg/kg) were injected intraperitoneal (Sans et al., 1999) during MCA occlusion at 15 minutes before reperfusion. Nontreated rats subjected to ischemia (n = 9) and nonoperated controls (n = 9) were also studied. Another group of rats was intravenously given either saline (n = 11) or vinblastine (Sigma) (0.5 mg/kg body weight) (n = 14) at day 0 (Batteur-Parmentier et al., 2000), and 4 days later ischemia was induced. At day zero, rats (saline or vinblastine) received the following antibiotics to prevent infection: 150,000 U/kg benzathinebenzylpenicillin (Benzatizil, Antibiotics Farma S.A.) and 10 mg/kg gentamicin (B. Braun S.A.). At days 0 and 4 (before MCA occlusion), arterial blood samples were withdrawn to count neutrophils (Sysmex SE-9000). Comparison of neutrophil counts in vinblastine-treated rats with rats receiving saline, as well as body weight and physiologic parameters, was made with the Mann-Whitney U-test.
Gel zymography
Frozen tissue samples were subjected to detergent extraction, purification of gelatinolytic activity, and zymography, as previously reported (Planas et al., 2001; Zhang and Gottschall, 1997). Frozen tissue was homogenized with lysis buffer containing 50 mM Tris-HCl pH 7.6, 150 mM NaCl, 5 mM CaCl2, 0.05% Brij-35, 0.02% NaN3, and 1% Triton X-100. All reagents, unless otherwise stated, were from Sigma. After homogenization and centrifugation 12,500 rpm for 5 minutes at 4°C, the supernatants were used for extraction of gelatinolytic activity. Protein (15 mg) in 500 μL was incubated with 50 μL of gelatin-sepharose (Gelatin Sepharose 4B, Amersham Biosciences, Uppsala, Sweden) under constant shaking at 4°C for 1 hour and centrifuged at 2,500 rpm for 2 minutes at 4°C. The pellet with gelatin-sepharose, which retained the gelatinases, was washed with 500 μL of washing buffer (containing the same components as the previous buffer, with the exception of Triton X-100) and each time centrifuged at 2,500 rpm for 2 minutes at 4°C before the separation of gelatinases with 150 μL of elution buffer (washing buffer containing 10% DMSO) by incubation at 4°C under constant shaking for 30 minutes followed by centrifugation, as described above. Extracted brain samples (3 μL; corresponding to 300 μg of protein in the supernatant obtained after tissue homogenization) were loaded in the gels, and a mixture of MMP-9 and MMP-2 (CC073, Chemicon) was used as the gelatinase standard. After electrophoresis, gels were incubated to allow gelatinase activity to take place (Planas et al., 2001). After staining, the gels were analyzed to determine intensity of the bands (Kds1D software, Kodak). Statistical analysis was carried out with two-way ANOVA by treatment (anti-ICAM-1 Ab versus anti-P-23 control Ab) and by MMP-9 band (95-kDa and 88-kDa).
Zymographic assay of plasma and neutrophils: isolation of neutrophils
Blood (2 mL) (citrate) was mixed with 2% dextran (Amersham) in saline and kept for 25 minutes at room temperature (RT). The supernatant (1 mL) was added to (0.5 mL) Ficoll (Biochrom KG, L6113, SeroMed), centrifuged (3,800 rpm 25 minutes, RT), and the resulting pellet (containing granulocytes) was washed (2 mL phosphate-buffered saline) and centrifuged (3,800 rpm, 10 minutes, RT). Any contaminant erythrocytes in this fraction were lysed with 10 mL 0.15M ammonium chloride for 10 minutes at 37°C and removed by centrifugation (2,500 rpm, 7 minutes) with two PBS washes. The pellet was mixed with 50 μL lysis buffer (as for gelatinase extraction) and sonicated, and the protein content was determined (Bradford, Bio-Rad). Zymography was performed with these samples and with 0.5 μL of plasma. We incubated the gels in the presence or absence of 50 mM EDTA, which inhibits the activity of gelatinases. In addition, we used two specific inhibitors for MMP-9: MMP-9/MMP-13 Inhibitor I (10 μM) and MMP-2/MMP-9 Inhibitor II (30 μM) (Calbiochem, San Diego, CA, U.S.A.).
Protein expression
Of the extracted brain samples, 75 μL was concentrated with trichloroacetic acid protein precipitation (Planas et al., 2001). The pellet was dissolved in loading buffer, and samples were run in a denaturing 10% polyacrylamide gel. Prestained SDS-PAGE molecular weight standards (Bio-Rad, Madrid, Spain) were run in one lane of each gel. Western blotting was carried out for MMP-9 with a mouse monoclonal antibody (mAb) (MAB 13420, Chemicon, 1:150) and for MMP-2 with a rabbit polyclonal Ab (pAb) (AB 809, Chemicon, 1:2000), as described previously (Planas et al., 2000, 2001).
Myeloperoxidase (MPO) expression (Wright et al., 1987) was studied by western blot (mAb M 1464, Menarini Diagnostics, 1:500) using crude brain homogenates obtained from the same animals used for studying MMP-9. In addition, the expression of an inducible heat shock protein (Hsp72) was examined in brain tissue by Western blot (mAb, Oncogene, 1:500) to check that the rats went through an episode of cerebral ischemia after MCA occlusion (Planas et al., 1997). Correct protein charge was tested with a rabbit pAb against actin (1:10,000) (Sigma).
Presence of myeloperoxidase in brain
MPO activity was determined (Batteur-Parmentier et al., 2000; Bradley et al., 1982) in nonischemic rats (n = 4) and rats subjected to ischemia (saline n = 4, and vinblastine n = 3) at 24 hours. Rats were anesthetized and perfused through the heart with saline. Frozen tissue samples were weighed and homogenized (Polytron PT3100) in 3 mL of 50 mM Tris-HCl pH 7.4 (10,000 rpm, 30 seconds on ice), and then 1 mL was mixed with 5 mL of 5 mM phosphate buffer pH 6.0 and centrifuged (22,000 rpm, 30 minutes, 4°C). The pellet was dissolved in 1 mL of 50 mM phosphate buffer pH 6.0 containing 0.5% hexadecyltrimethylammonium bromide. The mixture was frozen, unfrozen at 37°C, and sonicated for 10 seconds, and this procedure was repeated three times. Samples were kept on ice for 20 minutes, centrifuged (14,000 rpm, 15 minutes), and the supernatant was mixed with o-dianisidine dihydrochloride (Sigma) (0.167 mg/mL final concentration). MPO activity was determined with a spectrophotometer at 460 nm (Ultrospec 3000, Pharmacia Biotech) 3 minutes after the addition of 0.0005% H2O2. A standard curve with human MPO (Sigma) was used for calibration. Values obtained for the various groups of rats were compared with one-way ANOVA.
Immunohistochemistry
Immunohistochemistry was performed in paraffin sections (Planas et al., 2001) from rat brains obtained at 24 hours after MCA occlusion in groups of rats receiving mAb against ICAM-1 (n = 3), P-23 control mAb (n = 3), and nontreated (n = 3). The primary mouse mAb against MMP-9 were as follows: MAB 13420 (Chemicon) or Ab-10 (Oncogene), both diluted 1:50. After biotinylated anti-mouse Ab and the avidinbiotin complex (Vector Laboratories), the reaction was developed with diaminobenzidine (DAB) (brown stain). Several sections were counterstained with hematoxylin, and others were used for further staining with a rabbit pAb against MPO (A 0398, Dako). Double immunohistochemistry was performed as previously reported (Planas et al., 2001). A positive reaction gives a dark blue precipitate that appears as fine granules.
In addition, we performed immunohistochemistry with the 1A29 monoclonal antibody against ICAM-1 diluted (1:200) in control (n = 2) and ischemic brains at 24 hours (n = 3). In this experiment, we perfused the rats with 4% paraformaldehyde, kept the brains in the fixing solution overnight, and then obtained 50-μm thick sections with a vibratome. Sections were processed free-floating for immunohistochemistry using DAB to reveal anti-ICAM-1 staining and were then counterstained with hematoxylin to visualize cell nuclei.
In situ zymography
Frozen cryostat brain sections from ischemic rats (control n = 3 and vinblastine n = 3) were brought to RT and incubated with FITC-labeled DQ-gelatin (Molecular Probes) overnight at 37°C in a humidified chamber. Then, sections were washed with PBS and examined by fluorescence microscopy to reveal gelatinase activity at the cellular level. Some sections were also stained with a mAb against neuronal nuclei (NeuN, Chemicon, diluted 1:500) followed by a TRIC-labeled secondary Ab.
RESULTS
Several forms of MMP-9 increase in brain after ischemia: neutropenia prevents the ischemia-induced increase of the MMP-9 proform
Ischemia affected the cortex and striatum ipsilateral to MCA occlusion. Induction of 72-kDa heat shock protein (Hsp72) was used as an indicator of ischemia (Planas et al., 1997) (Fig. 1A). Hsp72 was hardly detected in nonoperated controls and in the contralateral hemisphere but was induced by ischemia (Fig. 1A). Ischemic rats showed MMP-9 in the ipsilateral cortex and striatum, as revealed by western blot (Fig. 1B). Zymographic analysis of these proteases revealed their activity on the gels with white bands where gelatin was degraded (Fig. 1C). The specific enzymes were identified by their molecular weight. Controls and the contralateral (C) hemisphere showed two faint bands of MMP-9, which were associated to molecular weights of 95 and 88 kDa and a major band corresponding to MMP-2 (Fig. 1C, lane 1). After ischemia, rats showed an increase of MMP-9 in the ischemic ipsilateral (I) cortex and striatum affecting both MMP-9 bands, particularly the 95-kDa band, whereas no main alteration in the MMP-2 was detected (Fig. 1C, lane 2).
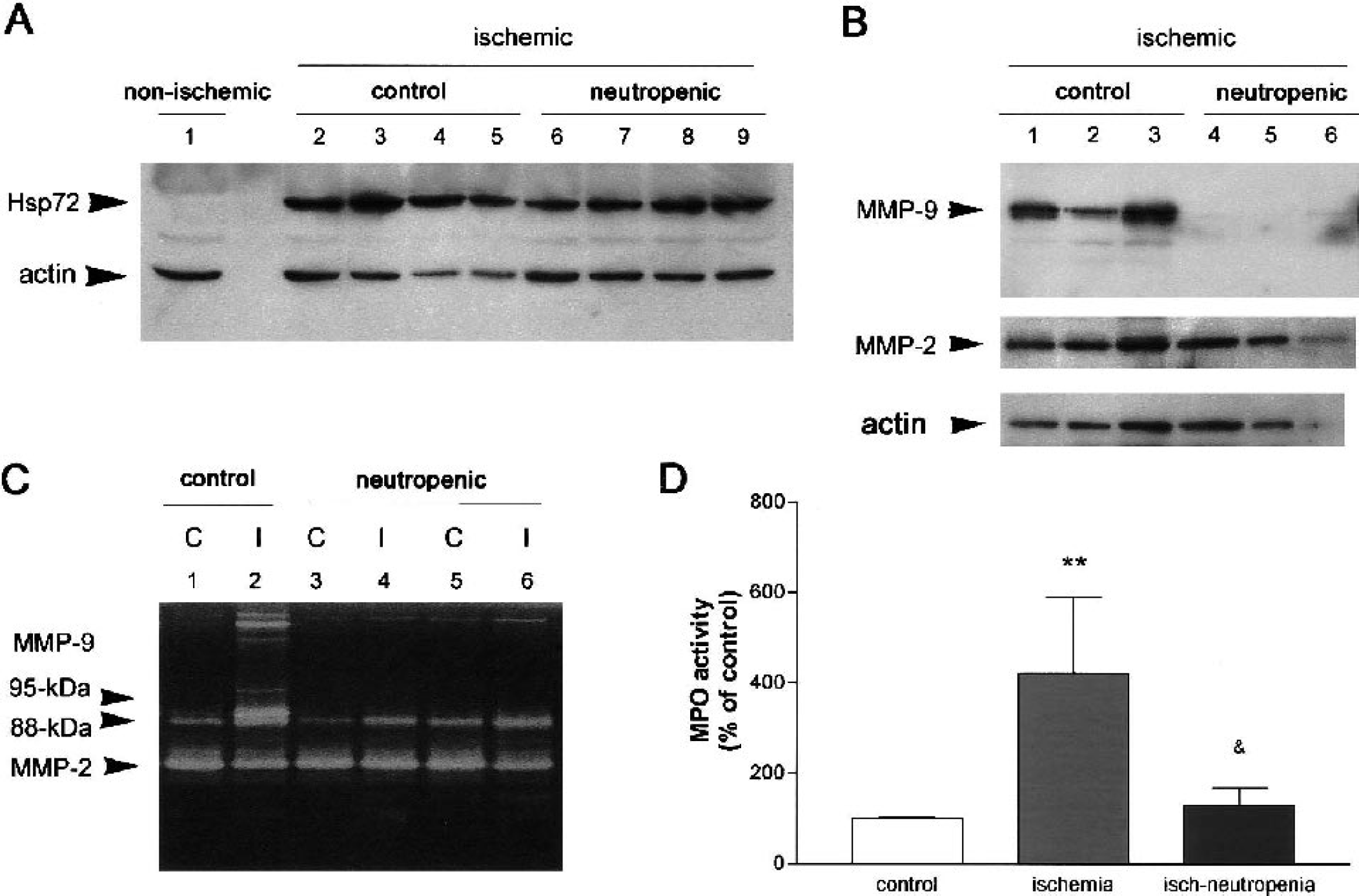
Effects of neutropenia.
The presence of neutrophils in blood was deeply reduced 4 days after vinblastine administration. Controls showed (mean ± SD neutrophils expressed as percent of total leukocyte counts) 14.28 ± 1.07% neutrophils, whereas vinblastine-treated showed 0.5 ± 0.36% neutrophils (P < 0.001) before ischemia. Body weight (mean ± SD) was 322.3 ± 18.9 g on day 1 and 329.3 ± 20.0 g on day 4 in saline-injected rats and 322.9 ± 14.1 on day 1 and 323.3 ± 13.6 on day 4 in vinblastine-treated rats. During surgery for MCA occlusion, mean ± SD rectal temperature was 36.88 ± 0.15°C in the saline and 36.95 ± 0.17°C in the vinblastine group. MABP was also monitored during surgery, and group means ± SD were as follows: 90.98 ± 3.65 mmHg and 93.11 ± 3.83 mmHg in the saline and vinblastine groups, respectively. No differences were found between groups for these physiologic variables. Neutrophil infiltration in the affected ischemic brain tissue was assessed by measurement of brain myeloperoxidase activity (MPO). Ischemia induced an increase (P < 0.01) in MPO activity in brain tissue as a result of neutrophil infiltration (Fig. 1D). Neutropenia significantly reduced (P < 0.05) the increase of MPO induced by ischemia (Fig. 1D). Likewise, the increase in the 95-kDa MMP-9 band that was induced by ischemia was completely prevented in neutropenic rats (Fig. 1C, lanes 4 and 6). Indeed, densitometric analysis of the gels showed a significant reduction in the intensity of the 95-kDa MMP-9 band (P < 0.01) (Fig. 2A). The effect of neutropenia was selective on the 95-kDa MMP-9 band, whereas ischemia-induced activation of the 88-kDa band was not abrogated by this treatment (Fig. 2A).
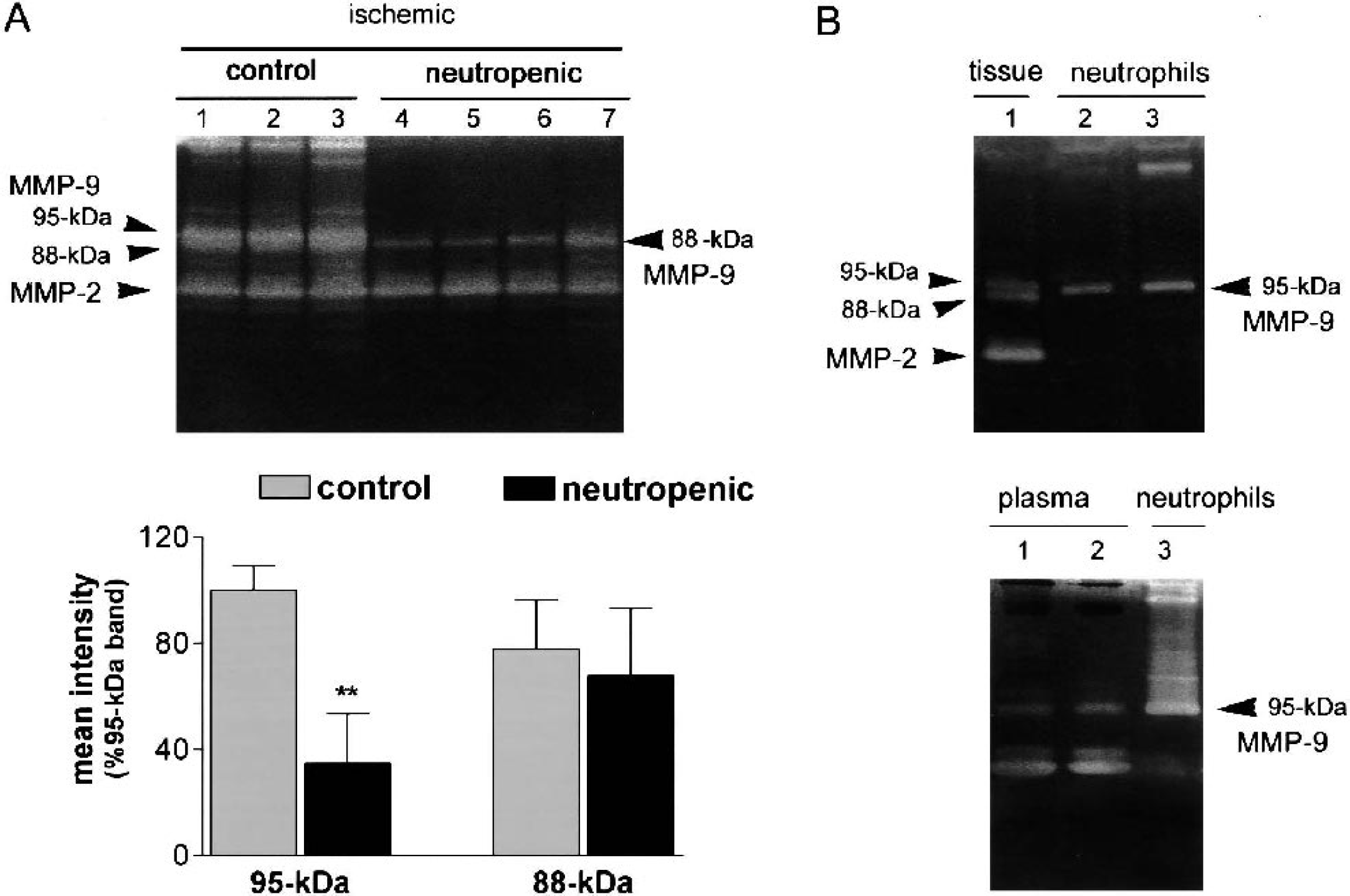
Neutropenia prevents the ischemia-induced raise of MMP-9 in the brain.
Neutrophils are rich in 95-kDa MMP-9 and dimers
Zymographic analysis of isolated blood granulocytes showed that these cells were rich in the 95-kDa MMP-9 proform and dimers (Figs. 2B and 3B). MMP-2 was less abundant than MMP-9 in neutrophils. In contrast, plasma was richer in MMP-2 than in MMP-9 (Fig. 2B). To verify that the 95-kDa band in the gel zymogram corresponded to gelatinase B (MMP-9), we incubated gels containing the same samples (MMP standard and neutrophils) in the presence or absence of EDTA to reveal gelatinase activity. This completely prevented band formation (not shown). In addition, we used two specific inhibitors for MMP-9: MMP-9/MMP-13 Inhibitor I and MMP-9/MMP-2 Inhibitor II. The band attributed to MMP-9 was no longer seen in the presence of inhibitors (Fig. 3A).
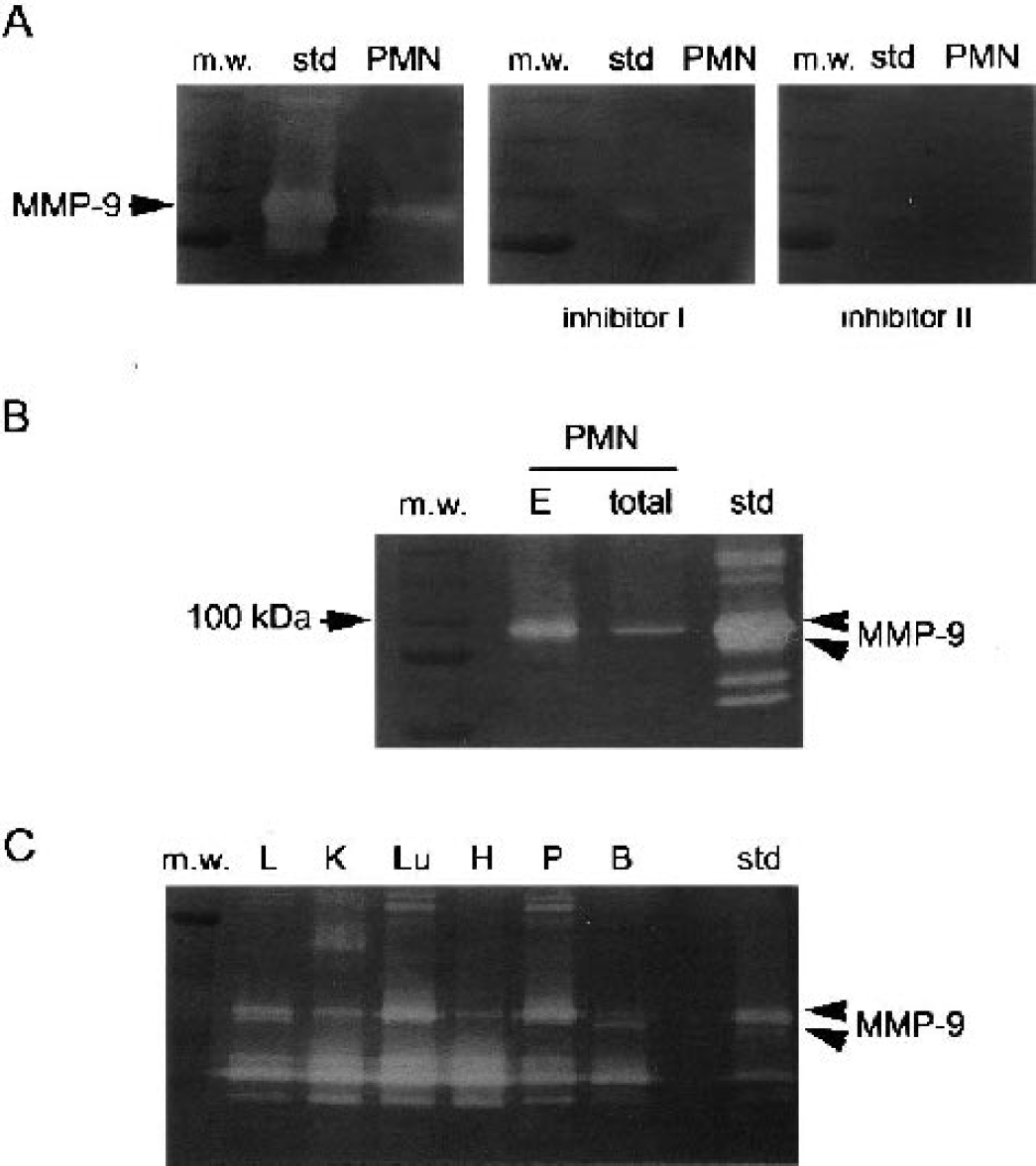
MMP-9 inhibitors prevent the formation of gelatinolytic bands. Neutrophils show a major 95-kDa MMP-9 band, whereas the 88-kDa band was associated to certain rat tissues.
Neutrophils showed the 95-kDa band, whereas the 88-kDa MMP-9 band found in brain tissue was not detected in granulocytes. We demonstrated that the 88-kDa band was not produced as an artifact during the extraction procedure by comparing MMP-9 bands in total neutrophil homogenates before extraction (total) with bands in the neutrophil gelatinolytic extracts (E) (Fig. 3B). Thus the 88-kDa band was associated to brain tissue. We then performed gelatinolytic extracts of various tissues in the control rat to find out whether the 88-kDa band was present. This band was found in the brain and liver, but was not so apparent in the kidney, lung, heart, or pancreas, showing a tissue-selective expression of the 88-kDa MMP-9.
Effect of treatment with anti-ICAM-1 mAb: reduction of MPO and MMP-9
Ischemia caused the induction of ICAM-1 expression in the endothelial vessel wall at 24 hours, as revealed by immunohistochemical studies with the 1A29 antibody (Fig. 4). This antibody was then administered systemically in vivo to block ischemia-induced ICAM-1 to prevent neutrophil adhesion to the endothelium and subsequent infiltration into brain parenchyma. Mean ± SD rectal temperature was 37.40 ± 0.12°C and 37.27 ± 0.11°C in the groups treated with the control and the anti-ICAM-1 mAbs, respectively. MABP was 100.80 ± 7.43 mmHg and 93.51 ± 4.16 mmHg for controls and anti-ICAM-1 groups. No differences were found between groups.
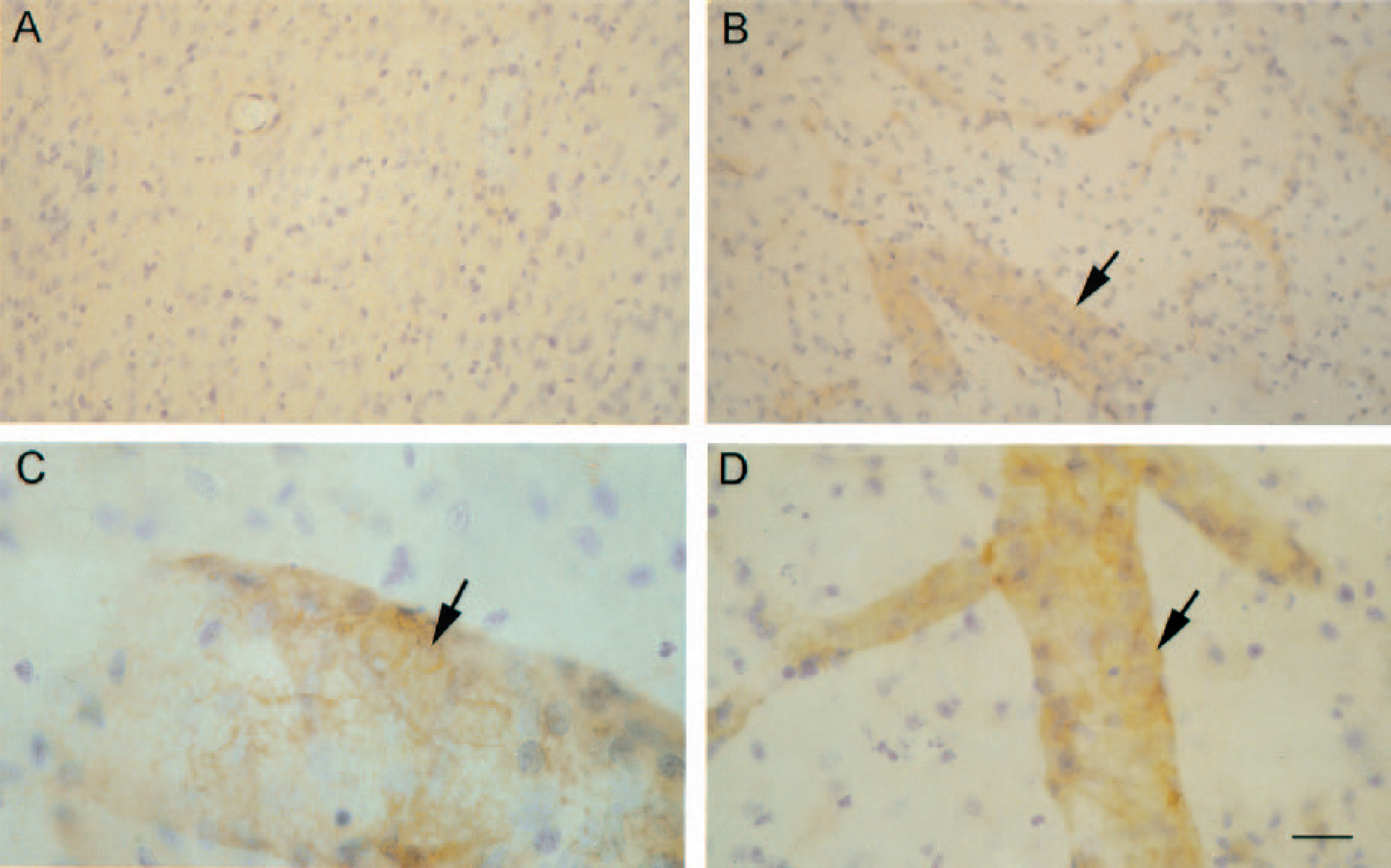
Induction of ICAM-1 after ischemia. Immunohistochemistry with 1A29 monoclonal antibody against ICAM-1 showing induction of ICAM-1 (brown) in the endothelial vessel wall at 24 hours after ischemia
Hsp72 expression was examined as a marker of ischemia-induced cellular stress. All ischemic rats included in this study showed Hsp72, regardless of the treatment they received (Fig. 5A).
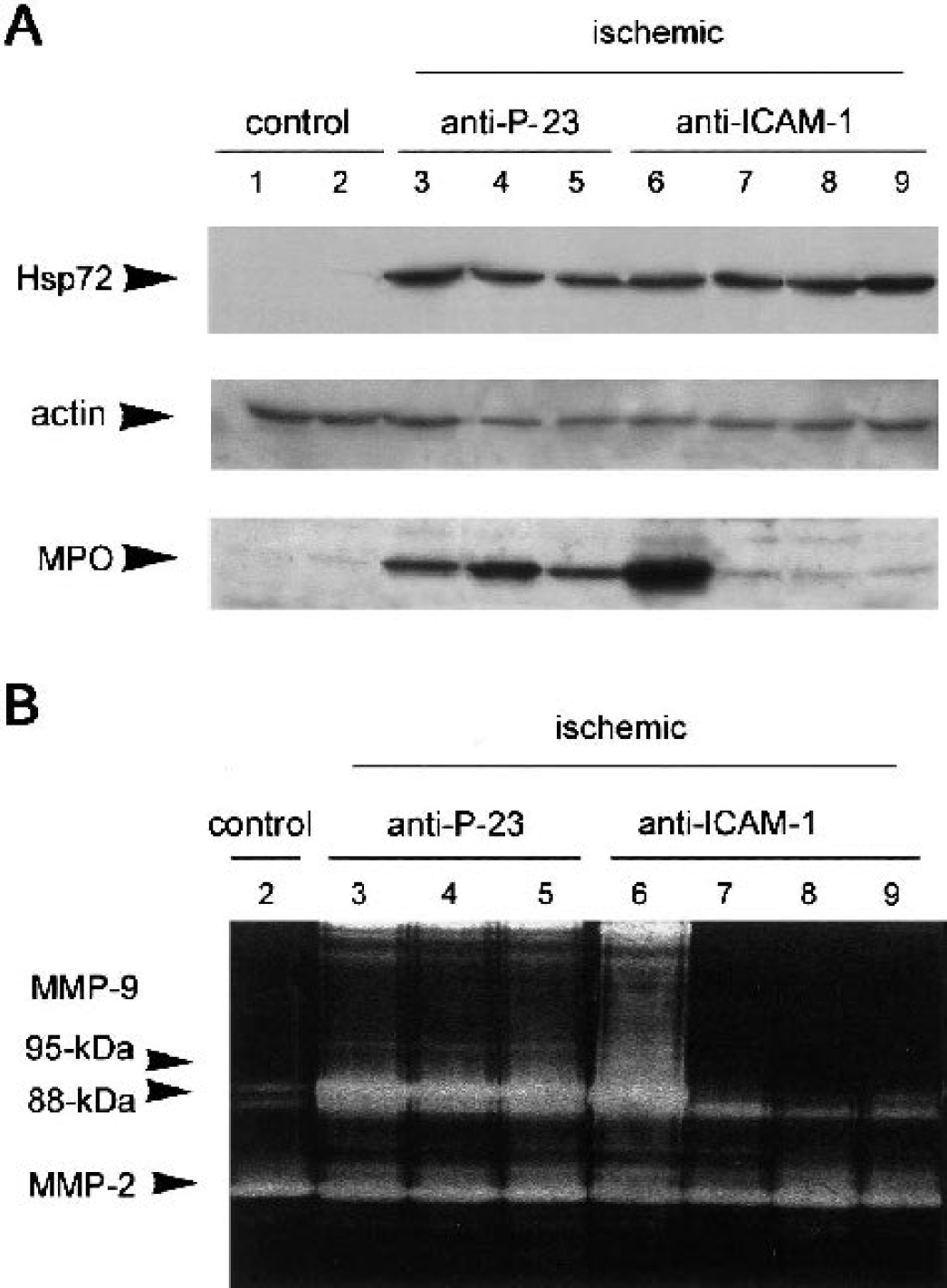
Effects of anti-ICAM-1 mAb.
We checked the effect of anti-ICAM-1 mAb treatment on neutrophil infiltration by determining the expression of MPO by western blot in the brain samples of the same animals as used for MMP-9 study. MPO was undetectable in the control brain (Fig. 5A, lanes 1 and 2) because rats were perfused with saline before brain extraction to wash out any remaining blood in the tissue. Ischemia/reperfusion increased MPO expression at 24 hours in the brains of nontreated rats and in rats receiving the control mAb (P-23), as expected because of neutrophil infiltration (Fig. 5A, lanes 3 to 5). Anti-ICAM-1 mAb preventedMPO increase in the ischemic tissue in six out of nine rats (Fig. 5A, lanes 7 to 9). The remaining three rats showed MPO expression indicative of neutrophil infiltration (Fig. 5A, lane 6).
MMP-9 activity (95-kDa and 88-kDa forms and dimeric forms of high molecular weight, around 210 kDa) increased 24 hours after MCA occlusion in the ipsilateral brain tissue (Fig. 5B, lanes 3 to 5) in relation to controls (Fig. 5B, lane 2), as revealed by zymography. Anti-ICAM-1 mAb, in rats in which neutrophil infiltration was prevented, strongly reduced the ischemia-induced rise in 95-kDa MMP-9 band and dimers (Fig. 5B, lanes 7 to 9). However, the three rats also treated with anti-ICAM-1 mAb but showing high MPO expression showed an increase in MMP-9 (Fig. 5B, lane 6) that was thus correlated with the presence of neutrophils. Measuring raw band intensity showed significant (P < 0.001) reduction of the 95-kDa MMP-9 form after blocking ICAM-1 (Fig. 6A). However, comparatively, the intensity of the 88-kD band was not significantly affected by this treatment (Fig. 6A).
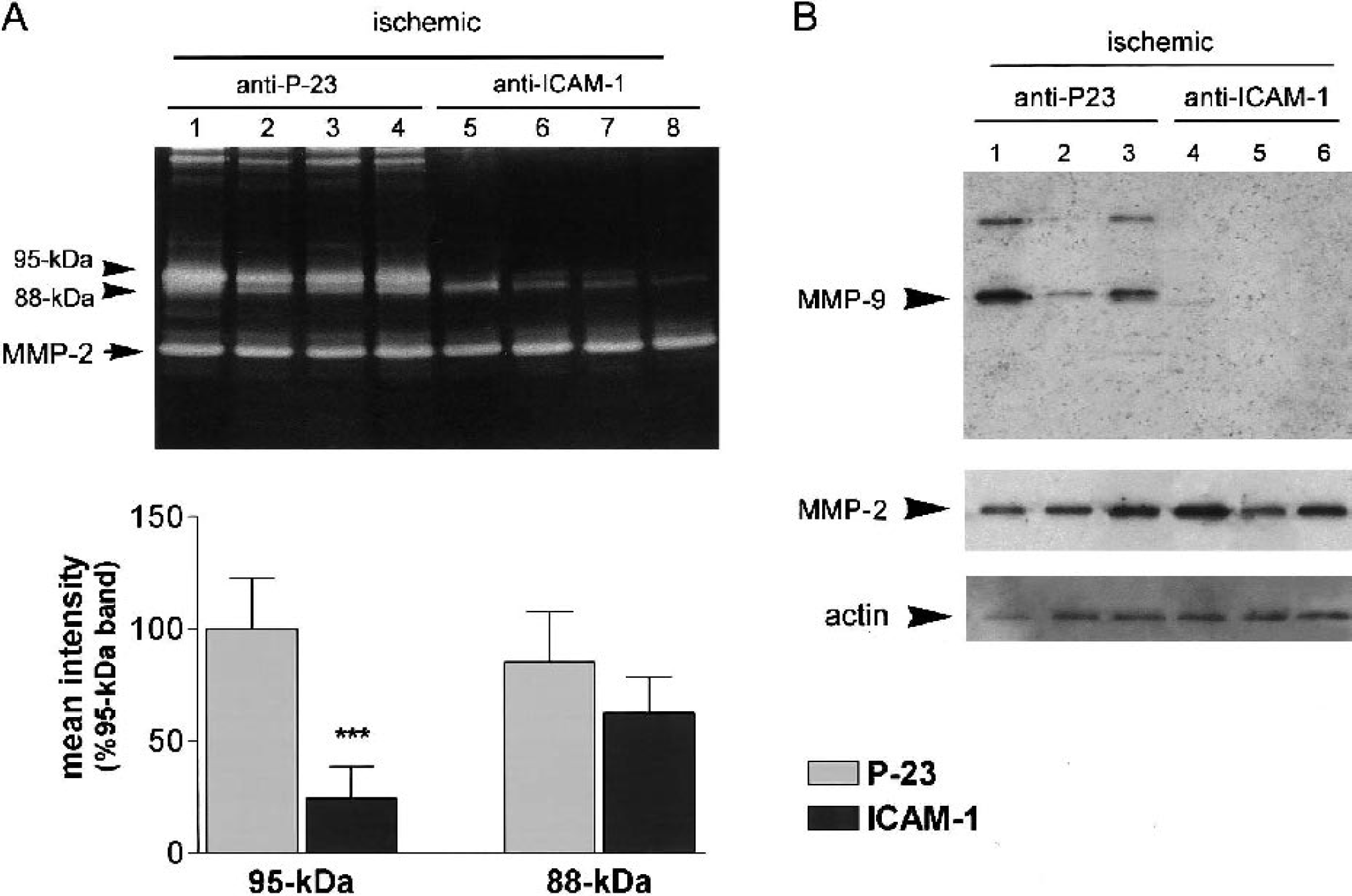
Anti-ICAM-1 mAb prevents the ischemia-induced raise of MMP-9 in brain.
The effect of ICAM-1 blockade on MMP-9 was further examined in the same animals by protein precipitation of samples and western blot analysis with a mAb against MMP-9. The expression of MMP-9 was highly increased 24 hours after MCA occlusion, but MMP-9 protein expression was dramatically reduced in rats treated with anti-ICAM-1 mAb compared with rats administered with the control mAb (Fig. 6B, lanes 4 to 6). However, treatment did not cause changes in MMP-2, as revealed by zymography and western blot (Figs. 5B, 6A, and 6B).
Cellular MMP-9 distribution
Tissue distribution of MMP-9 was examined by immunohistochemistry using two different monoclonal antibodies, which gave similar results. Basal expression of MMP-9 was detected in neuronal fibers (Fig. 7A). At 24 hours after MCA occlusion/reperfusion, the most striking feature was that the filamentous appearance of brown stain seen in controls turned to a more granular appearance (Figs. 7B and 7C), and occasionally, staining appeared in discrete enlargements in fibers. In addition, brown stain was detected in cells showing a morphology that was compatible with neutrophils, including polymorphous nucleus (arrowheads in Figs. 7B and 7C; inset in Fig. 7C) and in endothelial walls of small vessels. These later features were more abundantly seen at the edges of the ischemic core (particularly in the endopiriform cortex and surrounding zones). Double labeling with antibodies against MMP-9 (brown) and MPO (dark blue) revealed MMP-9 positive neutrophils in ischemic rats receiving control mAb (arrowheads in Figs. 7E and 7F; inset Fig. 7E). Ischemic rats treated with anti-ICAM-1 mAb (Fig. 7G) still showed the MMP-9 staining in fibers (Fig. 7D) and endothelium (Fig. 7G, arrow on the right), but they did not show the cellular MMP-9 stain that was attributed to neutrophils.
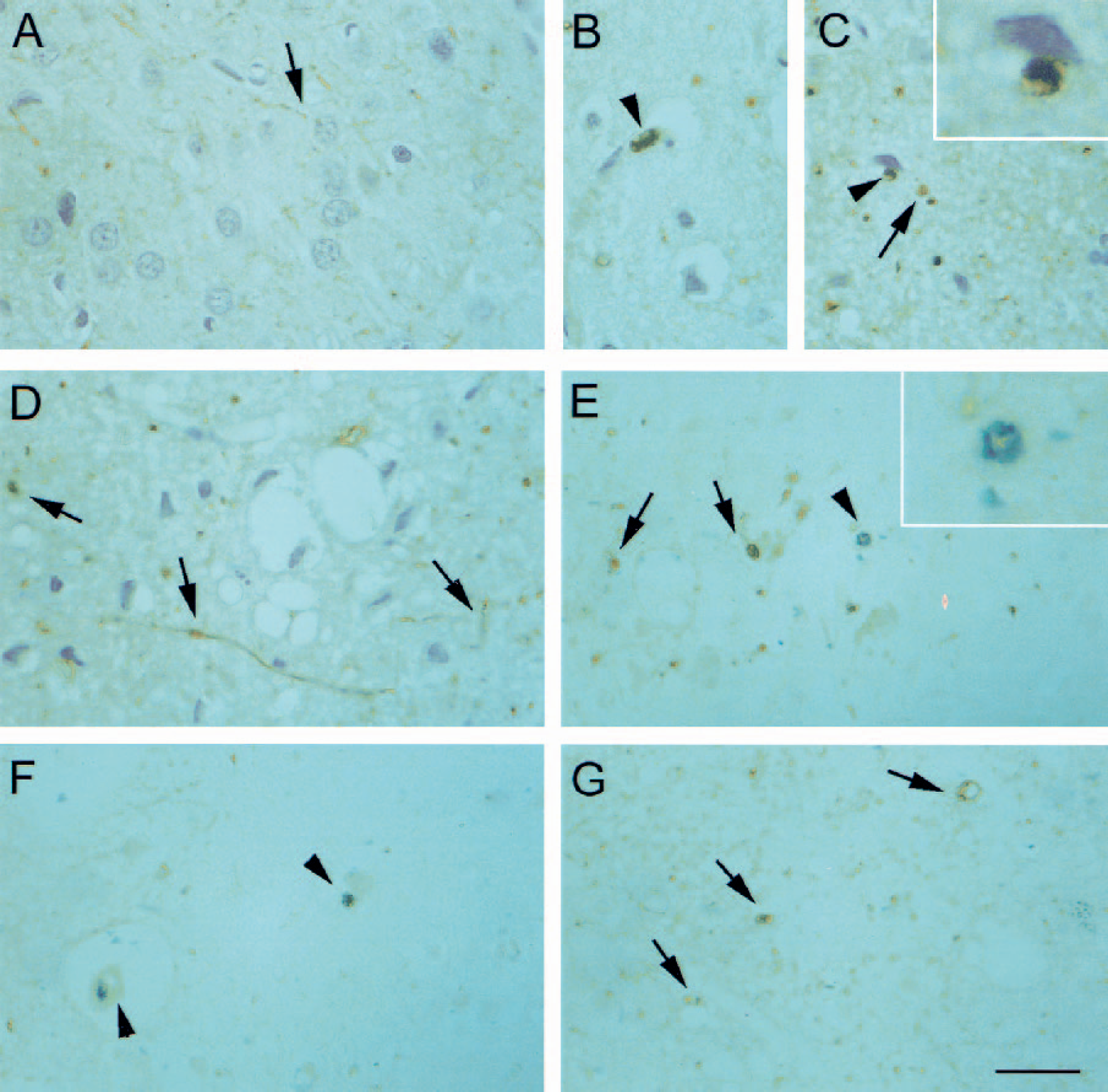
Cellular localization of MMP-9. Immunohistochemistry for MMP-9 in the cortex of control
Gelatinase activity increases in neural cells
The degradation of gelatin was increased in ischemic neurons located within the ischemic core in cortex and striatum at 24 hours after MCA occlusion/reperfusion (Fig. 8). The raise in gelatinase activity was restricted to the MCA territory in the ipsilateral hemisphere but did not affect the contralateral hemisphere (Figs. 8A and 8B). Gelatinase activity was found mainly in neural cells and some blood vessels (Fig. 8C), and many cells were identified as neurons with the neuronal NeuN marker (Fig. 8D). This effect was seen even after neutropenia (Figs. 8E and 8F), suggesting that the neuronal raise in gelatinase activity was not dependent upon neutrophil infiltration.
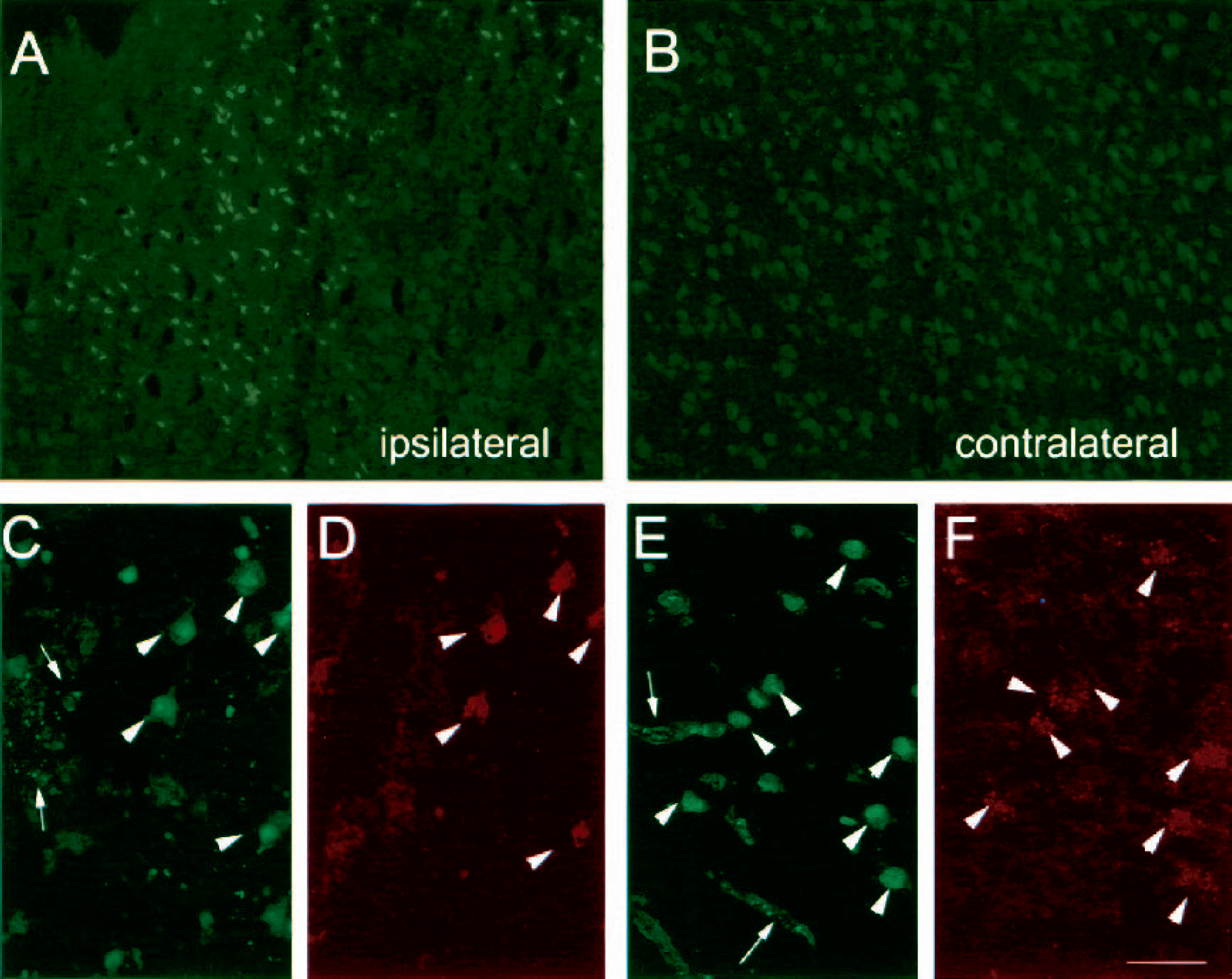
Cellular gelatinase activity increases in neurons and blood vessels after ischemia, regardless of neutrophil infiltration. Gelatinase activity
DISCUSSION
Ischemia/reperfusion induces a large increase in cerebral MMP-9 activity, which might participate in the pathogenesis of ischemic damage (Asahi et al., 2000). This study was addressed to find out whether neutrophil infiltration contributed to the enhanced MMP-9 activity in ischemia. We addressed this query because our previous findings showed release of MMP-9 proform in the extracellular space of the ischemic brain that was concomitant with neutrophil recruitment (Planas et al., 2002). To answer this question, we used the strategies of either preventing neutrophil adhesion to the endothelial vessel wall by systemic treatment with a mAb against ICAM-1 or depleting circulating neutrophils. The mAb against ICAM-1 that we used in the present study was administered to rodents in previous studies of focal cerebral ischemia (Chopp et al., 1996; Furuya et al., 2001; Zhang et al., 1994, 1995). Overall, treatments were effective; they abrogated the MPO rise in the ischemic brain at 24 hours. Rats in which treatment successfully prevented neutrophil infiltration showed a strongly reduced rise in MMP-9. Neutrophils are known to contain and to degranulate gelatinase granules, together with other types of granules, such as those stained with lactoferrin and azurophil granules identified by MPO (Borregaard et al., 1997). Those gelatinase granules have been described to contain and release MMP-9 (Pugin et al., 1999). Furthermore, we showed that the proforms of MMP-9 were abundant in neutrophils, and, in agreement with other authors (Romanic et al., 1998; Rosenberg et al., 2001), we detected MMP-9 staining in neutrophils within the ischemic brain tissue by immunohistochemistry. These findings led us to conclude that neutrophil infiltration into the ischemic parenchyma was necessary for the enhanced MMP-9 expression and activity (95-kDa proform and dimers) that occurs in the brain after ischemia. Besides neutrophils, MMP-9 was also found in plasma, but compared with neutrophils, the former was richer in MMP-2. Despite the fact that neutrophils might produce MMP-2 (Warner et al., 2000), the amounts are negligible compared with the MMP-2 content in the brain and plasma. This thus indicates that plasma was not the major source of the increase in MMP-9 induced by ischemia at 24 hours because an even greater increase in plasma-borne MMP-2 would be expected, whereas no MMP-2 alterations became apparent at this time point.
Ischemia-induced 88-kDa MMP-9-increase was still found after neutropenia and ICAM-1 blockade, thus suggesting that the increase in this form of MMP-9 was not derived from neutrophils but was intrinsic to the ischemic tissue. Also, we found constitutive MMP-9 protein expression in neuronal fibers and a change in MMP-9 distribution along fibers after ischemia, probably reflecting dynamic alterations in axons, together with increased MMP-9 staining in blood vessels, which is in agreement with previous reports (Mun-Bryce et al., 2002). Preventing neutrophil infiltration did not attenuate this pattern of MMP-9 expression after ischemia. In addition, we detected an increase in neuronal gelatinase activity at 24 hours after ischemia, which is in agreement with a previous report (Gu et al., 2002). Whether this neuronal gelatinase corresponds to MMP-9 remains to be demonstrated, but the neuronal increase in gelatinase activity was still found after abrogating neutrophil infiltration, suggesting an intrinsic activation of gelatinase in brain after ischemia. However, it is likely that the activation of MMP-9 occurs locally in the parenchyma of the injured tissue by a finely regulated process. For this reason, we cannot discard the possibility that, once in the tissue, neutrophils contribute to further enhancing the intrinsic cerebral MMP-9 response, either with protease activity or by releasing cytokines that might further induce MMP-9 gene expression, as occurs in myocardial ischemia/reperfusion (Lindsey et al., 2001).
In summary, ischemia/reperfusion induced a strong increase in several MMP-9 forms in the ischemic tissue. Among the increases in MMP-9 forms, we report that the increase in MMP-9 proform was essentially derived from infiltrating neutrophils, whereas the results suggest that other forms of gelatinase are increased in neural cells of the ischemic brain regardless of neutrophil infiltration. From the present results, we conclude that neutrophil infiltration contributes to the MMP-9 increase in the ischemic brain by releasing MMP-9 proform.
Footnotes
Acknowledgment:
We thank Mr. Josep Moreno for excellent technical assistance.