
Abstract
Calcium/calmodulin-dependent protein kinase II (CaM-kinase) is a central enzyme in regulating neuronal processes. Imbalances in the activity and distribution of this enzyme have been reported following in vivo ischemia, and sustained decreases in activity correlate with subsequent neuronal death. In this report, mice that had been rendered deficient in the alpha subunit of CaM-kinase using gene knock-out technology were utilized to determine whether this enzyme is causally related to ischemic damage. Using a focal model of cerebral ischemia, we showed that homozygous knock-out mice lacking the alpha subunit exhibited an infarct volume almost twice that of wild-type litter mates. Heterozygous mice exhibited slightly less damage following ischemia than did homozygous mice, but infarct volumes remained significantly larger than those of wild-type litter mates. We conclude that reduced amounts of the alpha subunit of CaM-kinase predisposes neurons to increased damage following ischemia and that any perturbation that decreases the amount or activity of the enzyme will produce enhanced susceptibility to neuronal damage.
Increased intracellular calcium is a common signaling mechanism essential for regulating many neuronal processes. Under normal physiological conditions, the concentration of intracellular calcium is stringently controlled. However, during energy compromised states, such as follows cessation of blood flow to the brain (ischemia), the concentration of intracellular calcium rises and remains elevated, sometimes initiating a chain of events that leads to irreversible neuronal damage (Siesjo and Bengtsson, 1989; Tymianski et al., 1993). How sustained increases in calcium leads to neuronal toxicity is a matter of much speculation. Several current hypotheses suggest that an increased concentration of intracellular calcium leads to excessive activation of calcium-dependent enzyme systems (Choi, 1990; Siesjo and Bengtsson, 1989). Enzyme hyper-activation can then produce damage either directly by intracellular breakdown (e.g., calcium-dependent proteases, lipases, and nucleases) or indirectly by producing an imbalance in neuronal homeostasis (e.g., through alterations in calcium-dependent protein kinases). Protein phosphorylation plays a major role in adapting a neuron's enzymatic machinery in response to environmental changes (Hanson and Schulman, 1992); thus, irreversible losses in protein kinase activity would be expected to significantly compromise a neuron's capacity to survive, particularly under extreme conditions such as cerebral ischemia.
We and others described that an irreversible loss of calcium/calmodulin-dependent protein kinase II (CaM-kinase) activity was correlated with neuronal damage following in vivo ischemia (Taft et al., 1988; Churn et al., 1990; Aronowski et al., 1992; Hanson et al., 1994; Shackelford et al., 1995). Based on these results, we postulated that decreased CaM-kinase activity leads to neuronal damage. Experimental attempts to address this question using administration of broad spectrum protein kinase inhibitors (e.g., staurosporine and H-7) have met with controversial results. Both increases (Madden et al., 1990) and decreases (Hara et al., 1990) have been reported for neuronal damage following in vivo ischemia after administration of these inhibitors. The reasons for these discrepancies are probably manifold; however, the lack of the inhibitors' efficacy or specificity may be a major factor. Such discrepancies can now be resolved using gene knock-out technology, an approach used recently to show that mice deficient in neuronal nitric oxide synthase exhibited increased resistance to damage following focal cerebral ischemia (Huang et al., 1994). Recently, mice lacking the alpha subunit of CaM-kinase (the alpha subunit is a neuron-specific isoform of this enzyme) were constructed using knock-out technology (Silva et al., 1992a; 1992b), thus providing a unique opportunity to investigate CaM-kinase's role in mediating neuronal damage following ischemia. Results presented in this report provide the first direct indication that targeted disruption of a single protein kinase, CaM-kinase, leads to increased neuronal damage following ischemia.
METHODOLOGY
Mice
Offspring bred from heterozygous CaM-kinase knock-outs were used for all of these studies. The original mutant mice in the 129 cvj background were backcrossed in the C57B1/6J strain to minimize complications of heterogeneous background. Every animal was genotyped using polymerase chain reaction (PCR) of a DNA sample from the tail. The (+/+) mice exhibited a fragment of 290 bp, the (−/−) mice exhibited a product of 320 bp, and the (+/−) mice exhibited a product at both lengths (data not shown).
Focal cerebral ischemia
Focal ischemia was induced in mice by a modification of a protocol established in rats (Aronowski et al., 1994). Between 5 and 7 weeks of age, animals were anesthetized with an intraperitoneal injection of chloral hydrate (0.35 g/kg), the skull exposed, and a small burr hole made over the middle cerebral artery. Body temperature was monitored using a rectal thermometer and maintained at 36.5 ± 0.35°C through use of a warming blanket for the entire period of occlusion and the first hour of reperfusion. Occlusion of the left middle cerebral artery was produced by sliding a 0.005 in diameter stainless steel wire under the exposed middle cerebral artery just distal to its origin from the internal carotid artery. When slightly elevated, the wire visibly occluded the lumen of the artery. The ipsilateral common carotid artery was then occluded by clamping with an atraumatic aneurysm clip. Cerebral perfusion at the surface of the cortex was measured 2 mm distal from the site of middle cerebral artery occlusion using a laser Doppler flowmeter (Model BPM2, Vesamedic, St. Paul, MN, U.S.A.). After 150 min of occlusion, reperfusion was established by reversing the procedure.
Infarct volume measurements
At ∼21 h following re-establishment of blood flow, mice were anesthetized with chloral hydrate and perfused with saline through the left ventricle. The brain was removed, sectioned into 1 mm coronal sections, and stained with 2% 2,3,5-triphenyltetrazolium chloride for 30 min at room temperature. Sections were then fixed in 10% buffered formalin. Total infarct volume was measured by capturing still video images of each section and outlining the unstained (infarcted) surface of each slice. Total volume was reconstructed by summing the surface areas of infarcted tissue from each 1 mm section. This process is almost identical to that described for analyzing infarct volumes in the rat model of focal ischemia (Aronowski et al., 1994). Both the investigator accomplishing the surgeries and the investigator measuring infarct volumes were blinded as to the genotype of the animals. Statistical significance was determined by analysis of variance (ANOVA) and the Newman-Keuls test; variability in the values reported in the text are as standard deviations (SDs).
SDS-PAGE and Western blot analysis of brain extracts
After 150 min of focal ischemia, mice were anesthetized with ether and decapitated. The brain was removed rapidly (within 15 s) and immersed in ice cold phosphate-buffered saline. The two hemispheres were separated, the cortexes isolated, and each cortex was extracted into 1 ml of homogenization buffer [50 mM HEPES (pH 7.4), 0.1% Triton X-100, 4 mM EGTA, 10 mM EDTA, 15 mM Na4P2O7, 100 mM β-glycerophosphate, 25 mM NaF, 0.1 mM leupeptin, 75 μM pepstatin, and 0.1 mg/ml aprotinin] with 15 strokes of a glass/Teflon homogenizer. Equal amounts of homogenate protein were solubilized in SDS-sample buffer and separated on a 10% SDS-polyacrylamide gel. After electrophoresis, the gel was transferred to nitrocellulose as described (Kolb et al., 1995) and stained with either monoclonal antibody 2D5 (specific to the alpha subunit of CaM-kinase), CB-β1 (specific to the beta subunit of CaM-kinase and supplied by Dr. Howard Schulman), or C4 (a monoclonal antibody specific to all isoforms of actin and supplied by Dr. Jim Lessard). Immunostained bands were visualized with alkaline-phosphatase conjugated goat anti-mouse (Promega, Madison, WI, U.S.A.), followed by 5-bromo-1-chloro-indolyl phosphate and nitro-blue tetrazolium (Promega).
RESULTS
To select a duration of ischemia for this comparative study, a dose–response curve was constructed of duration of ischemia versus infarct volume using wild-type C57B1/6J mice (the parent strain to the knock-outs) and employing an adaptation of the reversible left middle cerebral artery and ipsilateral common carotid artery (MCA/CCA) occlusion model (data not shown). A 150 min occlusion with 21 h of reperfusion in 5–6 week old wild-type mice provided a just submaximal infarct volume [21.9 ± 6.4 mm3 (n = 6)] and was selected for the comparative analysis. The lesion involves primarily cortical layers; however, some minor involvement of subcortical areas was also evident. All measurements reported in this study included only cortical infarct volumes.
Heterozygous CaM-kinase knock-out parents were mated and the resulting off-spring genotyped using PCR amplification of DNA isolated from the tail. At 5–7 weeks of age, animals were randomly grouped and subjected to 150 min of occlusion followed by 21 h of reperfusion. Littermate control mice (+/+) exhibited approximately the same volume of infarct [18.8 ± 5.1 mm3 (n = 6)] as the wild-type C57B1/6 mice. In contrast, significantly larger infarct volumes were found in the (+/−) mice (n = 5) (32.0 ± 8.1 mm3 p < 0.05) (Fig. 1A). Even larger infarct volumes were found in the mutant (−/−) mice (n = 5) (38.28 ± 11.0 mm3 p < 0.01) (Fig. 1A). Plotting the volume differences of (+/+), (+/−) and (−/−) mice over the rostral to caudal extent of the brain indicated that the volume differences were not significant until the fourth mm from the rostral pole (Fig. 1B). This is consistent with the extent of tissue supplied by the middle cerebral artery covering the lateral aspects of the cortex.
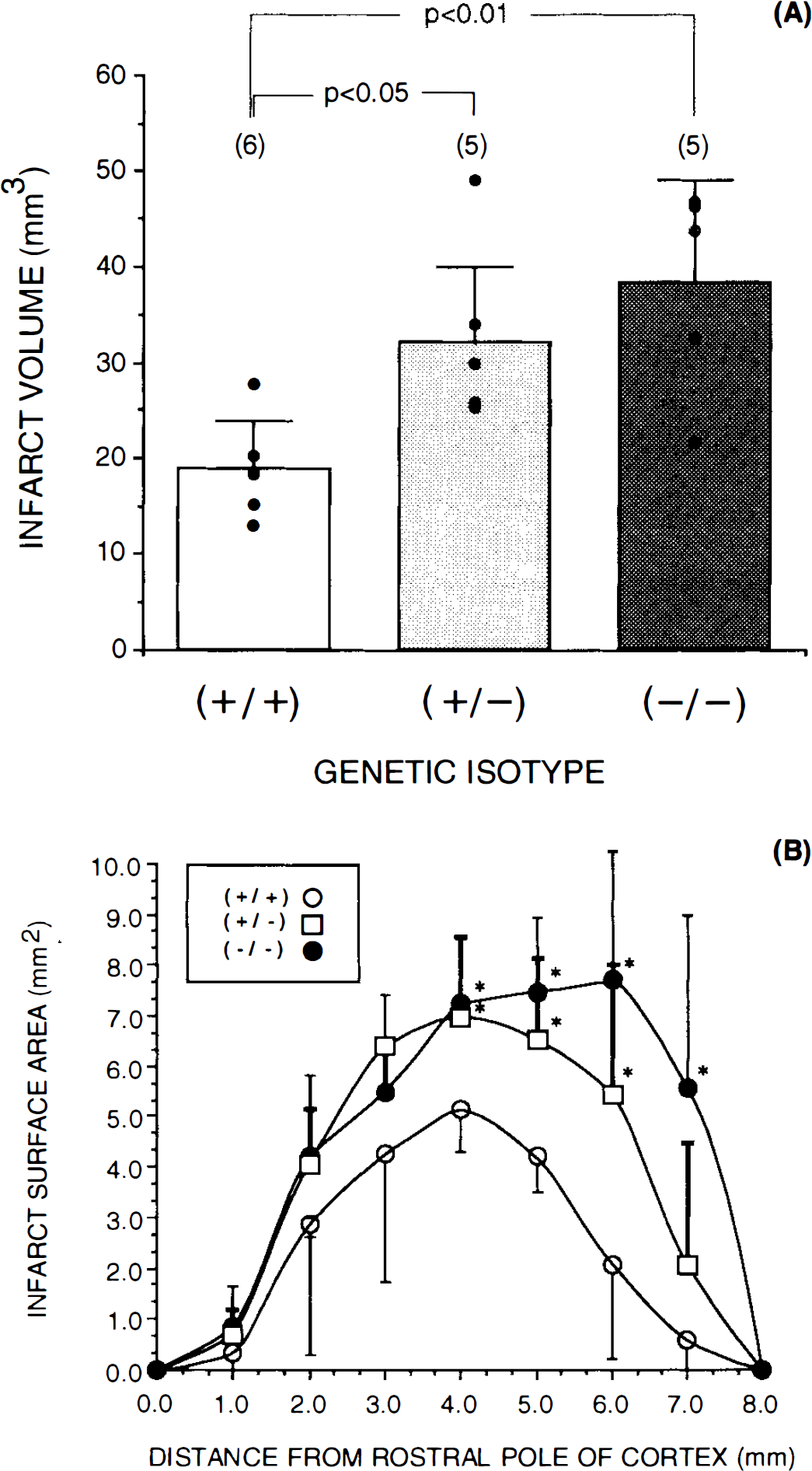
Preocclusion cerebral blood flow was not different between any of the groups and occlusion produced 93–95% reductions in every animal. Carbon black fills of the cerebrovasculature also indicated there was no macroscopic differences between the groups (data not shown). There was also no evidence of overt seizure activity before, during, or after induction of ischemia in any of these 5–7-week-old mice tested in our study.
Cortical tissue from the ipsilateral and contralateral cortex of a (+/+), (+/−), and (−/−) animal following 150 min of ischemia was analyzed by Western blot and is shown in Fig. 2. The cortex of each animal was separated into ipsilateral, I, (the side of the occlusion) and contralateral, C, (the side opposite occlusion) and processed as described in Methods. Constant amounts of homogenate protein were analyzed by SDS-PAGE and immunostaining using monoclonal antibodies specific to the alpha and beta subunits of CaM-kinase and actin. There was no detectable alpha subunit of CaM-kinase in (−/−) animals, while the (+/−) animals exhibited about half of the amount present in wild-type animals (+/+). These same extracts were also probed with a monoclonal antibody specific for the beta subunit of CaM-kinase The results show qualitatively (this colorimetric technique for visualizing immunostained proteins does not lend itself to accurate quantitation) that there was neither a large increase nor decrease in the amount of beta subunit in the (+/−) or (−/−) animals relative to the (+/+) animal. Finally, a monoclonal antibody specific to actin was used as an internal control, and no observable differences were found in actin levels among (+/+), (+/−), or (−/−) mice. Additionally, there was no detectable difference in the overall pattern of proteins on the Western blot, as revealed by Ponceau S staining (data not shown). Immunostaining for the alpha and beta subunits of CaM-kinase and actin was not appreciably different between the ipsilateral and contralateral sides of each animal's cortex after 150 min of ischemia (Fig. 2). These results are consistent with those from studies of the rat model of ischemia (Aronowski et al., 1992; Hanson et al., 1994) that indicate there is little loss in the amount of CaM-kinase enzyme due to proteolysis, at least up to 2 h of ischemia, in these mice. Our data are also consistent with previous reports (Silva et al., 1992b) indicating that (−/−) and (+/−) animals exhibit ∼50 and 71% of CaM-kinase activity relative to (+/+) mice, respectively.
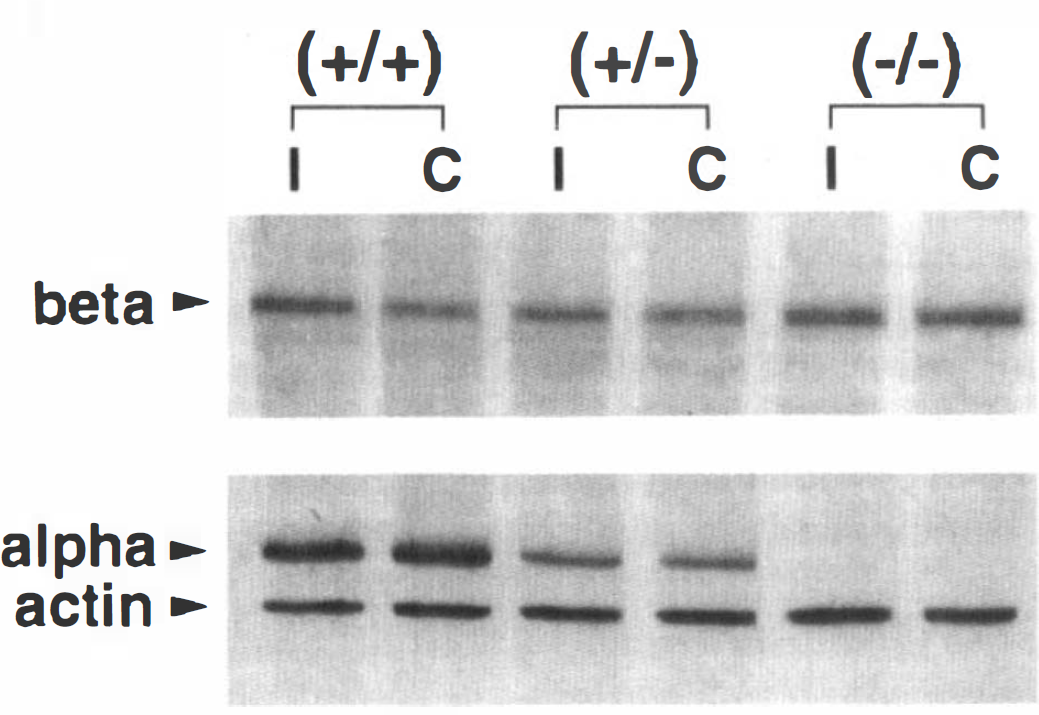
Western blot of cortical homogenates from knock-out mice and wild-type mice. Homogenates from wild-type (+/+), heterozygous (+/−), and homozygous (−/−) mice were prepared as described in Methods. These animals all received 150 min of unilateral focal ischemia, and the brains were harvested without reperfusion. I and C represent homogenates prepared from the ipsilateral and contralateral cortexes to the ischemic site, respectively. Beta subunit staining was accomplished with monoclonal antibody CB-β1, the alpha subunit with monoclonal antibody 2D5, and actin with monoclonal antibody C4. Beta staining was accomplished with 10 μg of total protein per lane and the alpha and actin immunostaining with 2.5 μg of total protein per lane.
DISCUSSION
Previous data provided strong correlative evidence that CaM-kinase might play a role in neuronal damage following ischemia (Taft et al., 1988; Churn et al., 1990; Aronowski et al., 1992; Hanson et al., 1994; Shackelford et al., 1995). A loss of CaM-kinase activity was detected immediately after ischemia in all brain regions studied; however, only in regions where the loss of activity was permanent was damage eventually manifest (Aronowski et al., 1992; Hanson et al., 1994). Congruent with these in vivo studies are two recent reports that showed that activation of the N-methyl-D-aspartate (NMDA) subtype of glutamate receptor in neuronal culture models also produced decreased CaM-kinase activity (Churn et al., 1995; Morioka et al., 1995) that further correlated with neuronal death (Churn et al., 1995). In the present study, mice engineered to be deficient in the alpha subunit of CaM-kinase were utilized to directly test the hypothesis that decreased CaM-kinase content and/or activity leads to increased neuronal damage following ischemia. Mutant mice exhibited no detectable alpha subunit mRNA or protein (Silva et al., 1992b); however, there remains significant CaM-kinase activity in brain homogenates of the (−/−) mice due to contribution from other CaM-kinase isoforms. This decreased activity is consistent with the immunostaining of the alpha subunit presented in Fig. 2. Apparently, the CaM-kinase activity remaining in the (+/−) and (−/−) animals is either quantitatively or qualitatively different than that in (+/+) animals and is insufficient to provide protection from ischemic damage. However, we cannot eliminate the possibility that other compensatory mechanisms (for example, up-regulation of another isoform of CaM-kinase) might influence the outcome to ischemia in these animals.
Light (Silva et al., 1992b) and electron microscopy (Comery et al., 1994) failed to find measurable differences in the cell number, morphology or synaptic organization in the hippocampus of (−/−) or (+/−) CaM-kinase knock-out mice relative to (+/+) leaving little evidence to support the hypothesis that structural differences in the neurons cause the increased susceptibility to ischemic damage. While we do not yet have quantitative data describing the light level morphology of the cortices of these animals, no gross morphologic differences were detected in the brains of any of the animals included in this study and we have no a priori reason to believe that synaptic connections will be different in the cortex than they are in the hippocampus. Finally, no apparent differences were detected in electro-physiologically measured synaptic responses of hippocampal slices prepared from (+/+)or(−/−) animals, suggesting basal excitatory and inhibitory synaptic transmission is intact in these animals (Silva et al., 1992b; Chapman et al., 1995). In particular, non-NMDA and NMDA mediated synaptic responses were indistinguishable from controls (Silva et al., 1992b). Therefore, the increased damage we observe is not likely due to a difference in basal glutamate receptor function. Recently, Chapman et al. (1995) showed that hippocampal slices from CaM-kinase knock-out mice exhibit greater posttetanic potentiation relative to their litter mates, suggesting that normal amounts of CaM-kinase alpha subunit restricts the amount of glutamate released after a tetanizing stimulus. It is possible that the increased damage detected in (+/−) and (−/−) animals following an ischemic insult is due to a similar excessive release of glutamate. This hypothesis is currently under investigation.
Earlier contentions suggested that plasticity and pathology might have underlying mechanistic similarities (Lynch and Seibert, 1989; Westage et al., 1994). Interestingly, CaM-kinase knock-out animals also exhibit defects in neuronal plasticity, specifically long-term potentiation (Silva et al., 1992b), and the induction of long-term potentiation was shown earlier to be prevented by CaM-kinase inhibitors (Malenka et al., 1989; Malinow et al., 1989). These animals also show deficits in certain spatial memory tasks (Silva et al., 1992a), have abnormal fear responses and exhibit aggressive behavior (Chen et al., 1994). Most recently, Butler et al. (1995) showed that these mice exhibited spontaneous seizure activity in late adulthood (>6 months of age). Apparently, as these mice age, an imbalance in the brain occurs favoring neuronal hyperexcitability that is eventually expressed as spontaneous seizure activity. Whether this spontaneous seizure activity complicates interpretations of the earlier studies demonstrating learning deficits in these mice is difficult to ascertain and is reviewed in Butler et al. (1995). However, seizure activity does not always lead to learning impairments (Holmes, 1991). Also unclear at this time is a mechanistic explanation for these observations. As cited above (see Chapman et al., 1995 for discussion), hippocampal slices from these animals appear deficient in their ability to control the release of glutamate suggesting CaM-kinase plays some role in down-regulating the release process under conditions of repetitive firing. An inherent difference in neuronal excitability, an imbalance in local feedback inhibition, or an alteration in the presynaptic release machinery itself could explain these results. In any case, for the first time, a single genetic abnormality (a deficiency in the alpha subunit of CaM-kinase) can be attributed to 1) a deficiency in learning and memory, 2) the production of spontaneous seizure activity and 3) enhanced susceptibility to ischemia. We suggest that CaM-kinase is one of the key molecules underlying yet to be defined mechanisms of both plasticity and pathology.
From results presented in this report, we conclude that CaM-kinase activity plays some neuroprotective role and when lost after ischemia (Churn et al., 1990; Aronowski et al., 1992; Hanson et al., 1994; Shackelford et al., 1995) or when inhibited by treatment with kinase inhibitor (Madden et al., 1990) or gene knock-outs, neuronal damage is manifest. These results further suggest that if losses of CaM-kinase activity could be prevented during ischemia, or induced to return following ischemia, neurons might recover. Determining the mechanism of the irreversible loss of the enzyme will likely provide new therapeutic avenues for future treatment of cerebral ischemia and possibly other neurodegenerative diseases.
Footnotes
Acknowledgment:
We thank Tara Vicknair for help with photography and Dr. Howard Schulman (Stanford University) and Dr. Jim Lessard (Children's Hospital Research Foundation, Cincinnati, OH, U.S.A.) for their generous gifts of beta subunit specific and actin specific monoclonal antibodies. This work was supported by grants from the National Institutes of Health (J.C.G. and M.N.W.) and the Whitehall, Klingenstein and Beckman Foundations (A.J.S.). M.N.W. is also supported by a Research Career Development Award from the National Institutes of Health.