
Abstract
Protein phosphorylation and dephosphorylation mediated by protein kinases and protein phosphatases, respectively, represent essential steps in a variety of vital neuronal processes that could affect susceptibility to ischemic stroke. In this study, the role of the neuron-specific γ isoform of protein kinase C (γPKC) in reversible focal ischemia was examined using mutant mice in which the gene for γPKC was knockedout (γPKC-KO). A period of 150 minutes of unilateral middle cerebral artery and common carotid artery (MCA/CCA) occlusion followed by 21.5 hours of reperfusion resulted in significantly larger (P < 0.005) infarct volumes (n = 10; 31.1 ± 4.2 mm3) in γPKC-KO than in wild-type (WT) animals (n = 12; 22.6 ± 7.4 mm3). To control for possible differences related to genetic background, the authors analyzed Balb/cJ, C57BL/6J, and 129SVJ WT in the MCA/CCA model of focal ischemia. No significant differences in stroke volume were detected between these WT strains. Impaired substrate phosphorylation as a consequence of γPKC-KO might be corrected by inhibition of protein dephosphorylation. To test this possibility, γPKC-KO mice were treated with the protein phosphatase 2B (calcineurin) inhibitor, FK-506, before ischemia. FK-506 reduced (P < 0.008) the infarct volume in γPKC-KO mice (n = 7; 24.6 ± 4.6 mm3), but at this dose in this model, had no effect on the infarct volume in WT mice (n = 7; 20.5 ± 10.7 mm3). These results indicate that γPKC plays some neuroprotective role in reversible focal ischemia.
Keywords
During ischemia and reperfusion, increases in intracellular Ca2+ occur at least in part in response to excitotoxicity (Choi, 1995). Ca2+ is an essential second messenger with a pivotal role in ischemic disease, and Ca2+ regulates the phosphorylation of many intracellular proteins (Hanson and Schulman, 1992). Signal transduction through Ca2+ is mediated in part by the activation of protein kinases, including protein kinase C (PKC), and protein phosphatases, including Ca2+/calmodulin (CaM)-dependent protein phosphatase 2B (calcineurin). Postranslational modification through protein phosphorylation plays an essential role in regulating various enzymes, ion channels, receptors, and cytoskeletal elements (Hanson and Schulman, 1992). Dysfunctional phosphorylation may, therefore, lead to loss of intracellular homeostasis and consequent collapse of cell integrity (Saitoh et al., 1991).
A member of the family of serine/threonine kinases, PKC is one of the most important multifunctional protein kinases in the brain. It is implicated in many vital aspects of CNS physiologic function, including synaptic plasticity, excitability, growth, proliferation, gene expression, and apoptosis (Tanaka and Nishizuka, 1994; Zhang et al., 1995). Protein kinase C consists of a family of closely related isoforms, most of which are detectable in the brain and are differentially activated by phospholipids and Ca2+. In response to cerebral ischemia, the catalytic activity of PKC in brain is greatly inhibited, and the enzyme is proteolyzed (Louis et al., 1988; Kochhar et al., 1989; Wieloch et al., 1991; Aronowski et al., 1992a; Cardell and Wieloch 1993). The γ isoform of PKC (γPKC) is highly enriched in neurons and is particularly susceptible to proteolytic degradation after ischemia (Huang et al., 1988; Wieloch et al., 1991; Zablocka et al., 1998). Irreversible inactivation and translocation of PKC always precedes neuronal damage, and is blocked by treatments that protect neurons from ischemic damage (Cardell et al., 1991; Aronowski et al., 1992b), implying a positive correlation between sustained activity of PKC and neuroprotection. On the other hand, Sieber et al. (1998) demonstrated that after global ischemia, expression and activity of PKC increases before delayed cell death in brain regions that are most susceptible to ischemic damage. Finally, results suggesting either a protective or deleterious role of PKC were obtained when ischemic damage was probed with inhibitors of protein kinases, staurosporine or 1-(5-isoquinolinesulphonyl)-2-methylpiperazine dihydrochloride (H-7), which also affect PKC activity. Hara et al (1990), injecting animals with staurosporine, observed neuroprotection after global ischemia, whereas Madden et al. (1991), using staurosporine or H-7, produced augmentation of ischemic damage using a model of spinal cord ischemia. There are many reasons for these discrepancies; however, the lack of these inhibitor's efficacy or specificity and different ischemic paradigms may be a factor.
Changes in intracellular Ca2+ also regulate dephosphorylation of intracellular substrates through activation of a ubiquitous Ca2+/CaM-dependent serine/threonine protein phosphatase, calcineurin. Calcineurin's effects are either by direct dephosphorylation of phosphosubstrates or by activation of protein phosphatase 1 in a reaction that involves dephosphorylation of a protein phosphatase 1 inhibitory protein (Klee, 1991; Mulkey et al., 1994). An inhibitor of calcineurin, FK506, has been shown to reduce ischemic damage after global (Drake et al., 1996) and focal (Sharkey and Butcher., 1994; Butcher et al., 1997) ischemia, suggesting that calcineurin substrates could be involved in ischemic disease.
Recently, γPKC knock-out mice (γPKC-KO) were used to characterize the role of this enzyme in synaptic plasticity. The γPKC-KO mice are fully viable, although they display abnormal gaits and mild deficits in spatial and contextual learning when subjected to behavioral testing (Abeliovich et al., 1993b). Mutant mice have little detectable difference in baseline synaptic transmission, exhibit normal long-term depression and paired-pulse facilitation, although they have diminished long-term potentiation (Abeliovich et al., 1993a).
We hypothesized that decreases in PKC phosphorylation resulting from ischemia may cause phosphorylation imbalances in favor of protein dephosphorylation, mediated at least in part by calcineurin, leading to increased damage. To test this hypothesis, we used γPKC-KO mice and demonstrated their elevated susceptibility to focal ischemia. Interestingly, the increased damage was reversed by FK-506 treatment, an inhibitor of calcineurin activity.
METHODS
Animals
The production of mice genetically deficient in the γ isoform of PKC was described previously (Abeliovich et al., 1993a). Briefly, a homologous recombination vector was constructed containing γPKC sequences harboring 2.5 kb deletion. Integration of this vector also containing neo resulted in a loss of an exon containing the nucleotide-binding domain required for catalytic activity. The vector was transfected into E14ES cells, and clones containing the desired homologous integration were identified by G418 selection and Southern blot hybridization. Chimeric males initially were mated to C57BL/6J females and were the same animals used in the study by Abeliovich et al. (1993a, 1993b). Finally, to improve the efficiency of breeding, mice were back-crossed eight times into the Balb/cJ background. Homozygous animals were bred to produce offspring (γPKC-KO) that were subjected to the experimental protocols described later. Mice were typed by polymerase chain reaction analysis with a set of primers as described by Abeliovich et al. (1993a). The average weight of γPKC-KO mice was 18.8 ± 1.4 g. Western blotting of brain extracts with an antibody specific to the γ isoform of PKC verified the absence of γPKC in these animals. Balb/cJ (18.6 ± 1.5 g), C57BL/6J (18.7 ± 2.0 g), and 129SVJ (19.2 ± 1.9 g) were obtained from Jackson Laboratories and housed in the institutional animal care facility for at least 72 hours before surgery. Both male and female mice were used. Each experimental group was built out of an equal number of mice of the same sex. In no group were the differences between males and females greater than 1. All animals were kept in a 12:12-hour light-dark cycle with free access to food and water. All procedures were in compliance with the National Institute of Health guidelines for the humane care of animals.
Focal cerebral ischemia
Unilateral middle cerebral artery (MCA) and common carotid artery (CCA) occlusion was performed exactly as described previously (Waxham et al., 1996). Briefly, mice starved overnight between 5 and 7 weeks of age were anesthetized with chloral hydrate (0.35 g/kg), the skull was exposed, and a small burr hole was produced over the MCA. A 0.003-cm (0.005-inch) diameter stainless steel wire (Small Parts, Inc., Miami, FL, U.S.A.) was placed underneath the left MCA rostral to the rhinal fissure, proximal to the major bifurcation of the MCA, and distal to the lenticulostriate arteries. The artery then was lifted and the wire rotated clockwise. The left CCA then was occluded using atraumatic Heifetz aneurysm clips. Reperfusion was established after 150 minutes of occlusion by first removing the aneurysm clips from the CCA, then rotating the wire counterclockwise, and removing it from beneath the MCA. This paradigm resulted in reproducible slightly submaximal infarct volume in our previous studies (Waxham et al., 1996). Interruption of flow through the MCA was inspected under the microscope and verified by cerebral perfusion measurement using a laser Doppler flowmeter (model BPM2, Vasamedics, Inc., St. Paul, MN, U.S.A.). To analyze animals with a similar level of ischemia, only mice that displayed reduction of cerebral perfusion after ischemia to less than 15% of their preischemic value in the center of the ischemic zone were included in the study. Core body temperature was monitored and maintained at 36.5 ± 0.35°C during ischemia and for the first hour of reperfusion using a feed-forward temperature controller (YSI Model 72, Yellow Springs, OH, U.S.A.) coupled to a heating lamp and warming blanket.
Infarct volume measurement
Twenty-four hours after the onset of ischemia, under chloral hydrate anesthesia, mice were perfused with 30 mL of saline through the ascending aorta. The brain was removed, sectioned into 2-mm thick sections, and stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC) in phosphate-buffered saline for 30 minutes at room temperature before fixing in 10% buffered formalin. Morphometric determination of infarct size was achieved using a computer-based image analyzer operated by “Brain” software (Drexel University, Philadelphia, PA, U.S.A.) as previously described (Waxham et al., 1996). Infarcts were restricted to cortical tissue. The infarct volume (in cubic millimeters) was calculated from the difference between the volume of contralateral cortex and the volume of the TTC-stained portion (nonischemic) of ipsilateral cortex of each mouse. This indirect measure of infarct volume, based on the assumption that the volume of the ipsilateral and contralateral cortex are the same before ischemia, corrects the total infarct volume for the edema component (Swanson et al., 1990). Statistical significance was determined by analysis of variance and the Newman-Keuls test.
Physiologic parameters
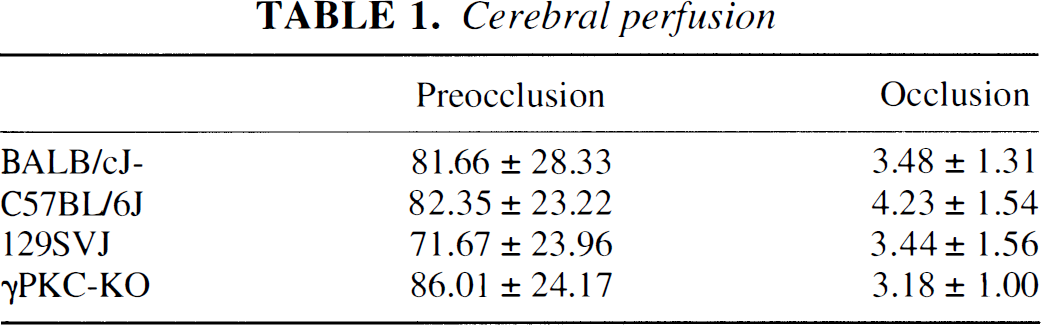
Body temperature in our study was controlled and sustained during ischemia and the initial hour of reperfusion. In a recent report, FK506 at the same dose used in this study (1 mg/kg) had no effect on brain and body temperature in experimental stroke in rats (Sharkey and Butcher, 1994; Butcher et al., 1997). In the current study, we did not measure PO2, PCO2, pH, and blood pressure. However, recent studies using rats and the same dose of FK506 showed no effect on blood gases, pH, and arterial blood pressure (Sharkey and Butcher, 1994; Butcher et al., 1997). Finally, using laser Doppler flowmeter to measure cerebral perfusion in the core of the infarction before and 15 minutes after ischemia, we did not find statistical cross-strain (including γPKC-KO) differences or treatment effect on cerebral perfusion (Table 1).
Cerebral perfusion
Other methods
FK506 (Fujisawa Pharmaceutical, Osaka, Japan) was dissolved in 100% ethanol, then diluted in saline (0.1% final concentration of ethanol) and administered at 1 mg/kg in 200 μL intraperitoneally 1 hour before vessel occlusion. This dose has been shown to produce protection from damage in the rat model of focal ischemia (Sharkey and Butcher., 1994). Because of the negligible amount of ethanol (equivalent to 0.2 μL of 100% ethanol) used during FK-506 injection, we used untreated animals as controls for FK-506.
Western blot analysis was performed on ipsilateral and contralateral cortex homogenates obtained from Balb/cJ and γPKC-KO mice as described previously (Waxham et al., 1996). Immunostaining of Western blots, as described previously (Aronowski et al., 1996), was performed using anti-γPKC-specific rabbit polyclonal antibody (C-19; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, U.S.A.), anti-α-subunit of CaM-KII monoclonal antibody (2D5), and a monoclonal antibody specific to all isoforms of actin (supplied by Dr. Jim Lessard). Immunopositive bands were visualized with alkaline phosphatase-labeled secondary antibody followed by 5-bromo-1-chloro-indolyl phosphate and nitro-blue tetrazolium (Promega, Madison, WI, U.S.A.).
Both CaM-KII and PKC catalytic activity were assayed using 3 μg of crude brain homogenate protein exactly as described previously (Aronowski et al., 1992a) using either synthetic peptide autocamtide-3 (KKALHRQETVDAL) (Aronowski et al., 1996) for CaM-KII or peptide NG(28-43) (AAAKIQASFRGHMAR) (Klann et al., 1998) for PKC.
RESULTS
Genetic background and susceptibility to ischemic damage
Gene knock-out approaches in mice have provided important new tools to investigate the genetic and molecular basis of brain damage after ischemia. However, an important concern in interpreting results from this approach is that of background genetic heterogeneity. This heterogeneity comes from three sources: (1) the embryonic stem cells used for “knocking out” the gene are from 129 mouse; (2) the founder mouse is typically C57BL/6; and (3) in the case of the current study, the strain used for generating mice for experimental analysis is BALB/c. Despite attempts to minimize heterogeneity by extensive back-crossing into the BALB/cJ strain, eliminating the possibility that the observed differences are caused by heterogeneity from either 129SV or C57BL/6 genetic carryover is difficult. This is particularly salient in studies of focal ischemia in mice, since genetic background has been shown to play a significant role in altering the extent of damage after the insult (Barone et al., 1993; Fujii et al., 1997; Maeda et al., 1999). To establish a database for the current and future work, we undertook a study to define the extent of damage after focal ischemia in each of the three mouse strains Balb/cJ, C57BL/6J, and 129/SVJ. Earlier work (Waxham et al., 1996) showed that 150 minutes of focal ischemia, followed by 21.5 hours of reperfusion, produced just submaximal infarct volume in the wild-type C57BL/6J strain of mice. A similar protocol was applied to evaluate the contribution of genetic background to infarct volume. These data are summarized in Fig. 1 and show that, overall, there is no statistical difference in infarct volume between the three strains of mice using this protocol. The average infarct volumes were 16.7 ± 4.9 mm3, 20.9 ± 6.3 mm3, and 22.6 ± 7.4 mm3 for 129/SVJ, C57BL/6J, and BALB/cJ, respectively. The infarct volume of commercially available C57BL/6J (20.9 ± 6.3 mm3) is nearly identical to wild-type litter mates bred from heterozygous α-subunit CaM-KII (αCaM-KII) knock-out mice in the C57BL/6 background (21.9 ± 6.4 mm3) (Waxham et al., 1996). Thus, the protocol we use for producing infarcts is consistent, and, overall, the genetic background contributes little to differences in infarct volume under the conditions used in our experiments.
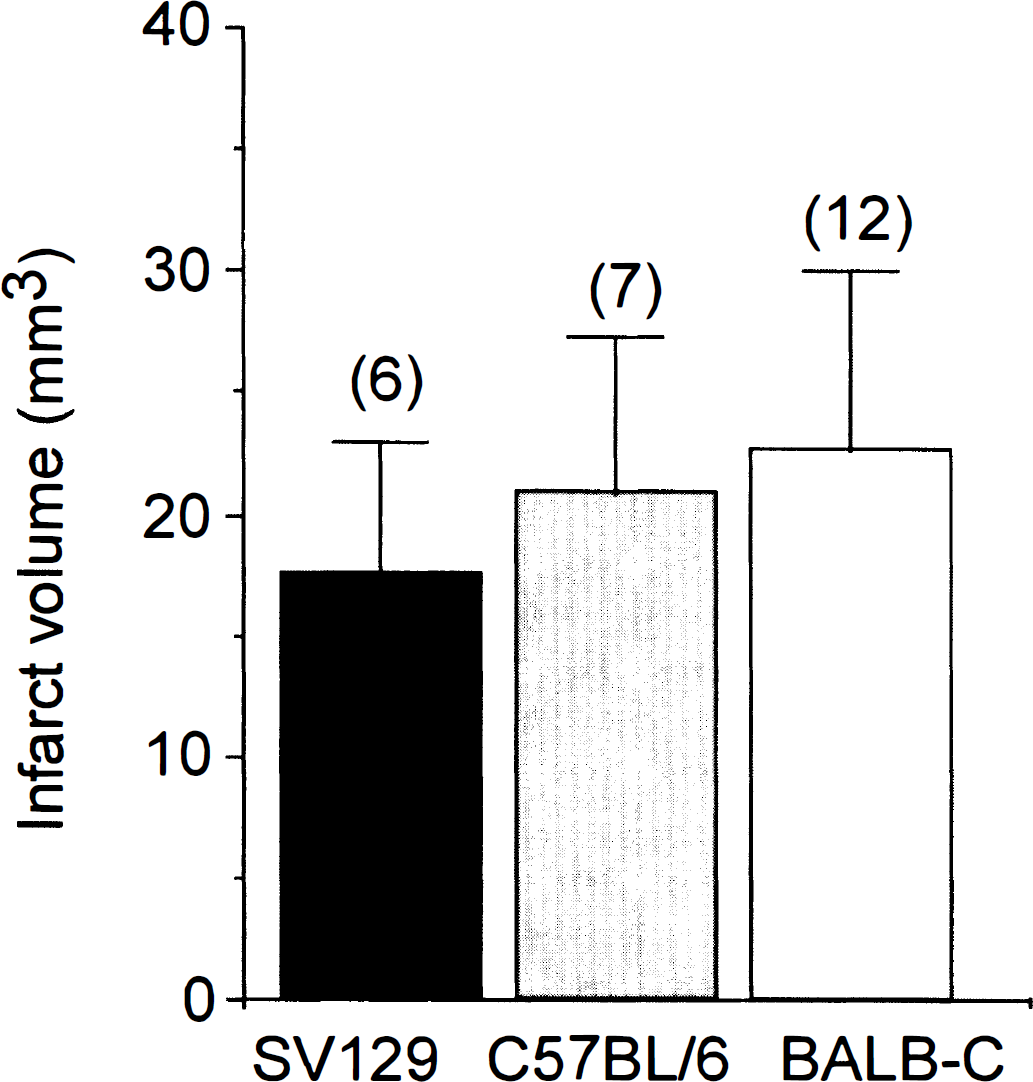
Infarct volume after 150 minutes of ischemia and 21.5 hours of reperfusion in 129/SVJ, C57BL/6J, and Balb/cJ mice. Statistical analysis was determined by analysis of variance and the Newman-Keuls test and showed no difference between the groups.
Genetic knock-out of γPKC increases susceptibility to ischemic damage
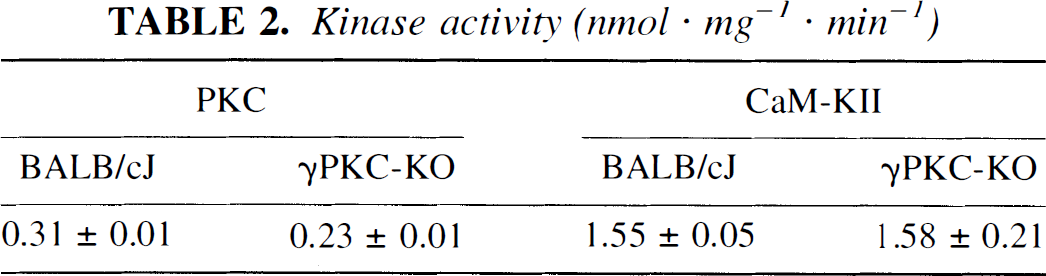
Because, as mentioned earlier, γPKC-KO mice were back-crossed into the BALB/c background, all remaining analyses were performed using BALB/cJ mice as controls. As anticipated, probing extracts from BALB/cJ and γPKC-KO brains, using antibody to γPKC, we detected an immunopositive band at approximately 87 kd in BALB/cJ, whereas no immunoreactivity was detected in γPKC-KO animals (Fig. 2). Probing the same blot with antibody to αCaM-KII and actin showed no difference in the quantity of these proteins between BALB/cJ and γPKC-KO animals (Fig. 2). Similar to the results of Abeliovich et al. (1993), γPKC-KO brain extracts displayed 74% of PKC activity relative to BALB/cJ. In contrast, there was no difference in CaM-KII activity between γPKC-KO and BALB/cJ mice (Table 2).
Kinase activity (nmol · mg−1 · min−1)
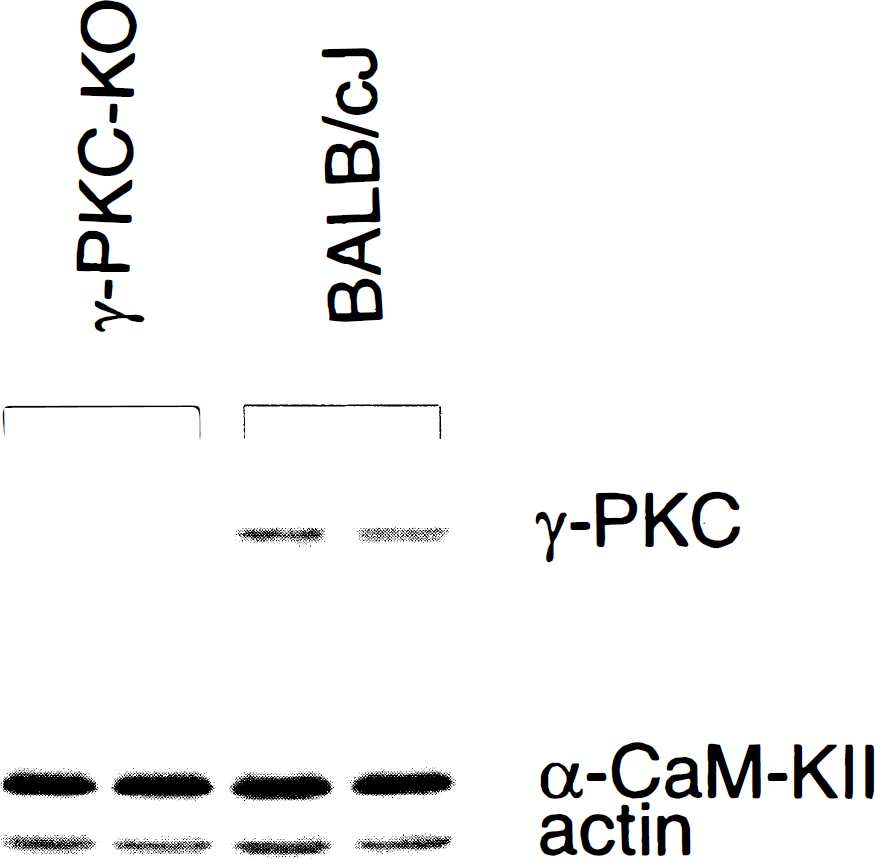
Immunoblot of γ isoform of protein kinase C (γPKC), α-subunit of Ca2+/calmodulin-dependent protein kinase II (CaM-KII), and actin. Forty micrograms of cortical homogenate protein from γPKC-KO and BALB/CJ mice were analyzed. Notice the absence of immunostaining of γPKC in γPKC-KO mice and no changes in immunostaining of CaM-KII and actin. Two representative γPKC-KO and BALB/CJ mice are shown.
To assess PKC's role in the pathobiologic mechanism of ischemia, we analyzed the susceptibility of γPKC-KO animals to damage produced by focal ischemia. Occlusion of the MCA/CCA in γPKC-KO mice resulted in infarct volumes of 31.1 ± 4.2 mm3 (n = 10) (Figs. 3 and 4), which were significantly larger than infarct volumes in BALB/cJ mice 22.6 ± 7.4 mm3 (n = 12) (P < 0.003) (Figs. 3 and 4). In general, the infarct volume between animals was consistent; however, one γPKC-KO animal developed whole ipsilateral hemisphere infarction (56.5 mm3). This type of infarction has been detected in only 4 of 300 mice subjected to this protocol, and the cause is not clear. We excluded this animal from the analysis to avoid overinflating the average infarct volume in the γPKC-KO group. We conclude that mice deficient in the γ isoform of PKC exhibit increased damage after focal ischemia.
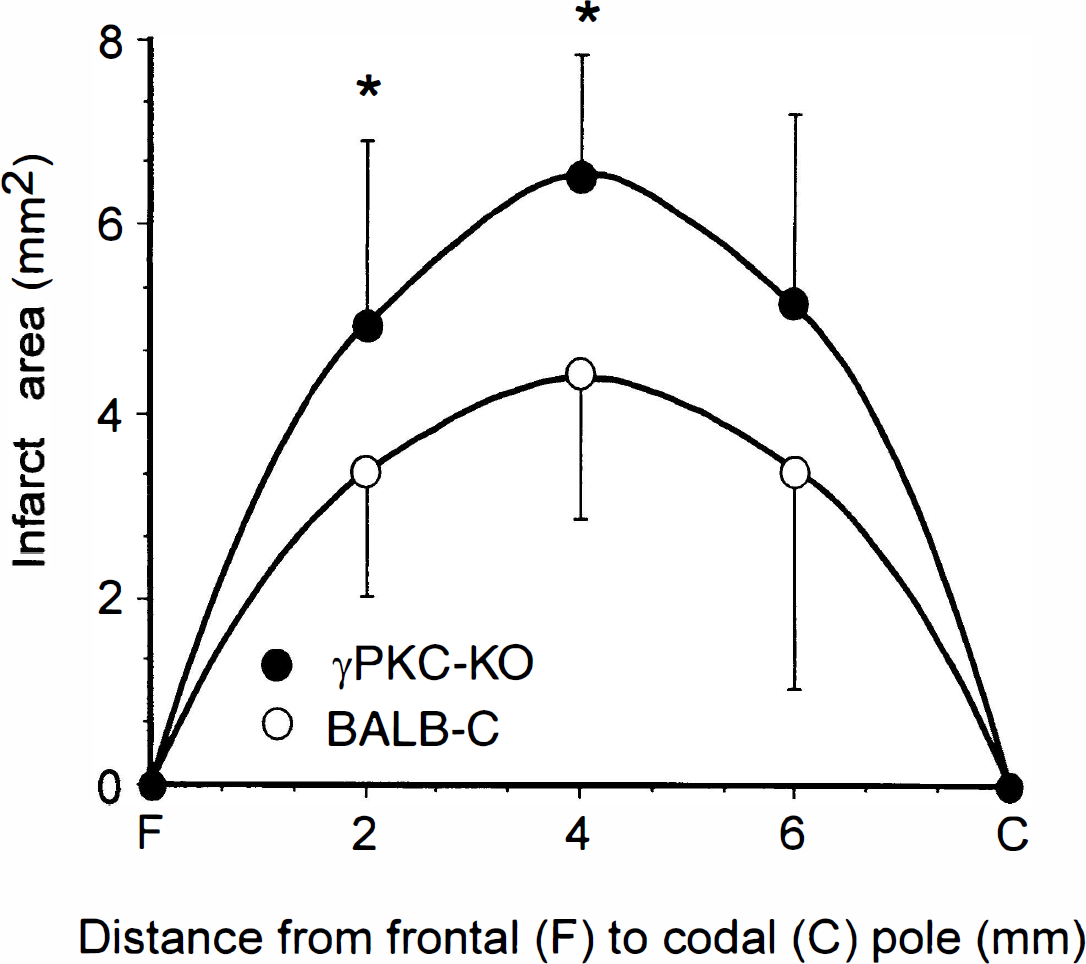
Reversible MCA/CCA occlusion resulted in larger infarct volumes in γPKC-KO mice. After 150 minutes of ischemia and 21.5 hours of reperfusion, animals were killed and infarct volume analyzed with triphenyltetrazolium chloride. Distribution of the infarcted areas is shown at different rostrocaudal levels in BALB/cJ and γPKC-KO mice. Infarct area is different (*P < 0.05) at the first two, but not the third, level as determined by the unpaired t-test.
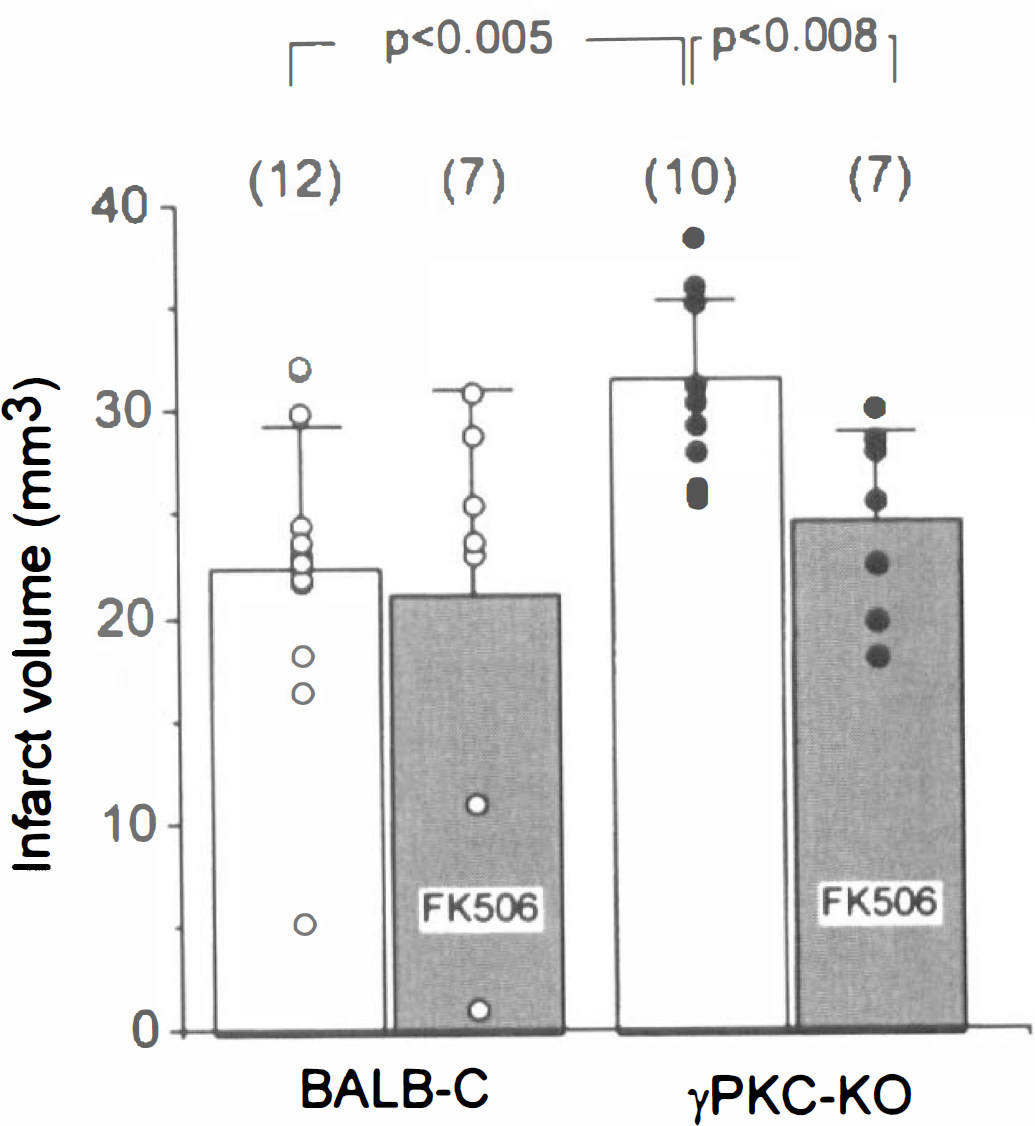
Neuroprotection studies exploring the efficacy of FK-506 to reduce infarct volume. Infarct volume (cubic millimeters, determined using triphenyltetrazolium choloride) after transient 150 minutes of left middle cerebral artery/common carotid artery (MCA/CCA) occlusion and 21.5 hours of reperfusion in γPKC-KO and BALB/cJ mice is illustrated. Mice were injected intraperitoneally with 1 mg/kg of FK506 1 hour before vessel occlusion. The number of animals in each group is indicated in parenthesis above each bar. Average infarct volume ± SD for animals are shown. Infarct volume from individual animals are indicated as dots within each group. Level of statistical significance was determined by analysis of variance and the Newman-Keuls test.
Selective attenuation of infarct volume in γPKC-KO animals by FK506
Because reduced PKC activity represents the likely explanation for increased susceptibility to ischemia in γPKC-KO mice, we explored the possibility that inhibition of protein dephosphorylation in these animals might be capable of attenuating their elevated ischemic vulnerability. Calcineurin is highly enriched in neurons and is postulated to reverse the phosphorylation of numerous PKC substrates, suggesting functional interplay between these two enzymes. To address this issue, the well-characterized brain-permeable calcineurin inhibitor FK506 was used. Treatment of γPKC-KO mice with FK506 1 hour before stroke resulted in significant (P < 0.01) reductions in infarct volume from 31.1 ± 4.2 (n = 10) to 24.6 ± 4.6 mm3 (n = 7). Similar treatment of wild-type BALB/cJ mice with FK506 did not significantly reduce ischemic infarct volume (Fig. 4). These results indicate that this dose of FK-506 can preferentially rescue neurons in mice missing the γ isoform of PKC. We propose that inhibition of calcineurin-mediated dephosphorylation is capable of compensating for impaired phosphorylation in γPKC-KO mice. These results also suggest that interplay between γPKC and calcineurin activity regulate ischemic vulnerability.
DISCUSSION
In the current study, we demonstrated that animals lacking the γ isoform of PKC displayed significantly greater susceptibility to brain damage produced by reversible focal ischemia, and this increase in damage was reversed by an inhibitor of calcineurin, FK506.
We were concerned with the genetic heterogeneity of these animals because strain differences in damage after ischemia have been documented in mice (Barone et al., 1993; Fujii et al., 1997). Our analysis showed different susceptibility to ischemia, although statistically insignificant, among BALB/cJ, C57BL/6J, and 129/SVJ, with BALB/cJ being most vulnerable and 129/SVJ being the least vulnerable. The gradient of strain vulnerability to ischemia was similar to that described in previous work (Barone et al., 1993; Fujii et al., 1997). When we compared susceptibility of all these mice to that of γPKC-KO, the γPKC-KO mice displayed significantly greater vulnerability to ischemia, strongly suggesting that the absence of functional γPKC contributed to the increased damage. This implies that γPKC plays an as yet undefined role in protecting neurons after an ischemic insult.
As mentioned earlier, studies on the role of PKC in ischemia have not established its relevance to ischemic damage. Both activation and inactivation of the enzyme by ischemia have been demonstrated, and both protective and detrimental effects of PKC have been postulated ((Louis et al., 1988; Kochhar et al., 1989; Wieloch et al., 1991; Aronowski et al., 1992; Cardell and Wieloch, 1993; Cardell et al., 1991; Aronowski et al., 1992b; Sieber et al., 1998; Hara et al., 1990; Madden et al., 1991). Protein kinase C is ubiquitously distributed throughout a variety of cell types, and in the brain PKC is present in neurons, glia, smooth muscles, endothelial cells, and other cells (Tanaka and Nishizuka, 1994). Protein kinase C consists of a family of at least 12 isoforms, with 4 isoforms (α, βI, βII, and γ) that require Ca2+ for their activation. Each isoform can be activated by a distinct subset of second messengers, can be expressed in distinct cell types, and has different intracellular localization (Tanaka and Nishizuka, 1994; Krizbai et al., 1995; Van der Zee et al., 1995; Erbrugger et al., 1997). Because each PKC isoform is likely to modulate distinct, potentially opposing functions, it could be misleading to draw conclusions about the role of PKC enzymes based on analysis of their (all isoforms together) generalized activity changes in whole brain. Additionally, compounds that inhibit all PKC isoform could lead to a different outcome than the selected loss of one isoform produced by gene knock-out.
Cerebrovascular tone has a direct impact on ischemic stroke outcome. Vasoconstriction resulting in reduced CBF can produce augmentation of ischemia and subsequent damage. Recently, activation of vascular PKC was demonstrated to produce vasoconstriction linked to the pathogenesis of cerebral vasospasm (Ohta et al., 1995). In addition, inhibition of PKC by the protein kinase inhibitor, staurosporine, was reported to inhibit vascular smooth muscle contractility (Asano et al., 1995), providing a possible explanation for the protective mechanism of staurosporine in ischemic models (Hara et al., 1990). A role for γPKC in regulating vascular contractility is unknown, and although absent from blood vessels (Mattila et al., 1994; Krizbai et al., 1995; Erdbrugger et al., 1997), γPKC could affect vascular tone through its innervation. γPKC is highly enriched in neurons, where it is postulated to regulate synaptic activity or gene expression in response to signal activating Ca2+ entry (Hata et al., 1993; Abeliovich et al., 1993a; Sakai et al., 1997). It is, therefore, logical to consider that increased vulnerability to ischemia in γPKC-KO animals, observed in the current study, results from γPKC mutation-induced neuronal dysfunction.
Neuronal PKC, similar to calcineurin, is involved in regulation of diverse aspects of signal transduction including neurotransmitter release, receptors, ion channels, enzyme regulation, and gene expression (Klee et al., 1988; Yakel, 1997; Ryves et al., 1996; Tanaka and Nishizuka, 1994). Calcineurin and γPKC share common substrates. Some of these include dynamin I, GAP-43, MARCKS, IP3 receptor, and nitric oxide synthase (Robinson et al., 1994; Robinson, 1991; Klee, 1991; Bredt et al., 1992; Dawson et al., 1993; Cameron, 1995; Mahoney et al., 1995). Deficient substrate phosphorylation by PKC, as that likely present in γPKC-KO mice, might, therefore, be corrected by inhibiting substrate dephosphorylation, including that by calcineurin.
One of the cell-permeable calcineurin inhibitors is the immunosuppressant drug FK506 (Liu, 1991). Similar to the effect of FK506 on infarct volume in γPKC-KO animals, FK506 was shown to protect rat brain from damage after reversible focal ischemia (Sharkey and Butcher, 1994; Butcher et al., 1997). This neuroprotective mechanism of FK506 was postulated to involve inhibition of calcineurin with subsequent inhibition of nitric oxide synthase and reduction of cytotoxic NO production (Bredt et al., 1992; Dawson et al., 1993). We find it surprising that the neuroprotective effect of FK506 was significant in γPKC-KO animals, whereas no effect in BALB/cJ mice was observed. It is generally accepted that the portion of ischemic brain most likely to be salvaged by therapeutic intervention is the ischemic penumbra, due to presence of collateral flow resulting in better perfusion than in the infarct core. Because γPKC-KO mice develop larger infarctions, it is possible that this is caused by lesser resistance of the γPKC mutated neurons to the level of hypoperfusion present in the penumbral region. The efficacy of FK506 in γPKC-KO may be linked to the possibility that there is a larger ischemic penumbra in these mice. The dose of FK506 used in the current study is the same as that which is optimal in producing immunosuppression and in protecting brain from focal ischemia in rats (Sharkey and Butcher, 1994). However, there is a difference in FK506 pharmacokinetics between rats and mice. The 1-mg/kg dose of FK506 used to produce immunosuppression may be suboptimal in mice (Iwata et al., 1993), since a three- to five-times larger dose is required to produce optimal immunosuppression and possibly neuroprotection in wild-type mice compared with rats (Bekersky I, personal communication, 1998). Because γPKC-KO mice may have impaired phosphorylation compared with wild-type animals, it is possible that a lower dose of FK506 is sufficient to correct deficient phosphorylation produced by the γPKC mutation.
In summary, γPKC-deficient mice are more susceptible to ischemia. This vulnerability may be mediated by imbalances between impaired PKC phosphorylation and unimpaired calcineurin-mediated protein dephosphorylation in the regulation of neuronal enzymes essential for cell homeostasis and survival.