
Abstract
Rats were subjected to the standard four-vessel occlusion model of transient cerebral ischemia (vertebral and carotid arteries). The effects of normothermic ischemia (37°C) followed or not by 30-minute reperfusion, as well as 30-minute postdecapitative ischemia, on translational rates were examined. Protein synthesis rate, as measured in a cell-free system, was significantly inhibited in ischemic rats, and the extent of inhibition strongly depended on duration and temperature, and less on the model of ischemia used. The ability of reinitiation in vitro (by using aurintricarboxylic acid) decreased after ischemia, suggesting a failure in the synthetic machinery at the initiation level. Eukaryotic initiation factor 2 (eIF-2) presented almost basal activity and levels after 30-minute normothermic ischemia, and the amount of phosphorylated eIF-2α in these samples, as well as in sham-control samples, was undetectable. The decrease in the levels of phosphorylated initiation factor 4E (eIF-4E) after 30-minute ischemia (from 32% to 16%) could explain, at least partially, the impairment of initiation during transient cerebral ischemia. After reperfusion, eIF-4E phosphorylation was almost completely restored to basal levels (29%), whereas the level of phosphorylated eIF-2α was higher (13%) than in controls and ischemic samples (both less than 2%). eIF-2α kinase activity in vitro as measured by phosphorylation of endogenous eIF-2 in the presence of ATP/Mg2+, was higher in ischemic samples (8%) than in controls (4%). It seems probable that the failure of the kinase in phosphorylating eIF-2 in vivo during ischemia is due to the depletion of ATP stores. The levels of the double-stranded activated eIF-2α kinase were slightly higher in ischemic animals than in controls. Our results suggest that the modulation of eIF-4E phosphorylation could be implicated in the regulation of translation during ischemia. On the contrary, phosphorylation of eIF-2α, by an eIF-2α kinase already activated during ischemia, represents a plausible mechanism for explaining the inhibition of translation during reperfusion
Keywords
Protein synthesis requires a number of specific and precise steps as well as the activity of many enzymes and plays a major role in cell function and viability. Protein synthesis suppression occurs in the brain as a consequence of ischemia and reperfusion (reviewed in Krause and Tiffany, 1993). Persistently reduced protein synthesis in certain vulnerable regions of the brain leads to neuronal necrosis (Thilmann et al., 1986; Krause and Tiffany, 1993), hence the importance of restored protein synthesis for complete functional and metabolic recovery of the brain during the postischemic period. This recovery has only been observed in such cases (Kleihues et al., 1975; Morimoto and Yanagihara, 1981; Bodsch et al., 1986).
Protein synthesis regulation is mainly exerted at the initiation step which depends on eukaryotic initiation factor 2 (eIF-2) because this factor is required to prime each 40S ribosomal subunit for every round of translation. Translational initiation is also regulated at the level of the binding of the 43S preinitiation complex to the 5′ end of mRNA, mediated by eIF-4E and other associated factors (reviewed in Sonenberg, 1994, and Pain, 1996). The initiation step is impaired during the postischemic period (Hu and Wieloch, 1993; Burda et al., 1994; DeGracia et al., 1996), and our laboratory has provided evidence of a tight correlation between the phosphorylation of eIF-2α and the inhibition of protein synthesis (Burda et al., 1994). Besides, partial fragmentation of eIF-4G after reperfusion has been reported (DeGracia et al., 1996).
Controversial results have been obtained when trying to measure protein synthesis during the intra-ischemic period, the main reason being the different in vitro methods used. Early studies from 1970 to the early 1980s, performed on the basis of relatively well preserved polyribosomal profiles and on incorporation of labelled amino acids to polypeptide chains, showed that neither the enzymes found as part of the cell sap, nor the ribosomes frozen as a consequence of the energy failure, lose their activity (Kleihues and Hossmann, 1971; Kleihues et al., 1975; Cooper et al., 1977; Burda et al., 1980; Morimoto and Yanahigara, 1981; Nowak et al., 1985). According to these results, the depletion of ATP/GTP stores could be considered as the cause of protein synthesis interruption during ischemia (Krause and Tiffany, 1993). Conversely, after intracerebral administration of 14C-leucine, the specific activity of brain proteins was found to be decreased to less than 5% of control values in complete ischemia conditions (Kleihues and Hossmann, 1971). Yanagihara (1978), using the model of carotid artery occlusion, noted a 50% decrease of in vitro protein synthesis after 30 minutes of ischemia, and Orunesu et al., (1980), using a gerbil model of unilateral carotid ligation, showed protein synthesis inhibition after only 5 minutes of ischemia. More recently, Erdogdu et al. (1993) have presented evidence of the activation of a translational inhibitor during ischemia using a cell-free system combining intact reticulocyte lysates with extracts from individual rat brain regions after four-vessel occlusion.
In the present study we show that the initiation step of translation is impaired both during the intra-ischemic and reperfusion periods. eIF-4E dephosphorylation could partially explain the inhibition found during the intra-ischemic period, whereas the eIF-2α phosphorylation, probably by an eIF-2α kinase activated during ischemia, could explain the translational inhibition during the reperfusion period.
EXPERIMENTAL PROCEDURES
Induction of ischemia
Adult Wistar rats (mean body weight 250 g) were used. The standard four-vessel occlusion model (Pulsinelli and Brierley, 1979) as modified by Schmidt Kastner et al. (1989) was used to produce incomplete forebrain ischemia, and severity of ischemia was verified by EEG and neurologic investigation. The experiments were approved by the Ethics Committee at the Institute of Neurobiology. On day 1 both vertebral arteries were irreversibly occluded by coagulation through the alar foramina after anesthesia with sodium pentobarbital (50 mg per kg of body weight, intraperitoneally) was administered, and on day 2 both common carotid arteries were prepared for occlusion under anesthesia induced by placing the rat in a jar with 2.5% halothane in a mixture of oxygen/nitrous oxide (30%/70%). Two minutes before the carotids were occluded for 5 to 30 minutes by small atraumatic clips, the halothane was omitted from the mixture. The recirculation of postischemic brain was verified by tracing the blood flow in carotid arteries. Normothermic conditions were monitored by using a microthermistor placed deeply in the ear and maintained during ischemia and reperfusion at approximately the same temperature (37°C) with a heating pad under a light bulb placed above the animal. Sham-control animals were prepared in the same way, but the carotids were not occluded. Animals were decapitated at the appropriate times and the brain was rapidly removed and processed. Postdecapitative ischemia was obtained by placing the rats in a jar ventilated with a mixture of 2.5% halothane in 30% oxygen and 70% nitrous oxide. Anesthetized rats were decapitated and whole unbroken heads were placed in thermostatically controlled chambers. After 30 minutes of postdecapitative ischemia, the brains were removed and processed.
Extract preparations
The neocortex from the brains obtained under the different experimental conditions were dissected and homogenized 1:2 with buffer H (50 mmol/L Hepes-Tris, pH 7.55, 140 mmol/L potassium acetate, 4 mmol/L magnesium acetate, 2.5 mmol/L dithiothreitol and 0.32 mol/L sucrose) and centrifuged at 11,000 × g for 10 minutes at 4°C to obtain the postmitochondrial supernatant (PMS). This was maintained at −60°C, or freeze-dried and resuspended in 175 μL of distilled water. Protein determination was as described by Bradford (1976).
In vitro translation assays. In vitro translation was assessed in a cell-free system (Burda et al., 1994), and the complete reactions (final volume, 100 μL) contained 50 mmol/L Hepes-Tris, pH 7.55; 150 mmol/L potassium acetate; 5 mmol/L magnesium acetate; 2.5 mmol/L dithiothreitol; 0.32 mol/L sucrose; 1 mmol/L ATP; 0.75 mmol/L GTP; 20 mmol/L phosphocreatine; 150 μg/mL creatine phosphokinase; 50 μmol/L amino acids, 200 μg of PMS (frozen or freeze-dried), and 5 μCi of L-[4,5-3H]leucine (25 μmol/L, 171 Ci/mmol). After 45-minute incubation at 30°C (triplicates), 50-μL samples were used to measure the radioactivity present in 25% trichloroacetic acid insoluble material.
eIF-2 Assay
The GTP-dependent binding of eIF-2 to the initiation form of methionyl-tRNA was measured in the freeze-dried PMS (30 μg) as previously described (Martín et al., 1993), in the presence of Mg2+ (1.5 mmol/L) and GTP-regenerating capacity (2.5 mmol/L phosphoenolpyruvate and 2 U/mL of phosphopyruvate kinase). The results were expressed as pmoles of Met-tRNAi incorporated per milligram of protein per 10-minute incubation.
Determination of the state of phosphorylation of eIF-2 and eIF-4E factors
Freeze-dried PMS fractions (75 μg) from sham-control and ischemic animals were resolved in horizontal isoelectric focusing slab gels (IEF) electrophoresis and analyzed by protein immunoblot as described previously (Martín et al., 1993; Garcia et al., 1994). The bands corresponding to eIF-2α, eIF-2α(P), eIF-4E, and eIF-4E(P) protein were stained on immunoblots that were performed with a polyclonal antiserum for eIF-2α (Martín et al., 1991) (1:200) purified by immunoaffinity and a commercial monoclonal anti-4E (Affiniti Research Products Limited, Mamhead Castle, Exeter, United Kingdom). Purified eIF-2 from calf brain (Alcázar et al., 1995), and partially purified eIF-4E from reticulocyte lysates were used as controls. Stained bands were scanned and quantitated with an image analyzer equipped with a software package (PDI, New York).
Double-stranded RNA-activated protein kinase levels
Human protein kinase (PKR) cDNA (Lee and Esteban, 1993) was expressed as a fusion protein with glutathione S-transferase by its subcloning in pGEX-2T vector (Pharmacia). Expression was carried out in competent BL21 (DE3) pLysS Escherichia coli cells and optimized to minimize degradation and increase solubility. The corresponding fusion protein (92 kd) was affinity-purified in native conditions in glutathione-sepharose (Pharmacia). The glutathione S-transferase PKR was used as immunogen in a standard immunization protocol (Harlow and Lane, 1988) to generate a rabbit polyclonal antiserum against human PKR. The antiserum recognized in Western blot human, rat, and mouse PKR and presented no cross-reactivity with any other protein kinase. Brain cortex from sham-control and ischemic animals kept frozen at −80°, were homogenized in glass to glass Elvehjem potters in lysis buffer: 20 mmol/L Hepes-KOH, pH 7.4; 120 mmol/L KCl; 5 mmol/L MgCl2; 1 mmol/L dithiothreitol; 1 mmol/L PMSF; 2 mmol/L benzamidine, 10 μg/mL pestatin, antipain, and leupeptin; 0.5% nonidet NP-40 and centrifuged at 17,000 × g for 30 minutes at 4°C. The supernatants were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) in 10% acrylamide-bisacrylamide gels, which were electrophoretically transferred to nitrocellulose paper in 25 mmol/L Tris-HCl, pH 8.3; 192 mmol/L glycine; 20% methanol for 2.5 hours at 0.8 mA/cm2 of gel. Bands corresponding to PKR were stained on immunoblots using the polyclonal antiserum and color was developed by using enhanced chemiluminescence kits containing reagents for horseradish peroxidase reaction (Amersham). Partially purified PKR from rabbit reticulocytes and rat liver were used as control.
eIF-2α Kinase assay
The eIF-2α kinase activity was measured by incubating freeze-dried PMS (200 μg of protein) from control and ischemic animals, both in presence and absence of 1 mmol/L ATP and 4 mmol/L magnesium acetate, for 15 minutes at 37°C. A 10- to 20-μL sample (up to 75 μg protein) of the incubation mixtures was resolved in IEF electrophoresis and analyzed by protein immunoblot as described above.
Statistical analysis
Multiple statistical comparisons were made by analysis of variance followed by Tukey-Kramer tests. Whenever a significant (P < .05) F value was identified, individual comparisons were made by Tukey-Kramer. Statistical comparisons were also made by Student' t-test (eIF-2α, eIF-4E, and PKR levels).
RESULTS
The cell-free system used to measure protein synthesis in the brain is based on our previous method (Fando and Wasterlain, 1980) and that described by Eisenstein and Harper (1984) and Cosgrove and Rapoport (1986) and is capable of efficient re-initiation. As shown in Table 1, leucine incorporation into PMS fractions obtained after the different periods of ischemia was substantially inhibited compared to controls. Inhibition increased gradually with the duration of ischemia and became significant (P < .05) after 15 minutes or more. Changes in leucine incorporation into PMS from brains kept at different temperatures of postdecapitative ischemia (30 minutes) are shown in Table 2. Almost no significant decrease in translation was observed in the brains maintained at 4°C or 20°C (not shown), whereas a significant decrease in protein synthesis occurred at 35°C and above. These results clearly indicate the importance of temperature during the ischemic episode.
Protein synthesis rate in sham-control and ischemic rats. Time effect on normothermic ischemia (four-vessel occlusion model)

Normothermic conditions (37°C) were monitored using a microthermistor placed deep in the ear and the temperature maintained with a heating-pad under a light bulb placed above the animal. Protein synthesis rate was measured by the incorporation of [3H]leucine into postmitochondrial supernatant (PMS) fractions from sham control and ischemic rats as. described in Experimental Procedures. Values represent the average ± SD of the different experiments. Significance between sham-control and ischemic rats
P < 0.05. (n), number of experiments.
Protein synthesis rate in sham-control and ischemic rats. Temperature effect on postdecapitative ischemia
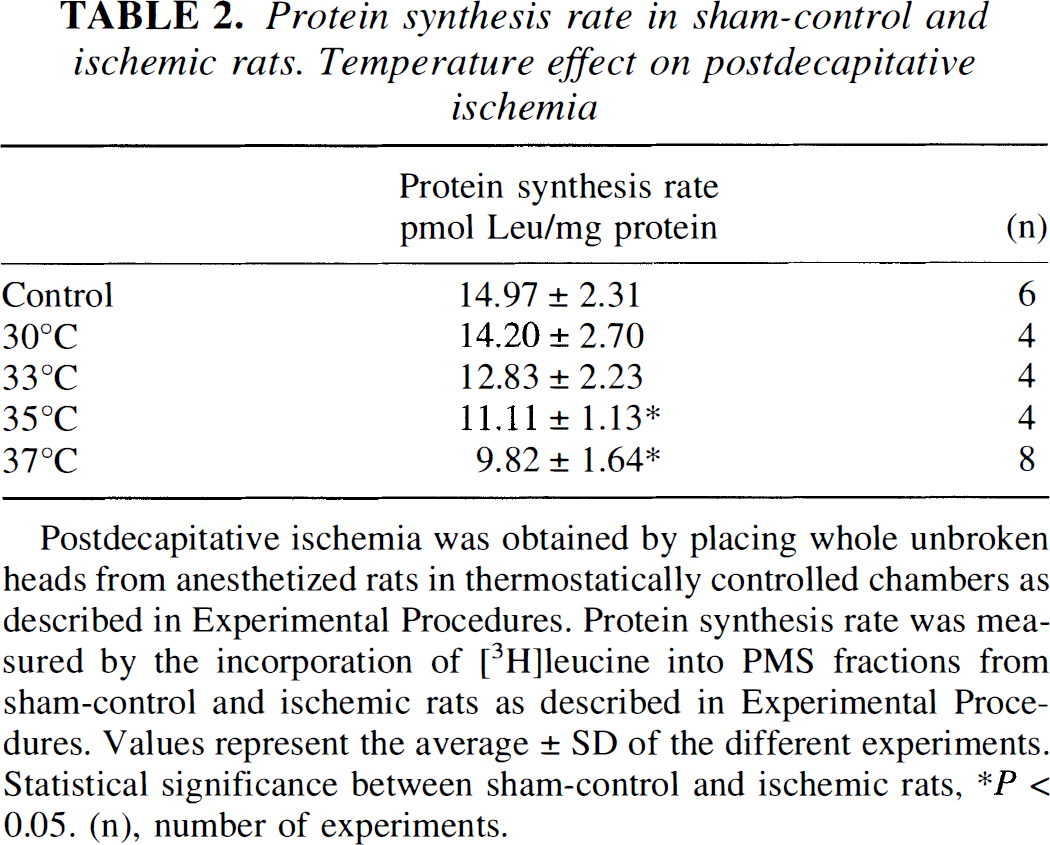
Postdecapitative ischemia was obtained by placing whole unbroken heads from anesthetized rats in thermostatically controlled chambers as described in Experimental Procedures. Protein synthesis rate was measured by the incorporation of [3H]leucine into PMS fractions from sham-control and ischemic rats as described in Experimental Procedures. Values represent the average ± SD of the different experiments. Statistical significance between sham-control and ischemic rats
P < 0.05. (n), number of experiments.
Newly initiated translation after ischemia and reperfusion was calculated by subtracting the data from in vitro translation reactions in the presence and absence of the initiation inhibitor, aurintricarboxylic acid. As Table 3 shows, reinitiation that was above 40% in control brains decreased to 8.7% after 30 minutes of ischemia and was completely blocked after 30 minutes of postischemic reperfusion. Ischemia alone had no significant effect either on eIF-2 activity or levels; however, significantly decreased activity of the factor after reperfusion was found (Table 3). Because eIF-2 activity was measured in the presence of Mg2+ and a GTP-regenerating capacity, a decreased eIF-2 activity in this assay points to an impairment of eIF-2B activity, possibly due to the presence of its inhibitor eIF-2(αP) (Rowlands et al., 1988; Burda et al., 1994). To determine eIF-2α levels, freeze-dried PMS samples were subjected to IEF electrophoresis and protein immunoblot analysis. As Table 3 shows, eIF-2(αP) was undetectable in sham-control and ischemic rats, whereas its levels clearly increased during reperfusion.
Changes in reinitiation ability and eIF-2 activity and levels in sham-control and ischemic rats
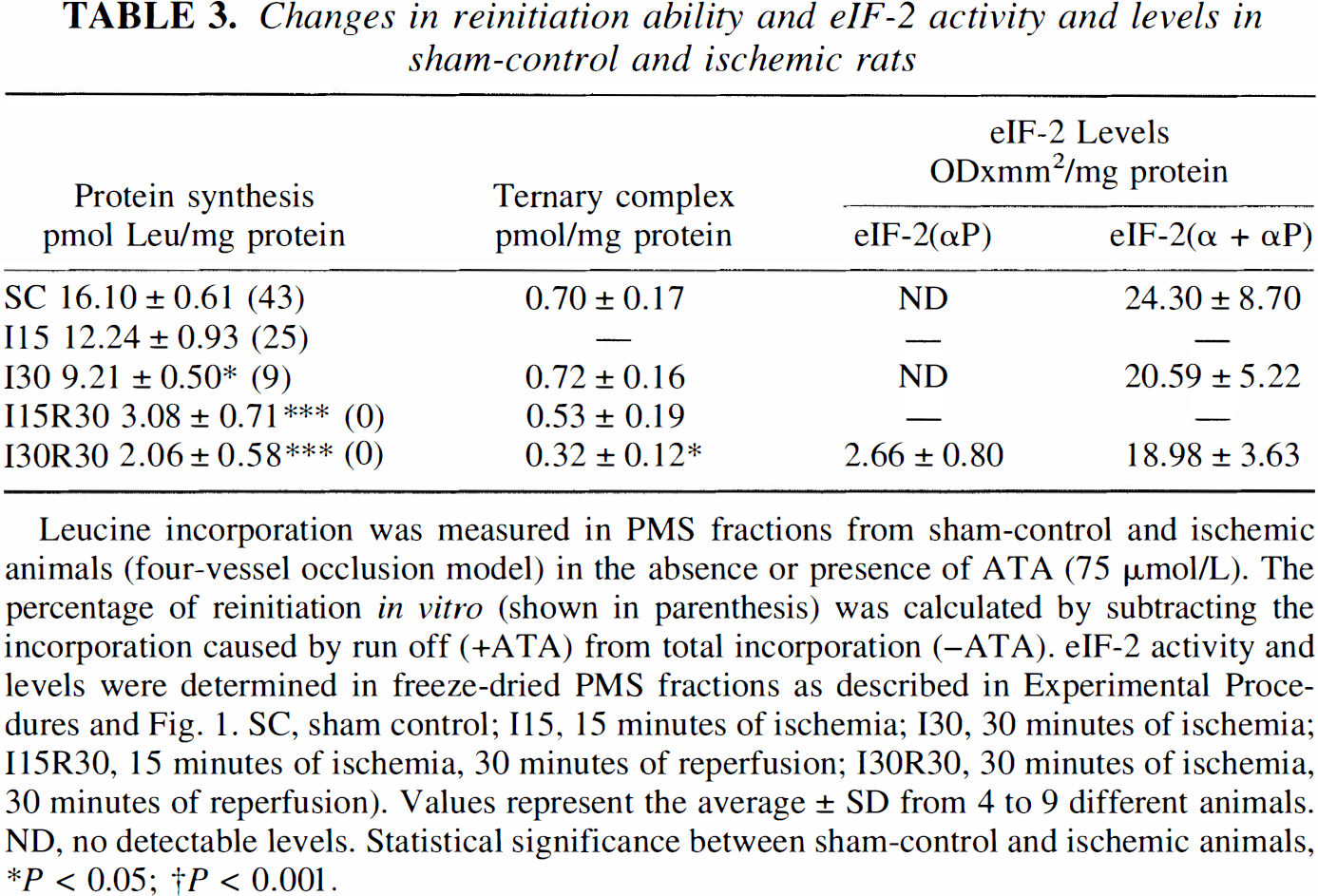
Leucine incorporation was measured in PMS fractions from sham-control and ischemic animals (four-vessel occlusion model) in the absence or presence of ATA (75 μmol/L). The percentage of reinitiation in vitro (shown in parenthesis) was calculated by subtracting the incorporation caused by run off (+ATA) from total incorporation (–ATA). eIF-2 activity and levels were determined in freeze-dried PMS fractions as described in Experimental Procedures and Fig. 1. SC, sham control; 115, 15 minutes of ischemia; 130, 30 minutes of ischemia; I15R30, 15 minutes of ischemia, 30 minutes of reperfusion; I30R30, 30 minutes of ischemia, 30 minutes of reperfusion). Values represent the average ± SD from 4 to 9 different animals. ND, no detectable levels. Statistical significance between sham-control and ischemic animals
P < 0.05;
P < 0.001.
Phosphorylation status of initiation factor 4E, under the same IEF electrophoresis conditions described for the study of eIF-2, was also assessed. As shown in Fig. 1, eIF-4E phosphorylation significantly decreased during the intraischemic period and experienced complete recovery after 30 minutes of reperfusion.
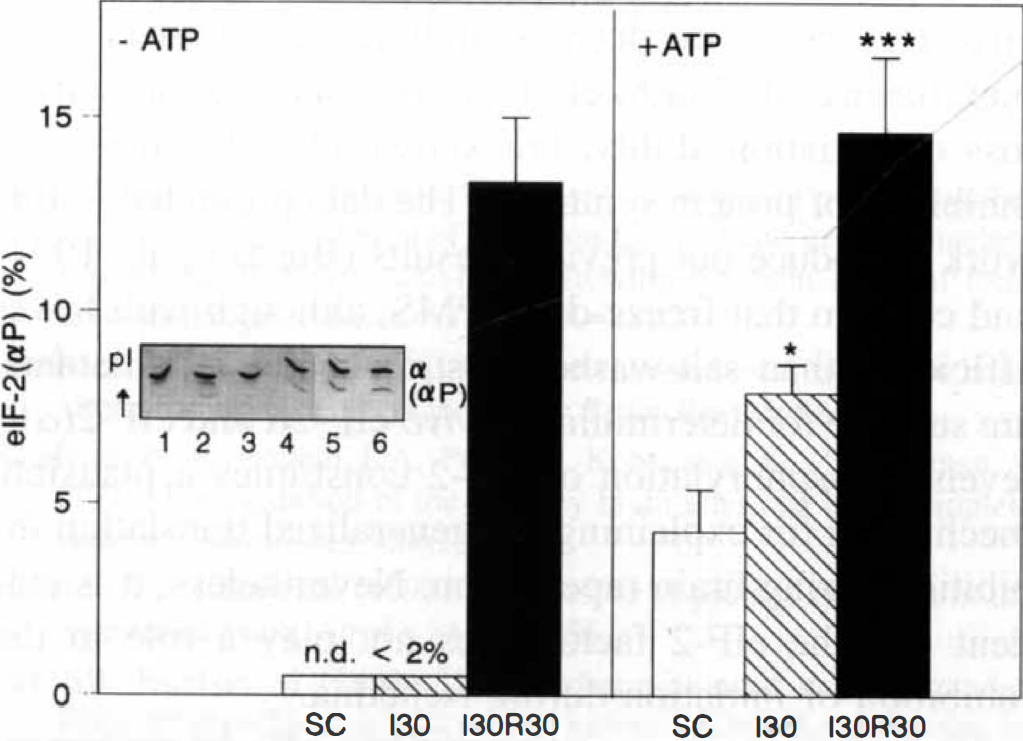
To ascertain whether the different levels of eIF-2(αP) in ischemia and reperfusion are reflecting changes in eIF-2α kinase activity, PMS fractions were incubated in the presence of ATP/Mg2+, and endogenous eIF-2 phosphorylation in vitro, was analyzed by IEF electrophoresis and protein immunoblot analysis. As shown in Figure 2, in the presence of ATP/Mg2+, the amount of eIF-2(αP) clearly increased in all samples indicating the presence of an eIF-2α kinase activity. This activity was significantly higher in intra-ischemic samples than in controls.
Changes in the levels and phosphorylation status of eIF-4E induced by ischemia/reperfusion. Freeze-dried PMS (up to 75 μg protein) from sham-controls and ischemic animals (four-vessel occlusion model) were subjected to IEF electrophoresis (0.75-mm thick, 5% acrylamide, 0.125% N,N′-methylenebisacrilamide, 8.5 mol/L urea, and 2% ampholines at pH 4–6) and analyzed by protein immunoblot as described in Experimental Procedures. The results represent the average ± SEM from five different animals. (Abbreviations: SC, Sham-control; I30, 30-minute ischemia; 130R30, 30-minute ischemia, 30-minute reperfusion.) Statistical significance between sham-control and ischemic animals, ***P < .001, *P < .05. Insert: Results of a typical experiment: Lane 1 and 5, SC; lane 2 and 6, I30; lane 3 and 7, I30R30; lane 4, eIF-4E partially purified from reticulocyte. Keys: eIF-4E, unphosphorylated eIF-4E; eIF-4E(P), phosphorylated eIF-4E. () percentage of phosphorylated eIF-4E. Changes in eIF-2α kinase activity induced by ischemia/reperfusion. The eIF-2α kinase activity was measured by incubating PMS fractions (200 μg) from sham-control and ischemic rats (four-vessel occlusion model) in the absence (lanes 1, 3, and 5) or the presence (lanes 2, 4, and 6) of 1 mmol/L ATP and 4 mmol/L magnesium acetate, for 15 minutes at 37°C. Aliquots of incubation mixtures (75 μg protein) were resolved in IEF gels (0.75-mm thick, 5% acrylamide, 0.125% N,N′-methylenebisacrilamide, 8,5 mol/L urea and 2% ampholines at pH 4–6) and analyzed by protein immunoblot. eIF-2α kinase activity is expressed as the percentage of eIF-2(αP) after the incubation with 1 mmol/L ATP. (Abbreviations: SC, Sham-control; I30: 30-minute ischemia; I30R30, 30-minute ischemia, 30-minute reperfusion.) Values represent the average ± SEM from five different animals. Statistical significance between sham-control and ischemic animals, ***P < .001, *P < .05. Insert: Results of a typical experiment: Lanes 1, 2 SC; lanes 3, 4:130; lanes 5, 6:130R30; Keys: α, unphosphorylated eIF-2; (αP), phosphorylated eIF-2α. n.d: not detectable levels.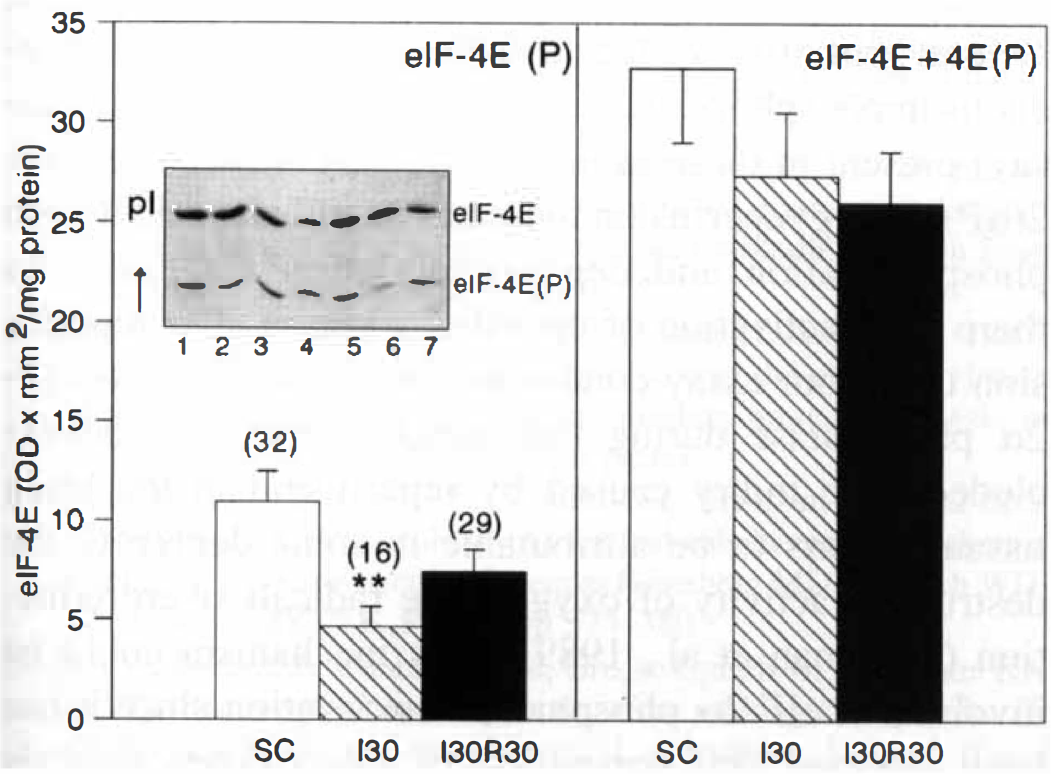
The levels of PKR, the eIF-2α kinase present in all tissues, were determined in rat brain lysates from sham-control and ischemic rats. PKR exhibits different electrophoretic mobility (60 to 68 kd) which likely results from its species origin and phosphorylation status (Hovanessian, 1989). In the rat brain lysates, the anti-PKR antibody recognized a majority band which migrated at 69 to 70 kd, exactly at the same position as PKR purified from rat liver (Fig. 3). Under our conditions, PKR purified from rabbit reticulocyte and recombinant human PKR migrated at 67 and 65 kd, respectively (not shown). PKR levels obtained by densitometric analysis of the bands were slightly higher in ischemic samples than in controls (Fig. 3).
PKR levels in sham-control and ischemic rats. Tissue lysates (up to 75 μg protein) prepared from sham-control and ischemic animals (four-vessel occlusion model) as described in Experimental Procedures, were subjected to SDS-PAGE electrophoresis and analyzed by protein immunoblot. (Abbreviations: SC, Sham-control; 130, 30-minute ischemia; I30R30, 30-minute ischemia, 30-minute reperfusion.) Values represent the average ± SEM from five different animals. Statistical significance between sham-control and ischemic animals, *P < .05 Insert: Lane 1, SC lane 2, I30; lane 3, I30R30; lane 4, PKR purified from rat liver.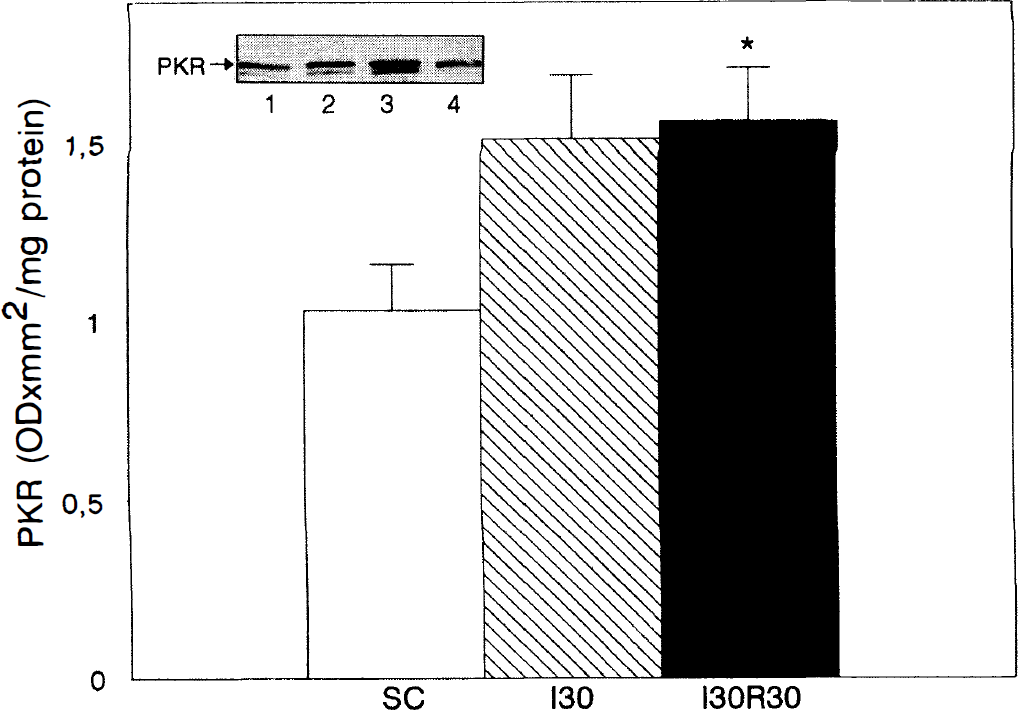
DISCUSSION
The cell-free system used in our study has been validated for the brain and the liver and accurately reproduces changes that occur in vivo (Martín et al., 1993; Burda et al., 1994; García et al., 1994). By using this method we clearly show that the degree of intra-ischemic disturbance in protein synthesis machinery strongly depends on the duration and temperature and less on residual blood flow. Residual blood flow gave rise to a slightly deeper deficit in brain protein synthesis activity when compared with postdecapitative normothermic ischemia. These differences had already been described and it is assumed that they are due to lactate accumulation under residual flow (Siesjo, 1992). Because the inhibition of translation in normothermic ischemia increases over time, it could be thought that this increase is related to incomplete ischemia of cortex because the Pulsinelli model and modifications there of do not guarantee complete ischemia. However, besides energy shortage and homeostasis failure, there might be other alterations produced during ischemia affecting protein synthesis. In fact, the protective effect of hypothermia on translation inhibition after postdecapitative ischemia, suggests the presence of injury mechanism/s sensitive to temperature.
Two facts could explain previous results showing preservation of translational activity during ischemia: (1) the different models of ischemia used, but more importantly, (2) the characteristics of the extracts used for in vitro incorporation, isolated polyribosomes versus microsomal or PMS fractions. Furthermore, it has been shown that the presence of chloride ions in buffers used for obtaining cell-free extracts inhibit the initiation step (Weber, et al., 1977), therefore, with these extracts, polysome run-off is measured instead of reinitiation, canceling possible differences between control and ischemic extracts (Cooper et al., 1977; Burda et al., 1980; Fando and Wasterlain, 1980).
It is generally accepted that protein synthesis is inhibited during the period of recirculation that follows transient cerebral ischemia. The precise mechanism of this inhibition, which resides in the block of initiation (Cooper et al., 1977; Hu and Wieloch, 1993), has been explained by demonstrating of the presence of eIF-2(αP) in vivo. This causes the decrease in binary and ternary complex formation (Burda et al., 1994) and consequently a loss of initiation ability, breakdown of polysomes, and inhibition of protein synthesis. The data presented in this work reproduce our previous results (Burda et al., 1994) and confirm that freeze-dried PMS, although with lower efficiency than salt-washed postribosomal supernatants, are suitable for determining in vivo eIF-2α and eIF-2(αP) levels. Phosphorylation of eIF-2 constitutes a plausible mechanism for explaining the generalized translation inhibition during brain reperfusion. Nevertheless, it is evident that the eIF-2 factor does not play a role in the inhibition of initiation during ischemia.
Under our experimental conditions, eIF-4E phosphorylation significantly decreased during the intra-ischemic period and experienced complete recovery after 30-minute reperfusion. Unchanged eIF-4E phosphorylation has been reported with a model of global brain ischemia and reperfusion by cardiac arrest and resuscitation (DeGracia et al., 1996). However, this study is based on immunoblots of SDS-PAGE –analyzed proteins and the phosphorylated/unphosphorylated forms of eIF-4E were not separated. In our study we also found a slight decrease in total eIF-4E levels associated with ischemia and reperfusion. Neumar et al. (1995) have reported lower levels than ours (57%), after 20-minute decapitation ischemia, nevertheless, DeGracia et al. (1996) did not reproduce their results with a different model of ischemia. Increased eIF-4E phosphorylation has frequently been observed when quiescent or dormant cells were stimulated by appropriate hormones, growth factors, or mitogens; conversely, decreased phosphorylation was seen in some states where translation is inhibited as in heat shock (Sonenberg, 1994; Flynn and Proud, 1996). It is not clear, however, which is the physiologic kinase of eIF-4E, protein kinase C being a possible candidate. Inhibition of protein kinase C has been reported during transient cerebral ischemia (Crumrine et al., 1990), which could be accountable for the decreased eIF-4E phosphorylation.
Although the phosphorylated eIF-2α levels are almost undetectable in vivo both in controls and ischemic samples, the activity of the eIF-2α kinase assayed in vitro in the presence of ATP/Mg2+ is, surprisingly, higher during ischemia. The depletion of ATP stores during ischemia may explain the apparent failure of the kinase to phosphorylate eIF-2α in vivo and, consequently, we cannot explain the inhibition of translation during this period by eIF-2α phosphorylation. It is probable, however, that the activated eIF-2α kinase could be responsible for eIF-2α phosphorylation during early reperfusion. If this increased activity corresponds to PKR, we can consider modulation of its activity and/or levels which we found slightly higher in ischemic animals than in controls.
After 30-minute reperfusion, very limited activity of the eIF-2α kinase was observed that could be explained, at least partially, by the lower concentration of endogenous unphosphorylated eIF-2α (nonsaturating in the assay) present in these samples. The accumulation of eIF-2(αP) during reperfusion indicates an imbalance between phosphorylation and dephosphorylation reactions. As there is no activation of the eIF-2α kinase after reperfusion under our assay conditions, inactivation of the eIF-2α phosphatase during that period should not be excluded. The injury caused by reperfusion in the brain tissue appears to be attributable in some degree to the destructive activity of oxygen-free radicals overproduction (Beckman et al., 1989). This mechanism could be involved in eIF-2α phosphatase inactivation since it has been reported that prevention of free oxygen radicals formation by graded reoxygenation or short-term hypoperfusion leads to a recovery in protein synthesis inhibition (Burda et al., 1991, 1995).
Two well-known eIF-2α kinases have been described in mammalian cells, the haemin controlled inhibitor and PKR and for the moment we cannot exclude the participation of either or even of other/s still unidentified kinases (Samuel, 1993, DeHaro et al., 1996) in the regulation of eIF-2 phosphorylation during ischemia and reperfusion. Putative positive modulators for these two kinases could be (1) heat-shock proteins, which interact with HCl and are induced after a wide range of injurious stresses including ischemia (Abe and Nowak, 1996), or (2) calcium ions which are known to accumulate to abnormally high levels in ischemic neurons before degeneration (Siesjo and Bengtsson, 1989; Krause and Tiffany, 1993) and can modulate PKR activity (Prosko et al., 1995; Alcázar et al., 1997).
Considering our results, we can conclude that high-energy phosphate depletion during ischemia cannot alone account for observed protein synthesis inhibition. Although ribosomes can tolerate prolonged periods of ischemia (Krause and Tiffany, 1993), as already suggested, some other components of the translation machinery might be altered (Morimoto and Yanagihara, 1981). These alterations could even contribute, to some extent, to the inhibition of translation during reperfusion. The modulation of eIF-4E phosphorylation could be implicated in the regulation of translation during ischemia. On the contrary, phosphorylation of eIF-2α, by an eIF-2α kinase already activated during ischemia, represents a plausible mechanism for explaining translation inhibition during reperfusion.