
Abstract
The complement anaphylatoxin C3a contributes to injury after cerebral ischemia in mice. This study assesses the effect of C3a receptor antagonist (C3aRA) on leukocyte infiltration into the ischemic zone. Transient or permanent middle cerebral artery occlusion (MCAO) was induced in wild-type C57BI/6 mice. Intraperitoneal C3aRA or vehicle was administered 45 mins before or 1 h after occlusion. Twenty-four hours after occlusion, we harvested brain tissue and purified inflammatory cells using flow cytometry. Soluble intercellular adhesion molecule (ICAM)-1 protein levels were assessed using enzyme-linked immunosorbent assays, and ICAM-1 and C3a receptor (C3aR) expression was confirmed via immunohistochemistry. In the transient MCAO model, animals receiving C3aRA showed smaller strokes, less upregulation of C3aR-positive granulocytes, and less ICAM-1 protein on endothelial cells than vehicle-treated animals; no significant differences in other inflammatory cell populations were observed. C3a receptor antagonist-treated and vehicle-treated animals showed no differences in stroke volume or inflammatory cell populations after permanent MCAO. These data suggest that blocking the binding of C3a to C3aR modulates tissue injury in reperfused stroke by inhibiting the recruitment of neutrophils to the ischemic zone. It further establishes antagonism of the C3a anaphylatoxin as a promising strategy for ameliorating injury after ischemia/reperfusion.
Introduction
Stroke is the third leading cause of death and the leading cause of disability in the industrialized world (Dirnagl et al, 1999). Existing clinical strategies involving pharmacologic or mechanical reperfusion, however, are successful in improving outcome in only a minority of patients (tissue plasminogen activator for acute ischemic stroke; The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group, 1995). This lack of success can be attributed, in part, to the complications of reperfusion involving edema, hemorrhage, and the formation of toxic oxygen free radicals (Kuroda and Siesjo, 1997). Pharmacologic strategies that preserve flow in ischemic but viable penumbral tissue are, therefore, needed to extend the therapeutic window of thrombolysis. Among numerous approaches, the inhibition of the complement cascade's proinflammatory effect in the acute poststroke period has recently become a focus of neuroprotective investigation.
Previous studies have shown that inhibition of the complement cascade reduces infarct volumes in animal stroke models (De Simoni et al, 2004; Vasthare et al, 1998). Much of this initial work employed pharmacologic complement inhibition using cobra venom factor, C1-esterase inhibitor, and soluble complement receptor-1. However, the lack of specificity of these agents left it unclear as to which complement components are most relevant in the pathogenesis of cerebral ischemia/reperfusion injury. Our group recently investigated the specific contribution of C1q, C3, and C5 to stroke pathology using genetic knockout strategies and showed that only C3 deficiency affords neuroprotection in mice (Mocco et al, 2006). This study suggested that C3 is the critical effector of complement-mediated ischemic neurotoxicity and showed a neuroprotective effect for the small-molecule inhibitor of C3a receptor (C3aR), SB 290157, also known as C3a receptor antagonist (C3aRA). A parallel study by another group showed a neuroprotective effect for the targeted inhibition of C3 convertase using intravenous CR2-Crry. This latter study showed that administration of CR2-Crry leads to a reduction of C3 deposition, neutrophil influx, and microthrombus formation in reperfused stroke (Atkinson et al, 2006).
Despite this encouraging work, the mechanism whereby inhibition of C3 exerts its neuroprotective effects remains unclear. We sought, therefore, to further investigate the mechanism of C3aRA as a way to assess the functional significance of C3a-C3aRA binding in the context of stroke pathology. Our previous work showed that C3aRA-treated mice exhibit a reduction in damage because of oxidative stress, as assessed by malondialdehyde assay (Mocco et al, 2006). The mechanism of this diminished oxidative damage, however, remains elusive. It also remains unclear whether the beneficial effects of C3aR antagonism are reperfusion dependent. In this study, we sought to further examine the mechanism of C3aR-mediated neuroprotection by characterizing inflammatory cell profile in ischemic brain tissue of animals treated with C3aRA. We hypothesized that blockade of C3a-C3aR interactions would attenuate neutrophil infiltration and reduce infarct volume in a murine model of reperfused stroke. Furthermore, we expected that this protective effect would be dependent on reperfusion in this model.
Materials and methods
Mice
The Columbia University Institutional Animal Care and Use Committee approved all the experiments presented in this manuscript. Adult male C57Bl/6 mice (8 to 12 weeks of age, 23 to 26 g) were obtained from Jackson Laboratories (Bar Harbor, ME, USA). They were housed in certified barrier facilities in microisolator cages with free access to food and water on a 12-h-light/12-h-dark cycle. C3a receptor antagonist (SB290157) purchased from Calbio-chem (Darmstadt, Germany), or an equal volume of vehicle (10% ethanol in water), was administered via intraperitoneal injection (1 mg/kg) in a masked fashion 45 mins before, or 1 h after, ischemic onset; we used a dosing strategy similar to those used in earlier studies (Ames et al, 2001; Mocco et al, 2006; Proctor et al, 2004).
Murine Model
Mice were subjected to middle cerebral artery occlusion (MCAO) as described previously (Connolly et al, 1996b; Li et al, 2005). Mice were anesthetized with 1.5% isoflurane delivered in a 70% N2O/30% O2 gas mixture. Middle cerebral artery occlusion was achieved by advancing a heat-blunted, silicon-coated 7-0 nylon monofilament from the common carotid artery to the middle cerebral artery origin. For the transient MCAO model, reperfusion was achieved by withdrawing the filament after 60 mins of ischemia. For the permanent MCAO model, the suture was permanently ligated around the common carotid artery. In the sham surgery cohorts, the occluding suture was inserted into the vessel and immediately withdrawn. For all animals, transcranial cerebral blood flow was measured using laser-Doppler flowmetry (Periflux System 5000; Perimed, Stockholm, Sweden) (Connolly et al, 1996a). Stroked animals were only included if occlusion caused a decrease in blood flow to 30% of the original blood flow, and reperfusion, when applicable, caused a doubling in Doppler blood flow. In addition, core temperature was monitored and held constant at 37.0° C ± 0.1° C during surgery using a Digi-Sense temperature controller (Cole-Parmer, Vernon Hills, IL, USA) via heat lamp coupled with a thermistor rectal probe model 400 (Yellow Springs Instruments Co. Inc., Yellow Springs, OH, USA). After surgery, an animal intensive care unit (Lyon Electric Company, Chula Vista, CA, USA) was used along with a Thermistemp temperature controller (Yellow Springs Instruments Co. Inc., Yellow Springs, OH, USA). This setup uses a digital thermometer and heating element to maintain a temperature of 37°C for 150 mins after induction of ischemia in both transient and permanent surgical models.
Physiologic Parameters
A separate cohort of animals was used to measure physiologic parameters (mean arterial blood pressure, arterial blood gases, and pH) 45 mins after injection of either C3aRA (n = 9) or vehicle (n = 10). The right common carotid artery was catheterized with PE-10 polyethylene tubing and connected to a blood pressure transducer (Stoelting, Wood Dale, IL, USA) for monitoring mean arterial blood pressure. Blood sample (0.1 mL) was obtained from the catheterized artery and measured for partial pressure of oxygen, partial pressure of carbon dioxide, oxygen saturation, and pH using an iSTAT portable clinical analyzer (East Windsor, NJ, USA).
Infarct Volume Assessment
Twenty-four hours after occlusion, C3aRA-treated (transient MCAO, n = 10; permanent MCAO, n = 9) and vehicle-treated mice dosed 45 mins before ischemia onset (transient MCAO, n = 12; permanent MCAO, n = 8) were euthanized, and 1 mm sections of brain were stained with 2,3,5-triphenyltetrazolium chloride. Staining with 2,3,5-triphenyltetrazolium chloride has been shown to be a highly reliable method for determining infarct volumes at 24 h after occlusion (Li et al, 1997). Infarct volumes were expressed as percentage of ipsilateral hemisphere occupied by infarct, as described previously (Connolly et al, 1996a). This same approach was used to calculate infarct volumes in mice treated 1h after ischemia (vehicle-treated, n = 14; C3aRA-treated, n = 11).
Preparation of Brains
Both permanent and transient MCAO model cohorts dosed 45 mins before stroke onset were analyzed for infiltrating inflammatory cells using flow cytometry, and 24 h after stroke onset, the mice were euthanized. After transcardiac perfusion with phosphate-buffered saline (PBS), brains were harvested, divided into ipsilateral and contralateral hemispheres, and minced in RPMI (Invitrogen, Carlsbad, CA, USA) containing 10% FBS (fetal bovine serum; Invitrogen, Carlsbad, CA, USA). The resulting suspension was passed through a microfilter (70 μm), pelleted, resuspended in 30% Percoll (Amersham, Piscataway, NJ, USA), and centrifuged at 27,000g for 30 mins. After centrifugation, the myelin layer was discarded and the remaining suspension was washed with Dulbecco's PBS containing 1% FBS.
Flow Cytometric Analysis
Antibodies were selected to isolate granulocytes, similar to a scheme described previously (Stevens et al, 2002). All antibodies used for flow cytometry were rat anti-mouse monoclonal antibodies (BD Pharmingen, Franklin Lakes, NJ, USA): fluorescein isothiocyanate (FITC)-conjugated anti-CD11b, R-phycoerythrin (PE)-conjugated anti-CD45, and PerCP-Cy5.5 (Peridinin chlorophyll protein conjugated with cyanine dye Cy5.5)-conjugated anti-Ly-6G/C. Antibodies were diluted in Dulbecco's PBS containing 1% FBS. Cell extracts were incubated simultaneously with the three antibodies and Fc block (anti-CD16/CD32) for 30 mins. Flow cytometric analysis was performed using a FACSCalibur (BD Biosciences, Franklin Lakes, NJ, USA), and the data were analyzed using Flowjo software (Tree Star, Ashland, OR, USA).
An antibody to CD45, a cell-surface marker expressed by all leukocytes as well as microglia, was used to exclude all other cell types. The CD11b marker, expressed by all non-lymphocyte leukocytes, was used to distinguish leukocytes from other cell types. Finally, the Ly-6G/C marker, expressed primarily by granulocytes and lymphocytes (Nagendra and Schlueter, 2004), was used to identify granulocytes. Using these three markers, we were able to separate the CD45-positive cells into the following three general subcategories: (1) lymphocytes (CD45-positive, CD11b negative, Ly-6G/C-positive), (2) microglia (CD45-positive, CD11b-positive, Ly-6G/C-negative), and (3) granulocytes (CD45-positive, CD11b-positive, Ly-6G/C-positive).
Each hemisphere was analyzed for leukocyte markers 24 h after the onset of ischemia for both permanent (C3aRA-treated, n = 9; vehicle-treated, n = 9) and transient (C3aRA-treated, n = 11; vehicle-treated, n = 9) ischemic MCAO models using flow cytometry. First, all non-leukocyte, non-microglial cells were eliminated from analysis by selecting CD45-positive cells. Then, the number of cells that expressed each leukocyte marker was compared with the percentage of the total CD45-positive cells to obtain the percentage concentration of each cell subtype, as described previously (Stevens et al, 2002). Sham-operated animals showed no differences in CD45-positive cell populations when comparing the ischemic and nonischemic hemispheres or across treatment groups (data not shown). Thus, we used the nonischemic (i.e., contralateral) hemisphere as a control for each animal, and normalized the expression of each marker in the ischemic hemisphere by comparing it with the expression level in the nonischemic hemisphere.
Enzyme-Linked Immunosorbent Assay
Adhesion molecules are intimately associated with the recruitment of inflammatory cells to the ischemic brain, and soluble intercellular adhesion molecule (ICAM)-1 is one of the most important adhesion molecules involved in leukocyte recruitment (Danton and Dietrich, 2003). We, therefore, sought to identify changes in ICAM-1 expression in the reperfused model to explore a possible mechanism for the observed change in leukocyte profile in the C3aRA-treated animals.
C3a receptor antagonist-treated (n = 13) and vehicle-treated mice (n = 10) were deeply anesthetized and perfused intracardially with 0.9% NaCl 24 h after reperfusion. Each hemisphere was homogenized and incubated in ice-cold lysis buffer (10mmol/L Tris, pH 8.0, 150 mmol/L NaCl, 1% Triton X-100, 1 mmol/L EDTA, and 5 μL/mL of protease inhibitor cocktail (Sigma P-8340, Sigma-Aldrich, St Louis, MO, USA)) for 1 h at 4°C. After 10 mins centrifugation at 2,000g, the supernatant was collected and aliquoted for enzyme-linked immunosorbent assay (ELISA), and the protein concentration was measured (Bio-Rad, Hercules, CA, USA). The levels of soluble ICAM-1 in each hemisphere were measured using an ELISA kit (Quantikine, R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions.
Immunohistochemistry
For all immunohistochemical analyses, mice were subjected to transient MCAO 45 mins after C3aRA or vehicle administration and were euthanized after 24 h. After transcardiac PBS perfusion, brains were rapidly harvested, fixed in 4% paraformaldehyde, and cryo-protected. Brains were frozen in optimal cutting temperature compound (Sakura Finetek US, Torrance, CA, USA), kept at −80°C, and cut into coronal sections of 20 μm using a cryostat. For immunofluorescent staining, sections were blocked with the secondary antibody—appropriate serum (10% donkey or goat serum) with PBS containing 0.2% Triton X-100 for 30 mins at room temperature. Sections were incubated in primary antibodies diluted in PBS containing 0.2% Triton X-100 overnight at 4°C. Sections were then washed and incubated with secondary antibodies for 1 h at room temperature. Nissl was used for counterstaining. Sections were then mounted on slides in Vectashield (Vector Laboratories, Burlingame, CA, USA) and visualized using a Bio-Rad 2000 confocal laser-scanning device (Bio-Rad, Hercules, CA, USA) with a Nikon E800 microscope (Nikon, San Diego, CA, USA).
Primary antibodies and reagents used for immunohistochemistry include rat anti-mouse monoclonal Ly-6G/C antibody (BD-Pharmingen, San Diego, CA, USA), 1:50 dilution; goat polyclonal anti-ICAM-1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), 1:100 dilution; mouse monoclonal anti-von Willebrand factor (vWF) antibody (Novocastra Laboratories, Newcastle, UK), 1:100 dilution; chicken monoclonal anti-mouse C3aR antibody (Accurate Chemical, Westbury, NY, USA), 1:100 dilution; and Neurotrace 640/660 fluorescent Nissl (Molecular Probes, Eugene, OR, USA). Secondary antibodies included Alexa Fluor 488 or 594-conjugated anti-mouse IgG, anti-rat IgG, anti-chicken IgG, and anti-goat IgG (Invitrogen, Carlsbad, CA, USA).
Intercellular Adhesion Molecule-1 Expression
The spatial expression of ICAM-1 in the ischemic hemisphere was assessed using immunohistochemistry. Vehicle-treated mice were subjected to transient MCAO as described above and were euthanized after 24 h. Tissue sections were prepared as detailed above using anti-vWF and anti-ICAM-1 primary antibodies.
C3a Receptor Expression
The number of C3aR-positive granulocytes was assessed using semiquantitative immunohistochemistry as previously described (Mocco et al, 2006). C3a receptor antagonist-treated (n = 3) and vehicle-treated mice (n = 3) were subjected to transient MCAO as described above and were euthanized after 24 h. Tissue sections were prepared as detailed above using anti-Ly-6G/C and anti-C3aR primary antibodies. An observer masked to the identity of the mice counted cells that were doubly positive for Ly-6G/C and C3aR in five representative medium-power fields (× 40 objective) in each of the four sections obtained from each mouse. The fields were chosen in the ischemic region in an anatomically consistent manner between mice. Results are reported as mean number of cells per field in the ipsilateral hemisphere. No double-positive cells were observed in the contralateral hemispheres of either cohort.
Statistical Analysis
All data are presented as mean ± s.e. Between-group differences were made using two-tailed unpaired Student's t-test. All values are considered to be significant when P < 0.05.
Results
Physiologic Parameters
No significant differences could be detected between C3aRA-treated and vehicle-treated cohorts for mean arterial pressure, pH, and partial pressure of CO2. However, the C3aRA-treated animals showed a significantly elevated partial pressure of O2 (C3aRA, 145 ± 8 mm Hg; vehicle, 117 ± 5 mm Hg; P < 0.05) and oxygen saturation (C3aRA, 99.1% ± 0.2%; vehicle, 97.6% ± 0.9%; P = 0.001) when compared with vehicle-treated animals (Table 1).
Physiological parameters 45 mins after injection
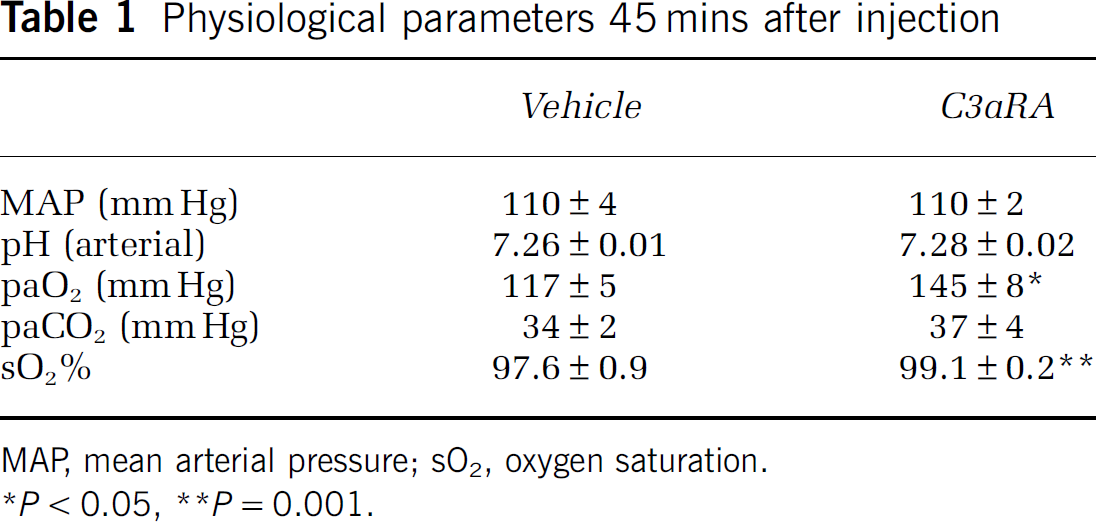
MAP, mean arterial pressure; sO2, oxygen saturation.
P < 0.05
P = 0.001.
Infarct Volume
In the pre-ischemia, treated, transient MCAO model, the C3aRA-treated (n = 12) cohort showed a large decrease in infarct volume relative to the vehicle-treated (n = 10) cohort (C3aRA, 28% ± 4%; vehicle, 48% ± 6%; P = 0.02) (Figure 1). When treated 1 h after ischemia, the C3aRA-treated (n = 11) cohort also showed a statistically significant decrease in infarct volume compared with the vehicle-treated (n = 14) cohort (C3aRA, 28% ± 5%; vehicle, 43% ± 4%; P = 0.03) (Figure 1). Conversely, in the permanent ischemia model, the C3aRA-treated (n = 9) cohort failed to show a difference in infarct volume or stroke distribution when compared with the vehicle-treated (n = 8) cohort (C3aRA, 47% ± 3%; vehicle, 50% ± 6%; P = NS) (Figure 1).
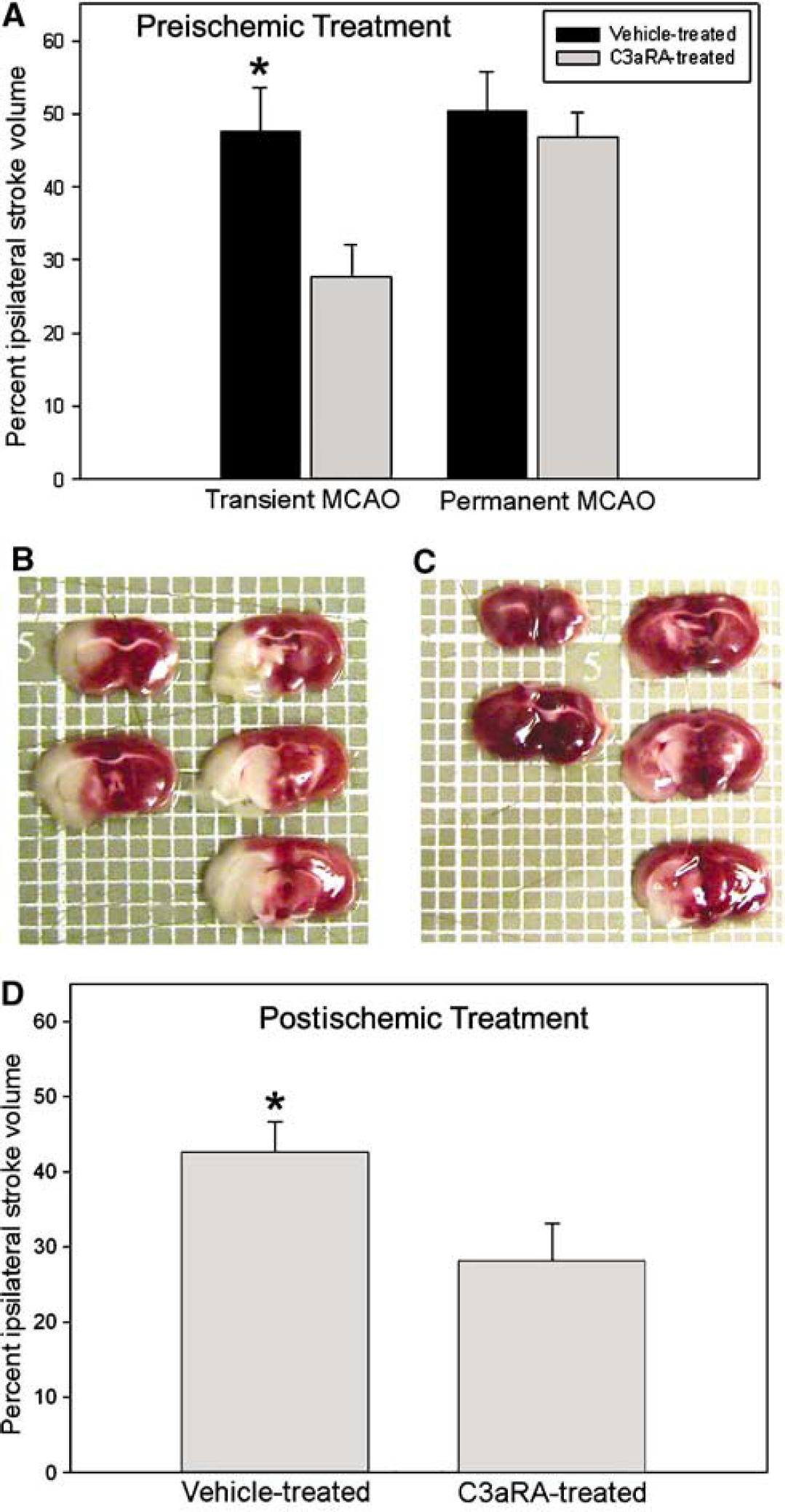
Infarct volume (percent ipsilateral hemisphere) for vehicle- and C3aRA-treated cohorts. When treated 45 mins before ischemia (
In both the pre- and postischemia, treated, transient MCAO experiments, the C3aRA-treated group showed a significant reduction in cortical and overall stroke volume compared with the vehicle-treated group. In the preischemia, treated groups, 56% of the total stroke volume was cortical in those treated with vehicle, whereas only 31% of the stroke volume was cortical in the C3aRA-treated group (P < 0.05). In the postischemia, treated, transient MCAO model, cortical ischemia represented 53% of the total stroke volume in the vehicle-treated group, compared with 35% in the C3aRA-treated group (P < 0.05).
The infarct area measurements were taken for both transient ischemia models in sections +1.8, +0.8, −0.2, −1.2, and −2.2 mm from the bregma. An additional section at −3.2 mm from the bregma was collected in the postischemia, treated group. In the preischemia, vehicle-treated cohort, the area measurements were 30%±7% at +1.8 mm, 40%±7% at +0.8 mm, 51%±6% at −0.2 mm, 53%±6% at −1.2 mm, and 51%±7% at −2.2 mm. In the C3aRA-treated group, the area measurements were 10%±4% at +1.8 mm, 22%±4% at +0.8 mm, 32%±4% at −0.2 mm, 35%±6% at −1.2 mm, and 34%±7% at −2.2 mm. In the postischemia, vehicle-treated group, the area measurements were 41%±5% at +1.8 mm, 56%±3% at +0.8 mm, 50%±4% at −0.2 mm, 53%±6% at −1.2 mm, 43%±7% at −2.2 mm, and 14%±5% at −3.2 mm. In the postischemia, C3aRA-treated cohort, the area measurements were 20%±6% at +1.8 mm, 39%±6% at +0.8 mm, 39%±6% at −0.2 mm, 37%±7% at −1.2 mm, 31%±8% at −2.2 mm, and 4%±4% at −3.2 mm.
Flow Cytometric Analysis
Leukocyte and microglia populations in treated animals of both transient and permanent MCAO models were analyzed in two steps (Figure 2). First, leukocyte/microglia populations were isolated by examining both CD45 and side-scatter profiles (Figures 2A, 2C, 2E, 2G, 2I, and 2K). Then, the CD45-positive cells were plotted for CD11b and Ly-6G/C (Figures 2B, 2D, 2F, 2H, 2J, and 2L). For the animals that underwent sham surgery, no significant differences could be discerned between ischemic and nonischemic hemispheres, or between C3aRA-treated and vehicle-treated cohorts (n = 4/group, data not shown).
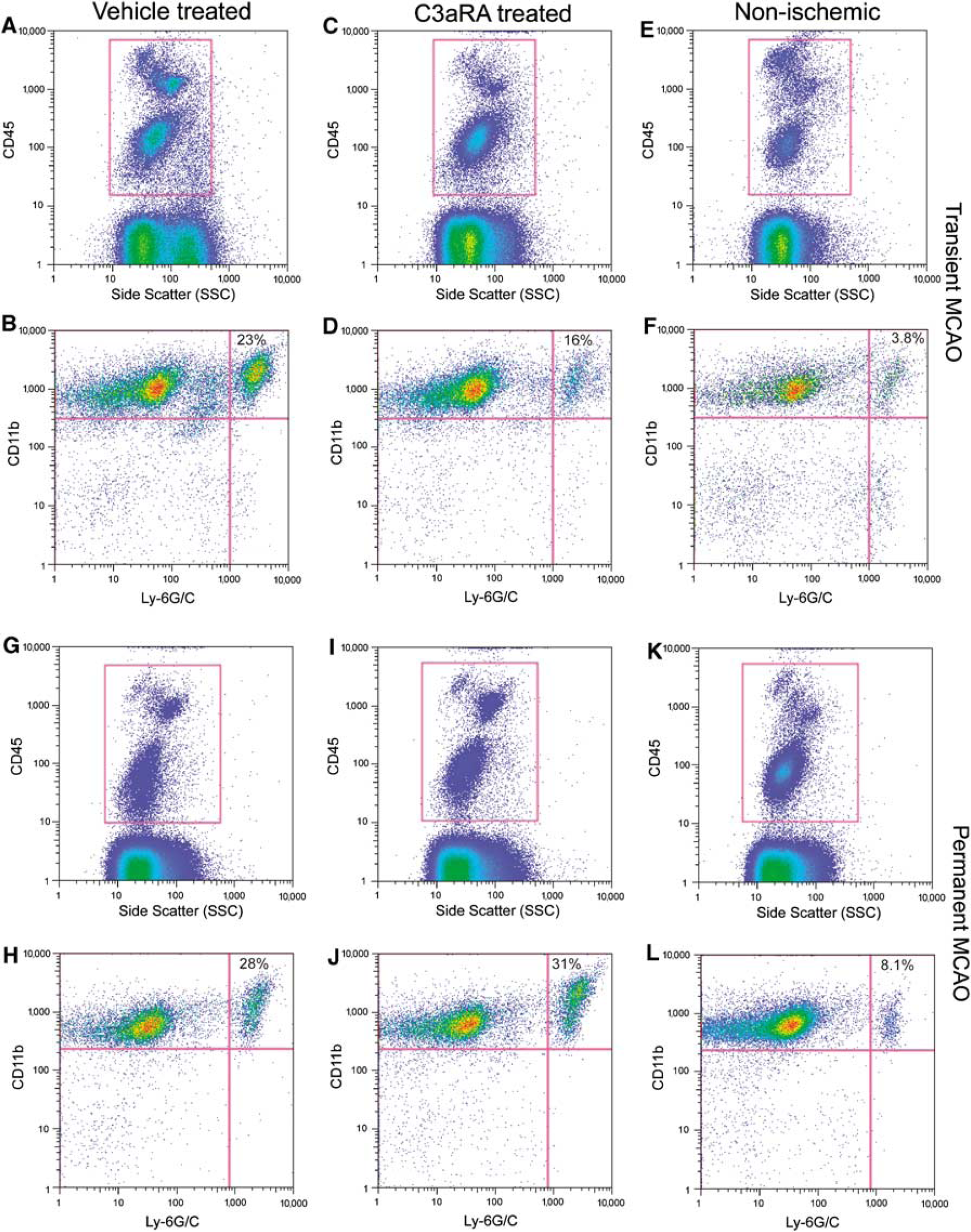
Representative flow cytometric plots of leukocyte profiles in transient MCAO (
Transient middle cerebral artery occlusion: For mice subjected to transient MCAO, both the C3aRA-treated and vehicle-treated cohorts showed a higher percentage of granulocytes in the ipsilateral hemisphere compared with the contralateral nonischemic hemisphere. This ratio of ipsilateral/contralateral granulocyte concentration was, however, significantly smaller in the C3aRA-treated cohort (ischemic, 16%±3.1%; nonischemic, 4.6%±0.8%; ratio ischemic/nonischemic, 3.7±0.6; n = 11) compared with vehicle-treated animals (ischemic, 23%±4.0%; nonischemic, 3.8%±0.7%; ratio ischemic/nonischemic, 6.7±0.8; n = 9; P < 0.01). At the same time, no differences could be discerned for microglia or lymphocyte populations (Figure 3A).
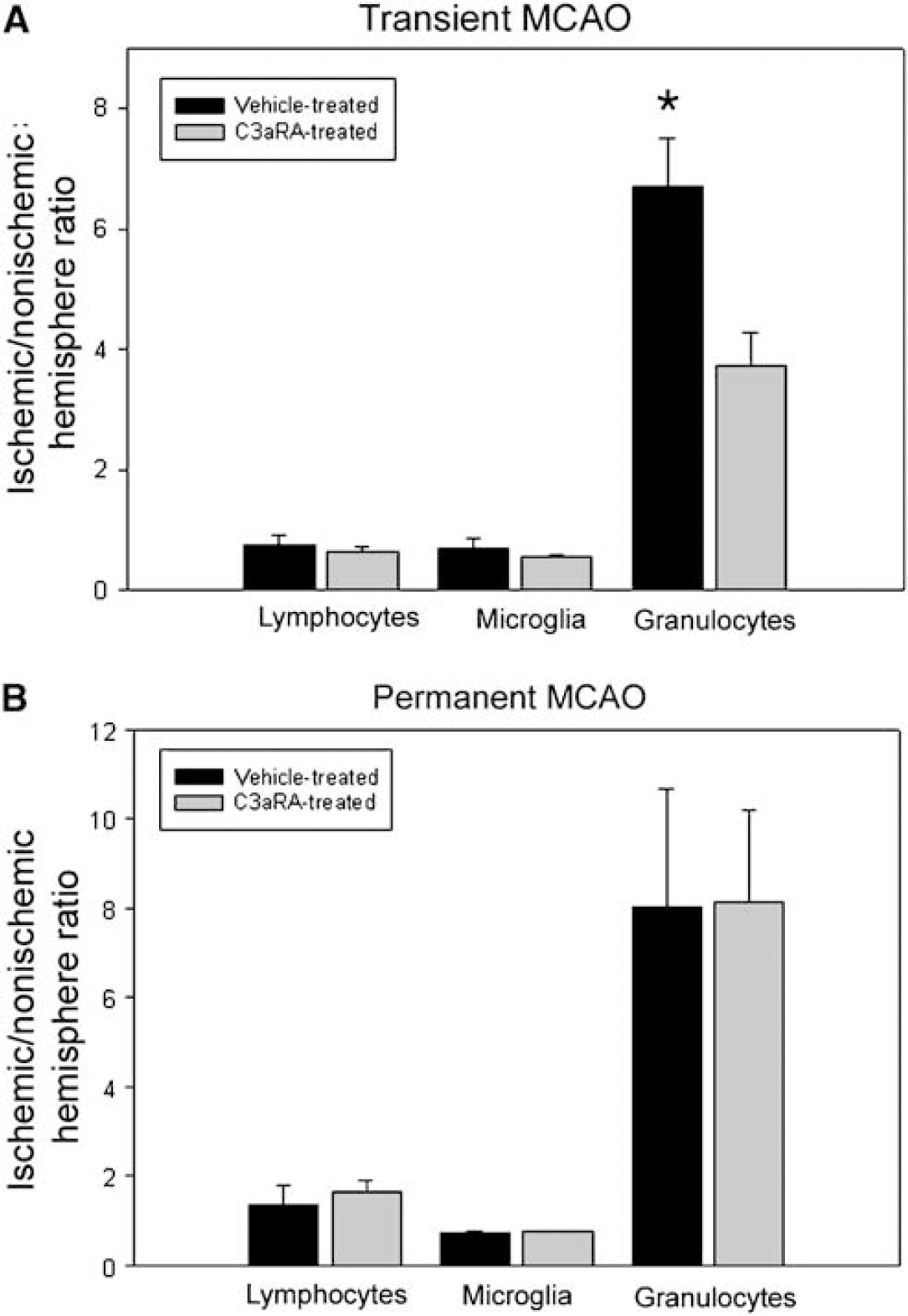
Ischemic versus nonischemic ratio of lymphocytes, microglia, and granulocytes. The raw number of each cell type in the ischemic hemisphere was counted and divided by the total number of CD45-positive cells to calculate the relative concentration of each cell. The cell concentrations of each animal's ischemic hemisphere were then normalized to their own contralateral (nonischemic) hemisphere by calculating the ischemic/nonischemic ratio. Granulocytes were found to be highly enriched in the ischemic hemisphere compared with the nonischemic hemisphere for all experimental groups. On comparing C3aRA- with vehicle-treated animals in the transient MCAO model, C3aRA-treated animals showed a significantly smaller increase in the percentage of granulocytes compared with vehicle-treated animals (
Permanent middle cerebral artery occlusion: For mice subjected to permanent MCAO, again, both the C3aRA-treated and vehicle-treated cohorts showed a higher percentage of granulocytes in the ipsilateral hemisphere relative to the contralateral nonischemic hemisphere. However, the C3aRA-treated group failed to show a reduction in the ipsilateral/contralateral ratio of granulocytes (ischemic, 25%±2.7%; nonischemic, 4.8%±1.3%; ratio ischemic/nonischemic, 8.0±2.6; n = 9) when compared with the vehicle-treated group (ischemic, 29%±5.0%; nonischemic, 7.0%±2.2%; ratio ischemic/nonischemic, 8.1±2.0; n = 9; P=NS) (Figure 3B). In addition, concordant with the findings from the transient ischemia cohort, the other cell subpopulations failed to display any significant differences between C3aRA-treated and vehicle-treated groups for the permanent model (Figure 3B).
C3a Receptor Expression: Transient Middle Cerebral Artery Occlusion
Using immunohistochemistry, we compared C3aR expression in the C3aRA-treated (n = 3) and vehicle-treated (n = 3) groups in transient MCAO cohorts (Figure 4). Costaining of Ly-6G/C and C3aR expression revealed many more cells per field that were positive for both markers in the ischemic hemisphere of the vehicle-treated group compared with the C3aRA-treated group (C3aRA, 0.27±0; vehicle, 1.83±0.27; P<0.01). Conversely, there were no significant differences between the number of granulocytes per field lacking C3aR expression in the vehicle-treated group (1.6±0.2) compared with the C3aRA-treated group (1.4±0.3). No double-positive cells were seen in the nonischemic hemisphere of either group. These data indicated that there was a reduction in granulocytes that expressed C3aR in the C3aRA-treated group.
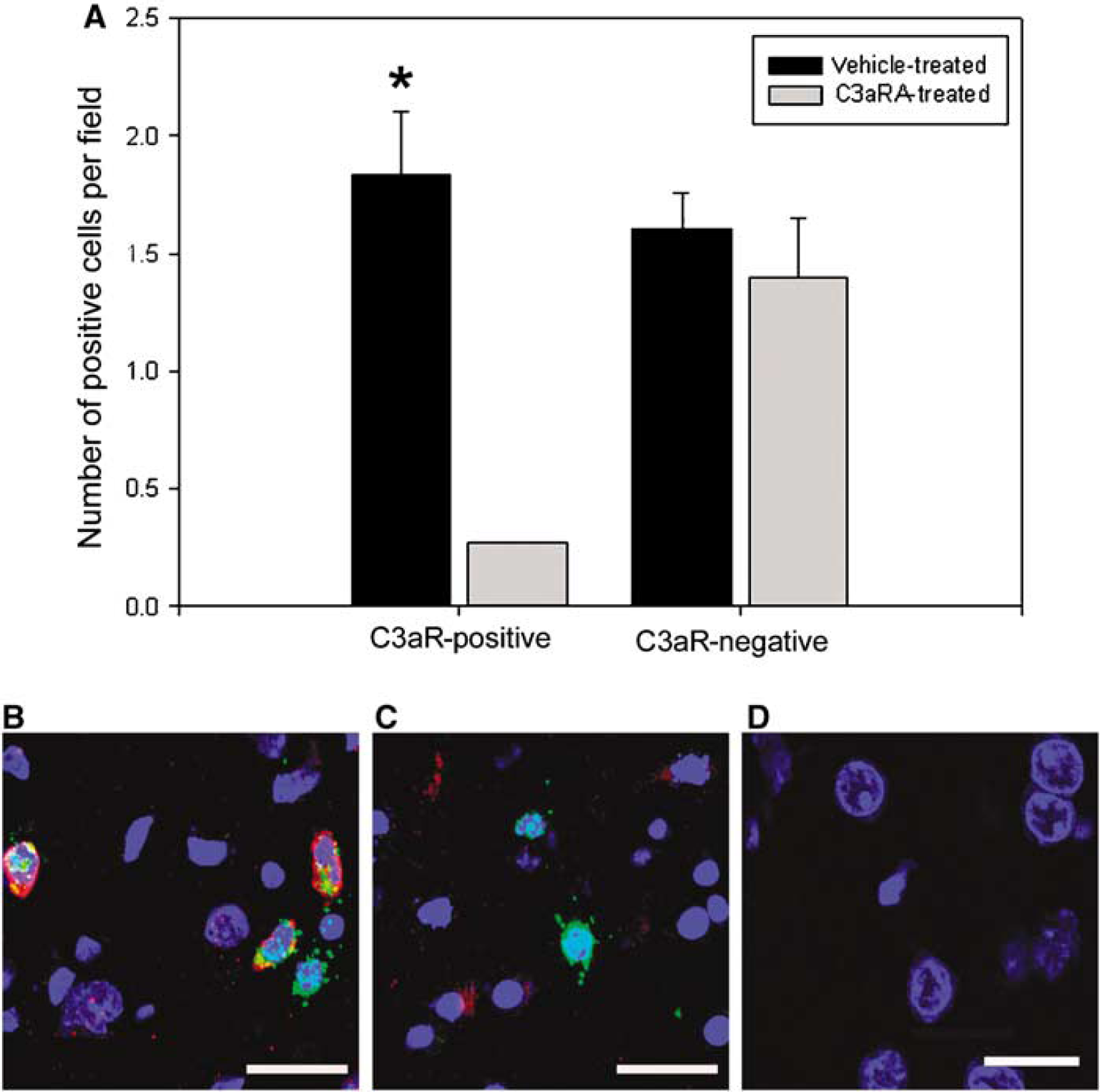
C3a receptor expression on granulocytes after transient MCAO (
Intercellular Adhesion Molecule 1
Using ELISA, ICAM-1 levels were compared in C3aRA-treated and vehicle-treated cohorts of mice subjected to transient MCAO (Figure 5A). The ischemic/nonischemic hemisphere ratio of soluble ICAM-1 was shown to be significantly reduced in the C3aRA-treated group (n = 13; ratio = 1.4±0.1) compared with the vehicle-treated group (n = 10; ratio = 1.7±0.1; P<0.05). Endothelial ICAM-1 expression was confirmed by costaining for ICAM-1 and vWF in a vehicle-treated animal (Figures 5B and 5C). Observable costaining of both markers was exhibited in the ischemic zone but not in the nonischemic areas.
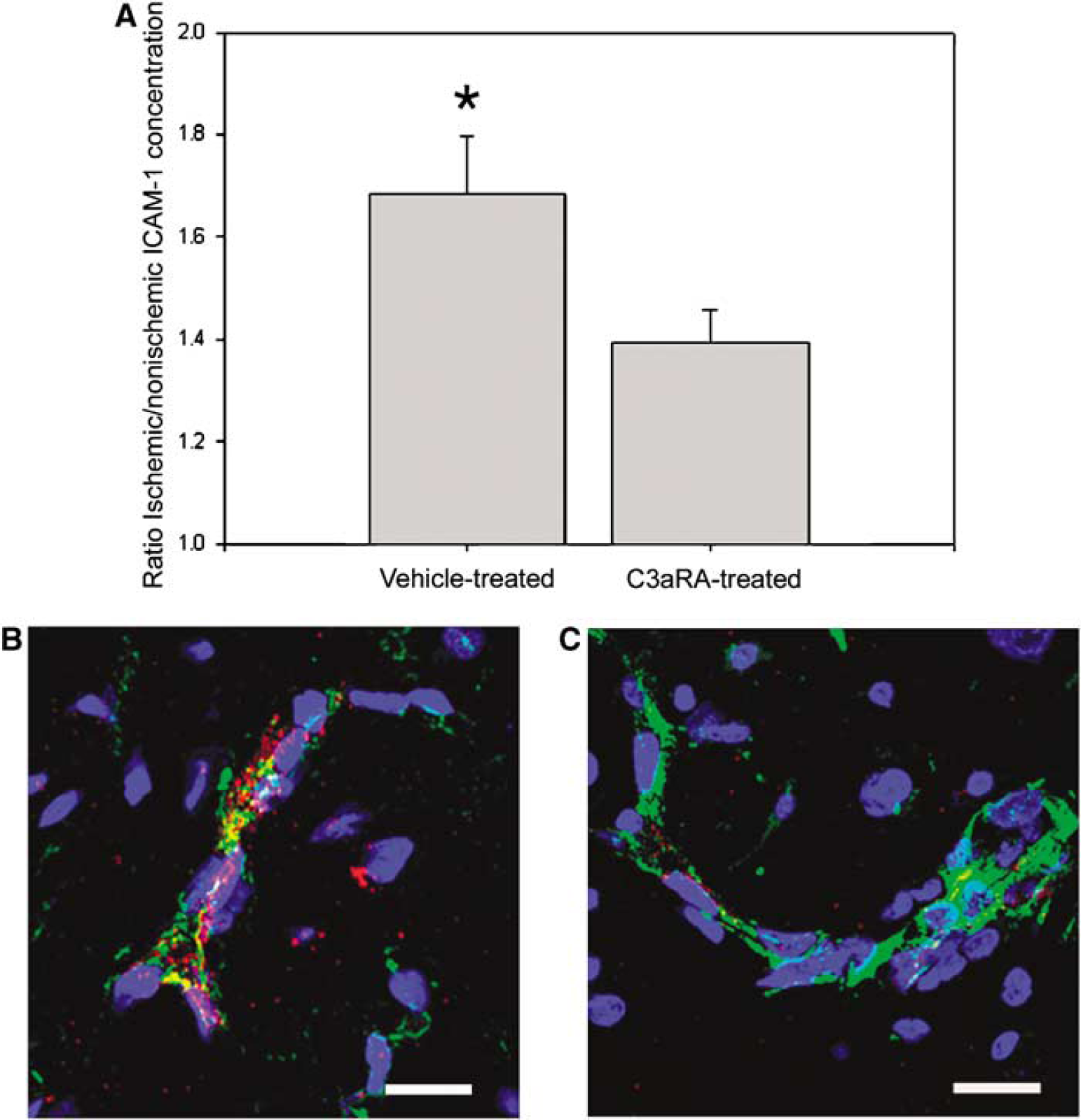
Intercellular adhesion molecule 1 expression after transient MCAO. Intercellular adhesion molecule 1 levels were quantified using ELISA; ICAM levels in each ischemic hemisphere were normalized to the nonischemic hemisphere to control for individual variability. Animals treated with C3aRA showed a significantly smaller increase in ICAM-1 levels in their ischemic hemisphere compared with vehicle-treated animals (
Discussion
Significant evidence has accumulated implicating complement component deposition in cerebral ischemia/reperfusion injury (Cowell et al, 2003; D'Ambrosio et al, 2001), and our recent work has identified C3 as the most influential mediator of ischemic neurologic injury in the complement cascade (Mocco et al, 2006). Inactive C3 becomes activated through its cleavage into the components C3a and C3b through the action of C3 convertase, and both cleavage products may be involved in cerebral ischemia/reperfusion injury (D'Ambrosio et al, 2001).
C3a along with C5a, known collectively as the anaphylatoxins, induces a variety of cellular responses, including Chemotaxis, degranulation, and oxidative burst, all involving granulocytes (Eisner et al, 1994; Soruri et al, 2003). Additionally, C3a is capable of inducing cytokine expression and release of adhesion molecule from endothelial cells, attracting leukocytes, and increasing vascular permeability (Foreman et al, 1996; Mulligan et al, 1997). The C3aR is constitutively expressed in the brain on astrocytes, microglia, neurons, oligodendrocytes, and endothelial cells. In addition, C3aR expression is known to be upregulated in the brain after cerebral ischemia primarily because of the influx of leukocytes (Barnum et al, 2002). Furthermore, C3a has been shown to bind to and increase intracellular calcium levels in human neutrophils (Barnum et al, 2002; Martin et al, 1997; Norgauer et al, 1993).
In this study, we investigated the neuroprotective mechanism of a small-molecule antagonist to C3aR in mice subjected to MCAO. We confirmed that administration of C3aRA before transient MCAO affords robust neuroprotection when assessed at 24 h after occlusion. We further showed that administration of C3aRA before ischemia in the absence of reperfusion does not yield any improvement in outcome. Together, these data suggest that the protective mechanism of C3aRA is dependent on a reperfusion event. We also have data suggesting that the C3aRA treatment preferentially protected the cortical tissue compared with the subcortical tissue in the reperfused MCAO model.
In addition, we compared the cerebral leukocyte/microglia profiles in mice treated with either C3aRA or vehicle before stroke onset. After flow cytometric analysis, C3aRA-treated mice showed a significantly lower ratio of ipsilateral/contralateral granulocyte concentration compared with vehicle-treated mice after ischemia/reperfusion but not after ischemia without reperfusion. These data suggest that C3aRA administration inhibited the influx of neutrophils into the ischemic hemisphere and thus attenuated inflammatory neurotoxicity.
Supporting this finding, using immunohistochemistry, we showed an abundance of granulocytes expressing C3aR in the ischemic hemisphere of the vehicle-treated group, but almost none in the ischemic hemisphere of the C3aRA-treated group. We also found a population of granulocytes lacking C3aR expression in the ischemic hemisphere of the vehicle group that were also found in the ischemic hemisphere of the C3aRA-treated group. Taken together, these data suggest that C3aRA may work by binding to a specific population of neutrophils receptive to C3a and preventing their influx, while at the same time not influencing those cells that do not express the C3aR. These data, suggesting a neutrophil-mediated mechanism, support our previous work, which showed a reduction in malondialdehyde levels in C3aRA-treated mice, indicating a reduction in oxidative damage (Mocco et al, 2006).
We further showed that treatment with C3aRA reduces ICAM-1 levels in the brain after ischemia/reperfusion. Intercellular adhesion molecule 1 is known to modulate neutrophil infiltration by promoting the binding of invading neutrophils to endothelial cells (Danton and Dietrich, 2003). Furthermore, a reduction in ICAM-1 leads to a reduction in neutrophil infiltration after stroke (Danton and Dietrich, 2003; Justicia et al, 2003). Therefore, in addition to stimulation of leukocyte recruitment via direct C3aR binding on granulocytes, C3aR may, in part, affect leukocyte migration through an upregulation of adhesion molecules. However, such a causal relationship has yet to be determined.
In addition, we have identified a novel physiologic consequence of C3aRA administration, as C3aRA-treated animals showed higher levels of oxygen in arterial blood. It is unclear as to what mechanism underlies this finding and whether this effect is generated through the interaction of C3aRA with C3aR. It is possible that this higher circulating oxygen level is contributing to the protective effect of C3aRA, as hyperoxia has been shown to be neuroprotective (Shin et al, 2007). Clearly, further research into the physiologic effects of C3aRA, both long- and short-term, is needed.
This study shows the profound impact that C3aRA can have on inflammatory cell migration in vivo and identifies neutrophil modulation as the likely source of C3aRA-mediated protection. These data are concordant with our group's previous studies, which showed that C3-knockout mice display robust neuroprotection whereas C5-knockout mice show no such effect (Mocco et al, 2006). Although it remains possible that C3a also modulates neutrophils indirectly through other cell types, our data strongly suggest that C3a modulates neutrophil activity directly.
Nevertheless, several questions relating to the role of C3a in stroke pathology remain unanswered. Firstly, although it seems that C3aR affects neutrophil infiltration, there is little evidence addressing how C3 itself becomes activated. In the classical complement cascade, the activation of C1 after antigen-antibody binding triggers the activation of C3. However, C1q-deficient mice show no reduction in C3 activation and no neuroprotection, indicating that C3 activation may occur through other means, such as the alternative and mannose-binding lectin pathways (Mocco et al, 2006). An alternative possibility is that C3 is cleaved by proteases such as thrombin, which is known to be upregulated in ischemic cerebral tissue (Xi et al, 2003).
Also unclear is the effect of C3aRA administration, and other anticomplement strategies, on long-term outcome. In fact, there is some evidence that complement cascade activation after stroke can yield beneficial effects through its role in the facilitation of the removal of apoptotic cellular debris (Fishelson et al, 2001; Mevorach et al, 1998). In addition, C3a has been shown to increase neighboring cells' responsiveness to growth factors, thus promoting neurogenesis, and C3 deficiency has been shown to inhibit neurogenesis (Rahpeymai et al, 2006; Ratajczak et al, 2004). In fact, a recent study suggests that mice deficient in C3 subjected to cerebral ischemia show similarly sized regions of infarction in the chronic stage of stroke recovery (Rahpeymai et al, 2006). Therefore, it is entirely possible that although acute inhibition of C3aR is beneficial, chronic inhibition may actually be detrimental. Further work for examining long-term outcomes in chronic versus acute treatment of C3aRA is critical for delineating the full range of mechanisms of function of C3a in ischemic brain.
We administered two different dosing regimens of C3aRA. The first involved injection immediately before the onset of ischemia. We chose to use a preischemic treatment protocol because administration of C3aRA is known to result in an acute period of neutropenia that largely recovers by 45 mins after injection (Proctor et al, 2004). Because it has not been verified whether this neutropenic effect is mediated by C3aRA-C3aR binding, our preischemic treatment eliminated the potentially confounding effect of this transient neutropenia. To assess the possible clinical relevance of C3aRA, we also compared drug and placebo treatment 1 h after the onset of ischemia. We again found substantial reductions in stroke volume, and in both experiments, C3aRA treatment provided better protection for cortical tissue compared with subcortical tissue. We have shown that C3aRA administration substantially reduces inflammation and cerebral damage after ischemia/reperfusion, which justifies further work to assess the utility of C3aRA and other complement-inhibiting agents in stroke therapy.
Our study highlights the importance of C3a in stroke pathology and elucidates the mechanism of C3a-mediated neurotoxicity by providing a direct link to neutrophil modulation. In addition, these data are significant because they suggest that complement-mediated neuroprotective strategies rely on a significant reperfusion event. This may, in part, explain the historic difficulty in clinical translation of effective preclinical antileukocyte strategies (Krams et al, 2003; Sughrue and Connolly, 2004).