
Abstract
We have previously shown that neuregulin-1 (NRG-1) protects neurons from ischemic brain injury if administered before focal stroke. Here, we examined the therapeutic window and functional recovery after NRG-1 treatment in rats subjected to 90 mins of middle cerebral artery occlusion (MCAO) and 24 h of reperfusion. Neuregulin-1 (2.5 ng/kg bolus, 1.25 ng/kg/min infusion) reduced infarct volume by 89.2% ± 41.9% (mean ± s.d.; n = 8; P < 0.01) if administered immediately after the onset of reperfusion. Neuroprotection was also evident if NRG-1 was administered 4 h (66.4% ± 52.6%; n = 7; P < 0.01) and 12 h (57.0% ± 20.8%; n = 8; P < 0.01) after reperfusion. Neuregulin-1 administration also resulted in a significant improvement of functional neurologic outcome compared with vehicle-treated animals (32.1% ± 5.7%; n 9; P < 0.01). The neuroprotective effect of the single administration of NRG-1 was seen as long as 2 weeks after treatment. Neurons labeled with the neurodegeneration marker dye Fluoro-JadeB were observed after MCAO in the cortex, but the numbers were significantly reduced after NRG-1 treatment. These results indicate that NRG-1 is a potent neuroprotective compound with an extended therapeutic window that has practical therapeutic potential in treating individuals after ischemic brain injury.
Introduction
Ischemic stroke occurs when the arterial blood supply to the brain is obstructed. The ischemic insult results in an early-onset neuronal death that begins within minutes after stroke. This area of brain injury (the infarct core) is characterized by low cerebral blood flow (CBF), energy failure and excitotoxicity (for reviews, see Barone and Feuerstein, 1999; Dirnagl et al, 1999). The resulting ischemic brain injury is also accompanied by increased synthesis of inflammatory molecules and cytokines in neurons, glia and in the cerebral vasculature, which endangers brain cells in a larger, surrounding area of brain tissue for which the blood supply is compromised but not completely interrupted (del Zoppo et al, 2000; Iadecola and Alexander, 2001; Stoll et al, 1998). A population of surviving, yet vulnerable, cells makes up the ischemic penumbra, which surrounds the core of the infarct (Dirnagl et al, 1999; Ferrer and Planas, 2003; Iadecola and Alexander, 2001; Touzani et al, 2001). In these brain regions, neurons can survive for several hours after stroke onset. The formation of the penumbra is a time-dependent situation, where tissues are capable of recovery; however, they may continue to progress towards infarction and subsequently injure the adjacent viable tissue without intervention.
Several studies suggest that there is a therapeutic window of opportunity between the onset of ischemia and irreversible neuronal death. Studies in animals and humans suggest that irreversible neuronal injury is not complete until ~6 h after ischemia (STAIR, 1999). Therefore, much of the brain tissue remains potentially salvageable even when neuroprotective agents are administered several hours after ischemic stroke. Interventions that are aimed at the mechanisms associated with the delayed, ischemiainduced neuronal injury in the penumbra consequently have potential for therapeutic treatment in human stroke.
Neuregulins are a family of growth factors that have been shown to be neuroprotective in vitro (Bermingham-McDonogh et al, 1996; Buonanno and Fischbach, 2001; Erlich et al, 2001; Goldshmit et al, 2001; Vaskovsky et al, 2000; Verdi et al, 1996). Two laboratories, including ours, have recently shown that administration of neuregulin-1 (NRG-1) reduces ischemic cortical damage after middle cerebral artery occlusion (MCAO) when administered before the onset of ischemia in rats (Shyu et al, 2004; Xu et al, 2004). We observed that a single intraarterial (IA) injection of a low dose of NRG-1 (2.5 ng/kg) prevented ischemic cortical damage after 1.5 h of MCAO when administered immediately before the onset of ischemia in rats (Xu et al, 2004). The neuroprotective effect of NRG-1 was coupled with an inhibition of apoptosis and proinflammatory responses, including glial activation and interleukin-lβ expression. This indicated that the effects of NRG-1 are associated with events that lead to delayed neuronal death. Therefore, it is plausible that an extended window of opportunity exists for NRG-1 to protect neurons after stroke. The goal of the present study is to investigate whether NRG-1 is neuroprotective after transient focal ischemia with an expanded therapeutic window for administration. We also examine whether the neuroprotective effect of NRG-1 results in improved neurologic outcome.
Materials and methods
Middle Cerebral Artery Occlusion
All surgical procedures were performed by sterile/aseptic techniques in accordance with institutional guidelines. Adult male Sprague–Dawley rats weighing 250 to 300 g were used for this study. A total of 164 rats were used in this study. Rats were anesthetized with a ketamine/xylazine solution (10 mg/kg, intraperitoneally) and subjected to left MCAO. Middle cerebral artery occlusion was induced by the intraluminal suture method as previously described (Belayev et al, 1995, 1996; Xu et al, 2004). To determine the effects of NRG-1 on ischemic stroke, rat were injected intravascularly with a single bolus 10-μL dose of vehicle (1% BSA in phosphate-buffered saline (PBS)) or NRG-1 (1 and 10 nmol/L NRG-1β (EGF-like domain, R&D Systems, Minneapolis, MN, USA) dissolved in 1% BSA/PBS) through a Hamilton syringe at a rate of 5 μL/min as described previously (Belayev et al, 1995, 1996; Xu et al, 2004). This resulted in the administration of 0.25 to 2.5 ng of NRG-1/kg body weight. Neuregulin-1 or vehicle was administered by bolus injection into the ICA through ECA. Solutions were administered either before MCAO or immediately after 1.5 h of MCAO and either 0, 4 or 12 h of reperfusion. Animals were killed 24 h after reperfusion or after 14 days for the long-term studies. All NRG-1 and vehicle treatment studies were performed in a double-blinded manner such that the individuals performing the surgery and calculating infarct volumes were unaware of whether the animal received vehicle or NRG-1 until after infarct volume was assessed.
Measurement of Infarct Formation, Neurologic Outcome
Animals were killed at the indicated time point and the brains were removed, sliced into 2 mm coronal sections (approximately +3.0 to −5.0 from bregma) using a brain matrix and stained with 2,3,5-triphenyltetrazolium chloride (TTC). Brain slices were incubated in a 2% TTC solution for 30 mins at 37°C and then transferred into a 10% formaldehyde solution for fixation. Infarct area of four slices of 2-mm coronal sections of each brain was calculated by capturing the images with a digital camera and using Image Pro software. The volume of infarction was calculated by an investigator who was masked to the experimental groups. Rats showing tremor and seizure (which occurred in >2% of all animals in this study) were excluded from studies of brain infarction to eliminate cerebral hemorrhage or brain trauma as potential variables in this study. Infarct volumes were analyzed by analysis of variance (ANOVA); P < 0.05 was regarded as significant. Neurologic score was determined in a double-blinded fashion using a five-point neurologic evaluation scale (Menzies et al, 1992) in rats treated with vehicle or NRG-1 4 h after reperfusion. All animals were tested before surgery (controls) and after treatment with NRG-1 or vehicle. Neurologic function was graded on a scale of 0 to 4 (normal score 0, maximal deficit score 4). Physiologic parameters were measured in six rats (three from each condition). Blood was withdrawn from the tail vein after MCAO and 30 mins after NRG-1 or vehicle injection. After the blood was heparinized, pH, CO2 and O2 concentrations, serum electrolytes and hemoglobin levels were analyzed by standard methods. Heart rate and blood pressure were measured before blood withdrawal.
Immunohistochemistry
After reperfusion for 24 h or 14 days, rats were deeply anesthetized and perfused transcardially with 0.9% saline solution, followed by 4% paraformaldehyde solution in PBS (pH 7.4). Coronal 20-μm frozen sections were used for immunohistochemistry. Infarction was measured using cresyl violet. Fluoro-Jade B (Chemicon, Temecula, CA, USA) labeling was performed according to the manufacturer's protocol. To assess the extent of neuronal injury in the sections, the number of Fluoro-JadeB (FJB)-positive cells per field was counted. Total cell number for each field was obtained in the ischemic cortical region or caudate putamen (striatum) for each section. In animals administered vehicle or NRG-1, cortex and striatum were examined in three 20-μm sections (+1 to −1 from bregma) per animal. To quantitatively estimate the immunoreactivity, digitalized microscope images were acquired at five random areas in the ischemic core and penumbra. Image Pro software was used for the measurement.
Statistical Analysis
All data are presented as mean ± s.e.m. or s.d. as indicated. Cell death and ischemic areas in each group were compared by one-way ANOVA. The difference for each comparison was considered significant at P < 0.05.
Results
Neuregulin-1 Reduces Neuronal Damage and Improves Neurologic Outcome After Middle Cerebral Artery Occlusion
Previously, we showed that a single 2.5 ng/kg IA administration of NRG-1 before MCAO prevented neuronal death after ischemia and reperfusion (Xu et al, 2004). To determine the maximally effective dose for neuroprotective treatment with NRG-1 in stroke, NRG-1 was injected as a 10-μL bolus at concentrations of 0 (n = 6), 0.25 (n = 4) and 2.5 ng/kg (n = 7) immediately before MCAO. Neuregulin-1 attenuated infarct volume in a dose-dependent manner. As we previously showed, pretreatment with 2.5 ng/kg NRG-1 reduced infarct volume by 89% after MCAO (Figure 1). Neuregulin-1 is also neuroprotective at a 10-fold lower dose; however, it is slightly less effective and reduced infarct volume by 67%. Neither dose resulted in overt symptoms, including seizures, cerebral hemorrhage, weight loss, ataxia or mortality.
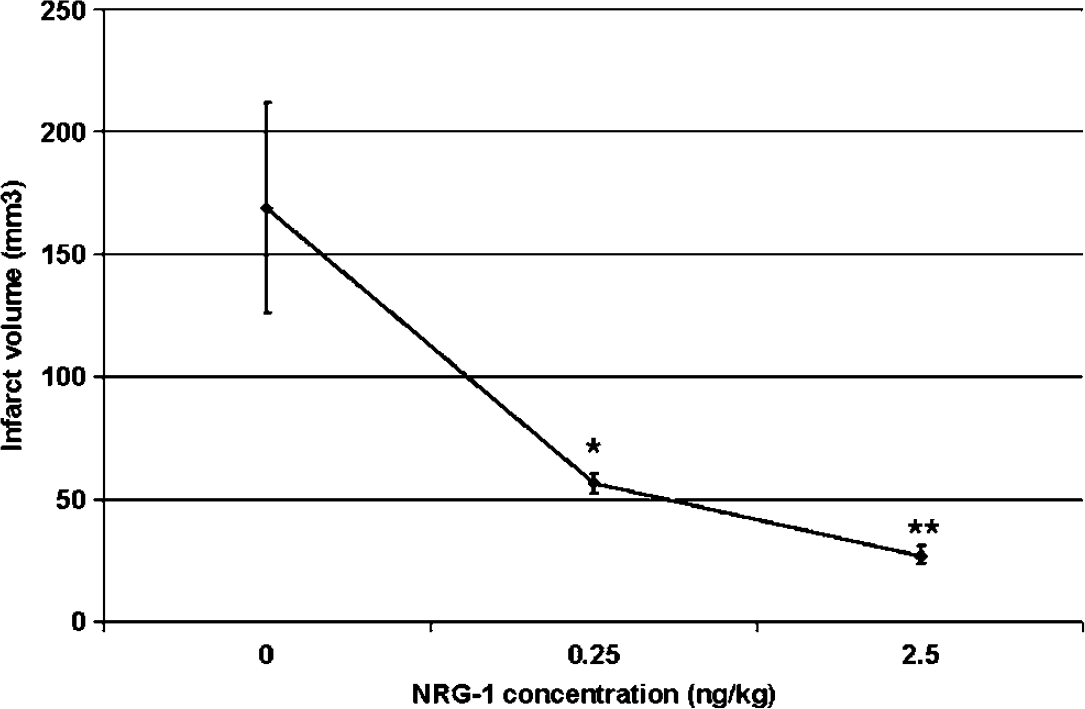
Dose–response curve for NRG-1 neuroprotection after MCAO. Neuregulin-1 was injected as a 10 μL bolus at concentrations of 0 (n = 6), 0.25 (n = 4) and 2.5 ng/kg (n = 7) immediately before MCAO. Infarct volume was determined after TTC staining as described in Materials and methods. 2,3,5-triphenyltetrazolium chloride, a colorless salt, is reduced to form an insoluble red formazan product in the presence of a functioning mitochondrial electron transport chain. Thus, the infarcted region lacks staining and appears white, whereas the normal noninfarcted tissue appears red. Values are presented as mean ± s.d. of all infarct volumes for each experimental condition; * denotes significantly different from respective vehicle-treated animals (*P < 0.05; **P < 0.01).
To determine the neuroprotective therapeutic window for NRG-1 after 1.5 h of MCAO, a single bolus injection of NRG-1 at a dose of 2.5 ng/kg was administered either 0, 4 or 12 h after reperfusion. There was a dramatic decrease in the size of the cortical and subcortical lesions in NRG-1-treated animals compared with controls. Results of representative TTC-stained sections show that NRG-1 treatment immediately after MCAO and the onset of reperfusion (n = 8) resulted in a complete inhibition of delayed infarct formation in the cortex when administered after 0 h (Figure 2B) or 4 h (Figure 2C) after reperfusion compared with rats administered only the vehicle (Figure 2A). The total infarct volume was reduced in NRG-1-treated rats by 90% when administered immediately after reperfusion (Figure 1D; R0). Infarction in the striatum was reduced by 78%, while cortical infarction was completely blocked. Neuregulin-1 also reduced infarct volume by nearly 70% when administered 4 h after reperfusion (n = 7; R4), and similarly blocked delayed neuronal death. Infarct formation was reduced by 57% when NRG-1 was administered 12 h after reperfusion (n = 8; R12). Neuregulin-1 treatment did not alter systemic blood pressure, heart rate, blood gases or serum electrolyte levels. Table 1 shows physiologic values for animals receiving vehicle or NRG-1 4 h after the onset of reperfusion.
Effect of NRG-1 on physiological parameters after MCAO
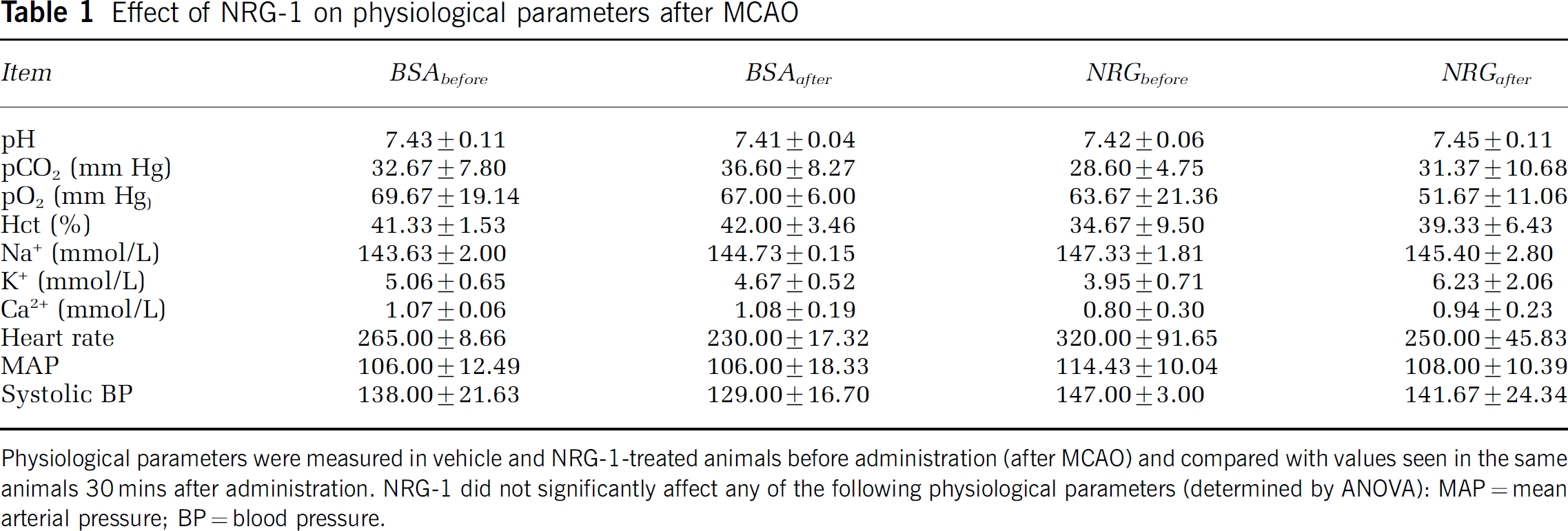
Physiological parameters were measured in vehicle and NRG-1-treated animals before administration (after MCAO) and compared with values seen in the same animals 30 mins after administration. NRG-1 did not significantly affect any of the following physiological parameters (determined by ANOVA): MAP = mean arterial pressure; BP = blood pressure.
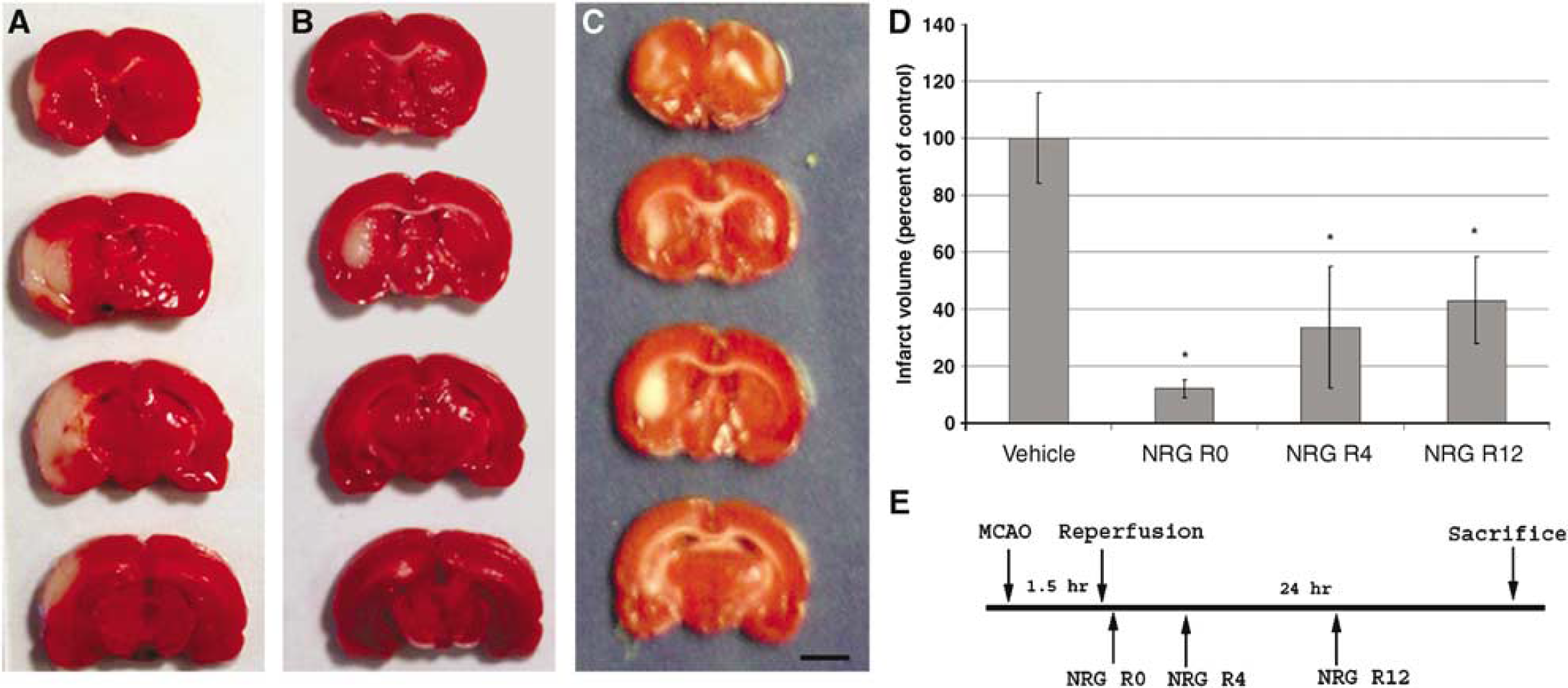
Neuregulin-1 treatment reduces MCAO/reperfusion-induced brain infarction. Representative TTC-stained coronal brain sections are shown, where rats were injected with vehicle (
Administration of Neuregulin-1 Improves Neurologic Function
Consistent with the infarct data, NRG-1 administration resulted in a significant improvement in neurologic function compared with vehicle-treated animals. In Figure 3, data are presented from animals treated with vehicle or NRG-1 4 h after reperfusion. All animals exhibited no neurologic impairment before surgery (all scores were zero). After MCAO, significant impairment of neurologic function was observed. When NRG-1 was administered after MCAO and 4 h of reperfusion, the NRG-1-treated group displayed a 32% improvement in neurologic score (2.4 ± 1.13; n = 9) compared with vehicle-treated rats (3.6 ± 0.55; n = 5). Similar results were seen when NRG-1 was administered at other time points (not shown).
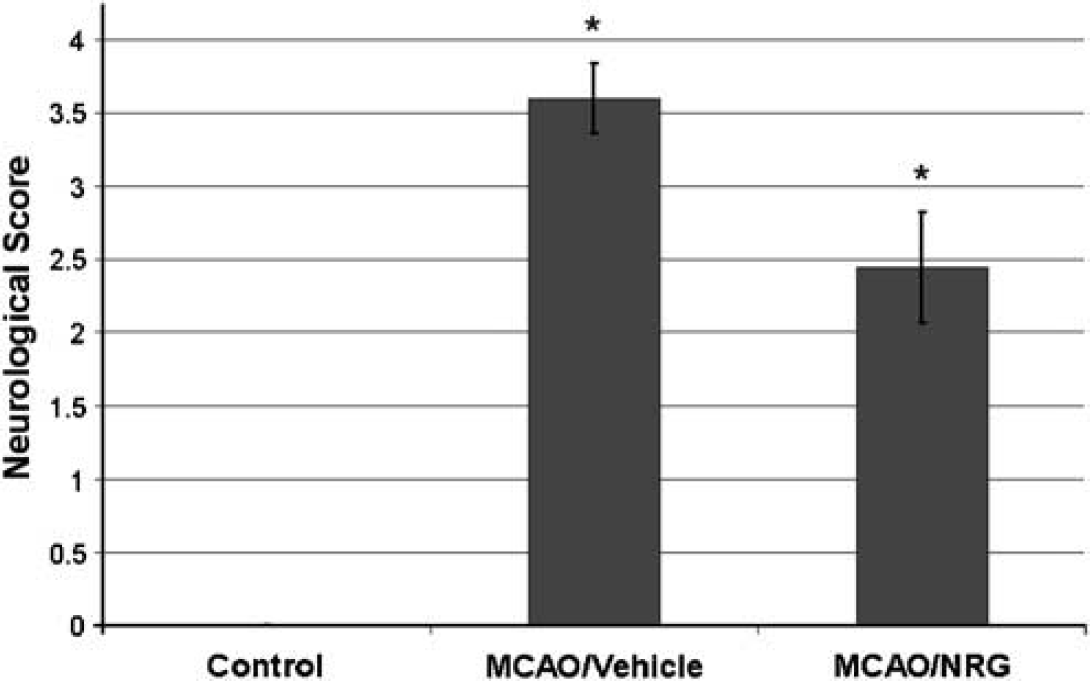
Neuregulin-1 administration resulted in a significant improvement in neurologic outcome (*denotes P < 0.01). Neuregulin-1 was administered after MCAO and 4 h of reperfusion. Neurologic function was graded on a scale of 0 to 4 (normal score 0, maximal deficit score 4). All animals were tested before surgery (controls; n = 14) and after treatment with NRG-1 or vehicle. The NRG-1-treated group (n = 9) displayed a 33% improvement in neurologic score compared with vehicle-treated rats (n = 5).
Neuregulin-1 Blocks ischemiainduced Neuronal Injury
To further examine the neuroprotective effect of NRG-1 after stroke, we stained sections of rat brain with Fluoro Jade B (FJB), a fluorochrome that binds specifically to degenerating fibers and cell bodies of neurons (Schmued and Hopkins, 2000). Results of a typical experiment are shown in Figure 4. Animals were treated with NRG-1 or vehicle 4 h after the beginning of reperfusion. The neuroprotective effect of NRG-1 was clearly indicated in the cerebral cortex after MCAO using FJB. After MGAO/reperfusion, numerous degenerating neurons were seen throughout the ipsilateral cortex and striatum in vehicle-treated animals, as indicated by FJB staining (Figure 4A and B). When animals were treated with NRG-1, 60% fewer degenerating neurons were observed in the cortex (Figure 4C and E) and a 55% reduction in FJB-positive cells was seen in the striatum (Figure 4D and E). As previously reported, similar results were seen with TUNEL labeling, indicating that NRG-1 blocked ischemiainduced apoptosis (not shown) (Shyu et al, 2004; Xu et al, 2004).
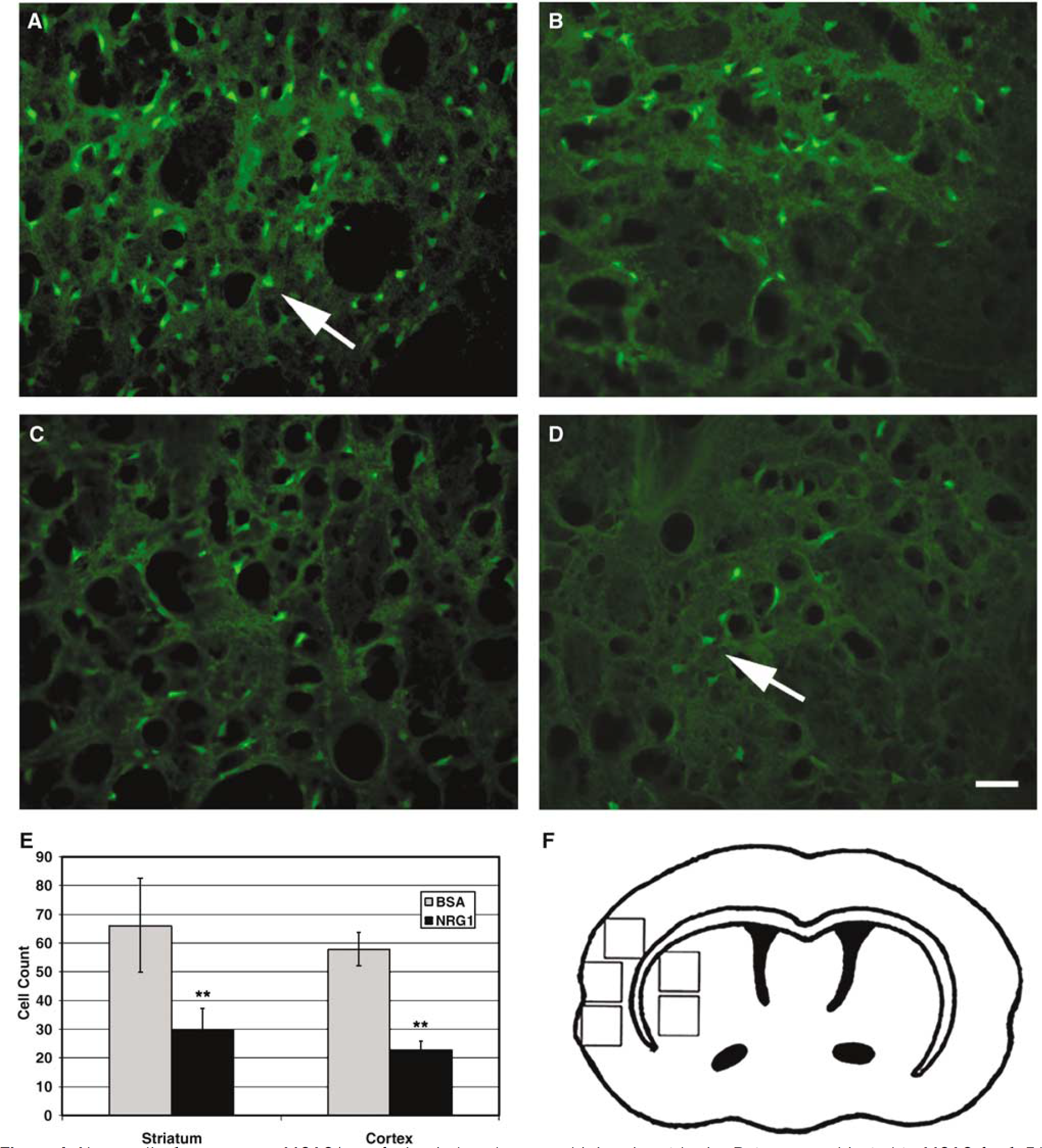
Neuregulin-1 suppresses MCAO/reperfusion-induced neuronal injury in rat brain. Rats were subjected to MCAO for 1.5 h, followed by reperfusion for 24 h. Animals were treated with NRG-1 or vehicle 4 h after the beginning of reperfusion. Fluoro Jade B (FJB) staining is found in the cortex (
Long-term Neuroprotective Effects of Neuregulin-1
The neuroprotective effect of the single NRG-1 injection was also examined 2 weeks after MCAO (n = 5). Figure 5A is a representative cresyl violet-stained section that shows a large subcortical infarct 14 days after NRG-1 treatment. However, there is still no cortical infarction at this time. This observation was consistent throughout these studies. In these rats killed two weeks after MCAO and treatment with NRG-1, high magnification of cresyl violet staining of sections from the ipsilateral striatum (Figure 5B) revealed a high density of relatively small-diameter cells (~10 μm). However, in the ipsilateral cortex (Figure 5C) and the contralateral cortex (Figure 5D) and striatum (not shown), we observed the presence of numerous large diameter cells (~20 μm) that were morphologically and immunologically determined to be neurons. This observation is consistent with previous studies showing hypercellularity in the ipsilateral hemisphere that results from an increase in glial cells labeled with the Nissl stain (Morioka et al, 1993). Quantitative analysis of neuronal density from the ipsilateral and contralateral cortices of animals killed 2 weeks after NRG-1 treatment indicated that there was no difference in neuronal density despite the massive damage to the ipsilateral striatum (Figure 5E). We also observed a large glial scar surrounding the subcortical infarct as evidenced by heavy GFAP labeling. However, there was no evidence of astrogliosis is seen in the cortex (Figure 5F). These results taken together suggest that delayed cortical neuronal death is prevented at least 2 weeks after the single injection of NRG-1 before MCAO.
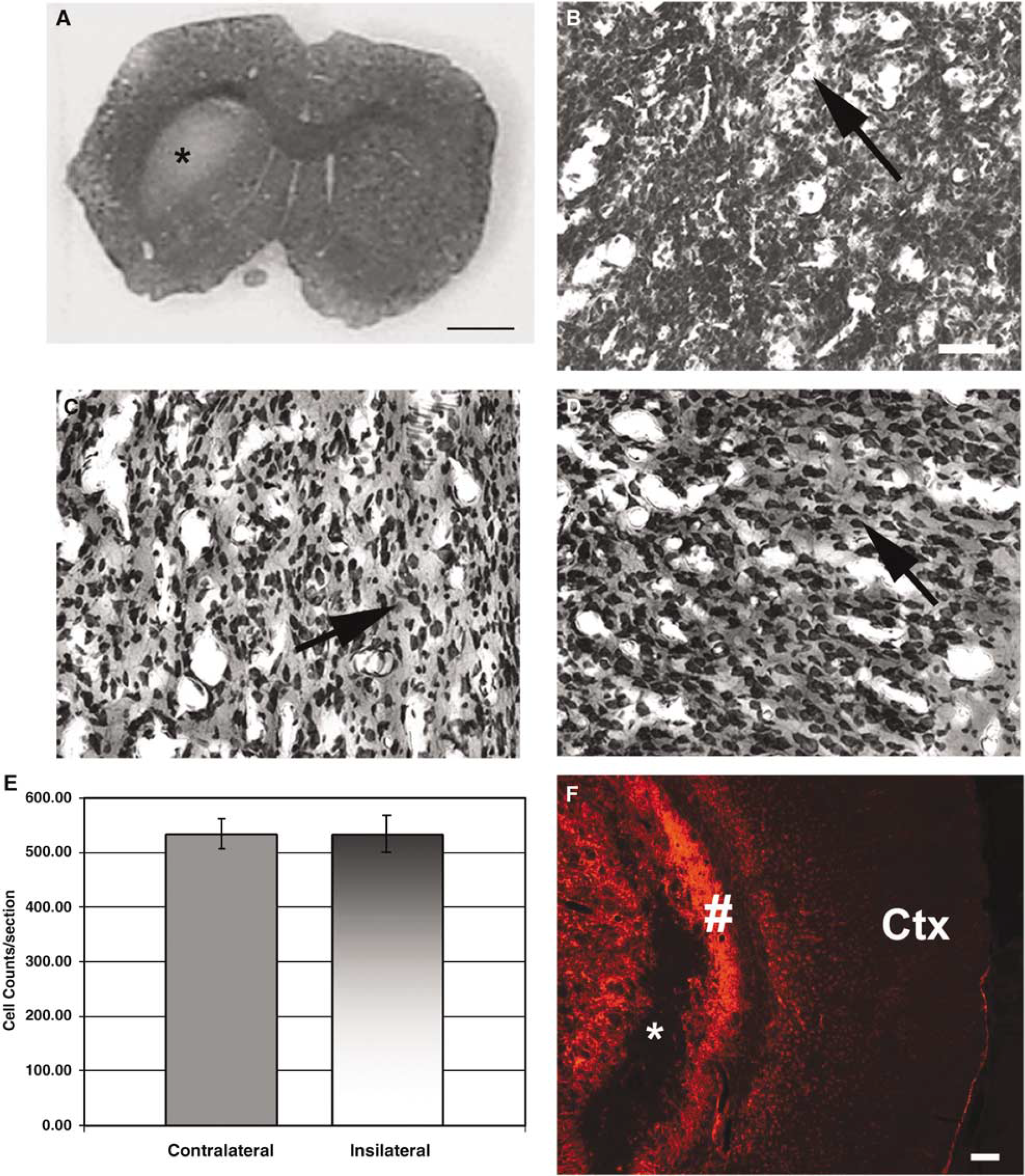
In this study, animals treated with NRG-1 were killed 2 weeks after MCAO. Infarction in the striatum is evident; however, no cortical neuronal death is observed in rats treated with NRG-1 (
Discussion
To address the numerous failures of clinical trials for neuroprotective drugs, a group of stroke scientists and clinicians formed the Stroke Therapy Academic Industry Roundtable (STAIR) to recommend guidelines for preclinical evaluation of neuroprotective drugs (STAIR, 1999). They proposed that studies performed at the basic science level should investigate a standard set of parameters before qualifying treatments for clinical trial, including functional outcome, the therapeutic window and long-term effects. The concept of the therapeutic window is indicated by studies in animals and humans suggesting that irreversible neuronal injury is not complete until ~6 h or perhaps days after ischemia. Therefore, much of the brain tissue remains potentially salvageable even when neuroprotective agents are administered several hours after ischemic stroke. Interventions that are aimed at the mechanisms associated with the delayed, ischemiainduced neuronal injury consequently have potential for therapeutic treatment in human stroke. A number of different agents, including calcium antagonists, glutamate receptor antagonists, free radical scavengers and antibodies against adhesion molecules, have been proposed for the treatment of stroke. However, all of the treatments used to date for stroke therapy have been unsuccessful in clinical trials. Administering growth factors could represent an alternative or combinatorial strategy for the treatment of ischemic brain injury and other neurologic disorders.
A recent report from our laboratory showed that pretreatment with NRG-1 prevented delayed neuronal injury and reduced proinflammatory responses (Xu et al, 2004). This study extends those findings to show that NRG-1 prevents delayed cortical neuronal death with an extended therapeutic window after ischemic brain injury. We further show that the neuroprotective effect of NRG-1 was associated with an inhibition of neuronal injury and resulted in a significant improvement of neurologic function. In our study, we have addressed several of the recommendations of the STAIR, including extension of the therapeutic window, histologic analysis, assessment of infarct volumes and functional recovery in a randomized, masked fashion. The recent report by Shyu et al showed that NRG-1 injected directly into the brain parenchyma also protected cortical neurons from ischemia damage without affecting other physiologic parameters, including blood pressure, heart rate, blood gases and blood chemistry (Shyu et al, 2004). Our results indicate that NRG-1 does not affect these functions in this model. Neuroprotective agents might also protect neurons by regulating CBF after ischemia. However, it has been documented that, in the transient MCAO model, CBF is restored to baseline values almost immediately after reperfusion (Tsuchidate et al, 1997; Tureyen et al, 2004). Therefore, although we cannot rule out effects on CBF, it is unlikely that the effects of NRG-1 several hours after reperfusion are due to regulating blood flow. We plan to look at the direct effects of NRG-1 on CBF in future studies. With regard to IA administration, there is clear precedence for its utility in a number of neurologic disorders, including stroke (Connors, 2002; Higashida et al, 2003). In fact, t-PA, the only drug approved for stroke therapy, is administered IA. This also allows the drug to be delivered directly to the brain and minimized the effects of biodegradation by the liver. Larger doses of the drug would be required for intravenous delivery, which could result in undesired complication. Nonetheless, we will investigate the use of additional delivery mechanisms in future studies.
Another concern raised by the STAIR was whether the neuroprotective drug could cross the blood–brain barrier (BBB). A recent study showed that NRG-1 enters the brain and spinal cord by a saturable receptor-mediated transport mechanism (Kastin et al, 2004). We recently provided evidence suggesting an important role for erbB receptors in the neuroprotective effects of NRG-1 in stroke and the potential specificity of the compound after injury. We showed that erbB4 was upregulated in the periinfarct regions of the ipsilateral cortex after MCAO (Xu and Ford, 2005). We showed that erbB4 was upregulated in FJB-positive cells, suggesting that erbB receptors are induced in injured neurons. The increase in erbB receptors was also seen in a subpopulation of macrophages/microglia. We propose that the induction of erbB receptors in the periinfarct neurons and macrophages/microglia is an adaptive response that increases the capacity of neurons to respond to NRG-1 and acts to prevent neuronal injury. Taken together, these data suggest that exogenously administered NRG-1 can enter the brain across the BBB and the specific upregulation of erbB receptors in regions of brain injury may provide potency and specificity to NRG-1 effects after injury. However, NRG-1 might also enter the brain through breaches in the BBB, which is damaged after ischemic stroke.
The combined in vivo and in vitro evidence provide compelling evidence for a neuroprotective role for neuregulins in ischemic brain injury. Our data show that NRG-1 protects neurons from delayed, ischemiainduced cell death with an extended therapeutic time window. We acknowledge that previously neuroprotective strategies successful in rodents have failed in human clinical trials. However, there are advantages to NRG-1 mechanisms that make it an attractive therapeutic prospect. These include the following: (1) many previous therapeutic strategies manage the early, excitotoxic pathways, while NRG-1 blocks the mechanisms associated with delayed neuronal death; (2) the extended therapeutic window makes NRG-1 a practical treatment for neuroprotection and (3) NRG-1 recognizes the glial contribution to acute stroke and prevents inflammatory mechanism initiated by glial cells (Xu et al, 2004). Future studies will continue to examine the suitability of neuregulins for clinical trials. Clinical studies using NRG-1 alone or in conjunction with other therapies, such as inhibitors of excitotoxicity or thrombolytic compounds, might be beneficial in the treatment of patients with acute stroke.