
Abstract
Na+–K+–Cl− cotransporter isoform 1 (NKCC1) and Na+/Ca2+ exchanger isoform 1 (NCX1) were expressed in cortical neurons. Three hours of oxygen and glucose deprivation (OGD) significantly increased expression of full-length NCX1 protein (∼116 kDa), which remained elevated during 1 to 21 h reoxygenation (REOX) and was accompanied with concurrent cleavage of NCX1. Na+/Ca2+ exchanger isoform 1 heterozygous (NCX1+/−) neurons with ∼50% less of NCX1 protein exhibited ∼64% reduction in NCX-mediated Ca2+ influx. Expression of NCX1 and NKCC1 proteins was reduced in double heterozygous (NCX1+/−/NKCC1+/−) neurons. NCX-mediated Ca2+ influx was nearly abolished in these neurons. Three-hour OGD and 21-h REOX caused ∼80% mortality rate in NCX1+/+ neurons and in NCX1+/− neurons. In contrast, NKCC1+/− neurons exhibited ∼45% less cell death. The lowest mortality rate was found in NCX1+/−/NKCC1+/− neurons (∼65% less neuronal death). The increased tolerance to ischemic damage was also observed in NCX1+/−/NKCC1+/− brains after transient cerebral ischemia. NCX1+/−/NKCC1+/− mice had a significantly reduced infarct volume at 24 and 72 h reperfusion. In conclusion, these data suggest that NKCC1 in conjunction with NCX1 plays a role in reperfusion-induced brain injury after ischemia.
Introduction
The electroneutral Na+–K+–Cl− cotransporter transports Na+, K+, and Cl− into cells under physiological conditions with a stoichiometry of 1Na+:1K+:2Cl− (Russell, 2000). Na+–K+–Cl− cotransporter isoform 1 (NKCC1) has a broad tissue distribution, whereas NKCC2 is found only in the vertebrate kidney (Delpire and Mount, 2002). NKCC serves multiple functions, including ion and fluid movements in secreting or reabsorbing epithelia and cell volume regulation (Delpire and Mount, 2002; Russell, 2000). We reported that NKCC1 activity, reflected by protein phosphorylation, is increased in the cortex and striatum during 0 to 8 h reperfusion after 2-h transient middle cerebral artery occlusion in rat (tMCAO, Yan et al, 2003). Either pharmacological blockage of NKCC1 or genetic ablation of NKCC1 significantly reduces infarct volume after tMCAO (Chen et al, 2005; Yan et al, 2001). Administration of NKCC inhibitor bumetanide intravenously before occlusion reduces edema formation in rats subjected to permanent MCAO (pMCAO, O'Donnell et al, 2004). These findings suggest that NKCC1 activity is involved in cerebral ischemic damage. Moreover, our in vitro studies in astrocytes or in oligodendrocytes show that NKCC1 activation leads to Na+ overload, which triggers reverse mode operation of Na+/Ca2+ exchanger (NCXrev) after ischemia and excitotoxicity (Chen et al, 2007; Kintner et al, 2007; Lenart et al, 2004). Although these findings support that NKCC1 and NCXrev may collectively contribute to ischemic cell damage, it has not yet been tested in in vivo ischemic models.
NCXrev causes Ca2+ influx after NMDA (N-methyl-
To investigate further the role of NKCC1 in conjunction with NCX1rev in ischemic neuronal death, we examined whether reduction of NKCC1 and NCX1 could provide neuroprotection after in vitro or in vivo ischemia. Preliminary data in this study were reported previously (Luo et al, 2007).
Materials and methods
Materials
Eagle's modified essential medium and Hanks balanced salt solution were from Mediatech Cellgro (Herndon, VA, USA). Fetal bovine serum was obtained from Valley Biomedical (Winchester, VA, USA). Horse serum was obtained from Hyclone Laboratories (Logan, UT, USA). 1-[6-Amino-2-(5-carboxy-2-oxazolyl)-5-benzofuranyloxy]-2-(2-amino-5-methylphenoxy)ethane-N,N,N′,N′-tetraacetic acid (fura-2 AM) was obtained from Invitrogen (Carlsbad, CA, USA). KB-R7943 was from Tocris (Ellisville, MO, USA). Antibody for β-tubulin type III was from Promega (Madison, WI, USA). Na+–K+–Cl− cotransporter monoclonal antibody (T4) was from Developmental Studies Hybridoma Bank (Iowa City, IA, USA). Anti-Na+/Ca2+ exchanger isoform 1 antibody was from Swant (Benllinzona, Switzerland). FragEL™ DNA Fragmentation Detection Kit was from Calbiochem (La Jolla, CA, USA).
Animal Preparation
The NCX1 transgenic mouse (SV129/Black Swiss) and NKCC1 transgenic mouse (SV129/Black Swiss) were established previously (Flagella et al, 1999; Reuter et al, 2002). The genotype of each mouse was determined by a PCR of DNA from tail biopsies as described before. NKCC1+/− and NCX+/− breeders were set up to obtain a double NCX1+/−/NKCC1+/− heterozygous mouse in the study. NCX1−/− mouse was embryonically lethal and could not be used in the study (Cho et al, 2000).
Primary Cultures of Mouse Cortical Neurons
E14-16 pregnant mice were anesthetized with 5% halothane and euthanized as described in our recent study (Chen et al, 2005). Fetuses were removed and rinsed in cold Hanks balanced salt solution. Each mouse fetus was genotyped using fetus tail biopsies with the PCR method (Chen et al, 2005). The cortices were removed and minced. The tissues were treated with trypsin at 37°C for 25 mins. The cell suspension after centrifugation was diluted in Eagle's modified essential medium containing 5% fetal bovine serum and 5% horse serum (HS). Cells from individual fetal cortices were seeded separately in poly-
Oxygen and Glucose Deprivation Treatment
DIV 10 to 15 neuronal cultures were rinsed with an isotonic OGD solution (pH 7.4), as described before (Luo et al, 2005). Cells were incubated in 0.5 mL of the OGD solution in a hypoxic incubator (model 3130; Thermo Forma, Marietta, OH, USA) containing 94% N2, 1% O2, and 5% CO2. The oxygen level in the medium of cultured cells in 24-well plates was decreased to ∼2 to 3% after 60 mins in the hypoxic incubator (Beck et al, 2003). The OGD incubation was 3 h and for reoxygenation (REOX), the cells were incubated for 21 h in 0.5 mL of Eagle's modified essential medium containing 5.5 mmol/L glucose at 37°C in the incubator with 5% CO2 and atmospheric air. Normoxic control cells for cell viability assay were performed in sister cultures (Luo et al, 2005). In our pilot studies, no significant differences were found in the neuronal toxicity assay after either 2 or 3 h OGD and 21 or 22 h REOX.
Measurement of Cell Death
Cell viability was assessed by propidium iodide (PI) uptake and retention of calcein using a Nikon TE 300 inverted epifluorescence microscope (Tokyo, Japan). Cultured neurons were rinsed with HEPES-MEM ((4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)-minimal essential medium) (composition described previously in Su et al, 2002). Cells were incubated with 1 μg/mL calcein-AM and 10 μg/mL PI in the same buffer at 37°C for 30 mins. For cell counting, cells were rinsed with the isotonic control buffer and visualized using a Nikon × 20 objective lens. Calcein and PI fluorescences were visualized using FITC filters and Texas Red filters as described before (Beck et al, 2003). Images were collected using a Princeton Instruments (Trenton, NJ, USA) MicroMax CCD camera. In a blind manner, a total of 1,000 cells/condition were counted using MetaMorph image-processing software (Universal Imaging Corp., Downingtown, PA, USA). Cell mortality was expressed as the ratio of PI-positive cells to the sum of calcein- and PI-positive cells.
Gel Electrophoresis and Western Blotting
Cells were scraped from the plates and lysed in phosphate-buffered saline (pH 7.4) containing 2 mmol/L EDTA and protease inhibitors by 30 secs sonication at 4°C (Kintner et al, 2004). Brain homogenates and crude membrane fractions were prepared as described before (Yan et al, 2003). Protein content in each sample was determined by the bicinchoninic acid (BCA) method. Protein samples (15 μg/lane) and prestained molecular mass markers (Bio-Rad, Hercules, CA, USA) were denatured in 2 × sodium dodecyl sulfate reducing buffer. The samples were then electrophoretically separated on 6% sodium dodecyl sulfate gels, and the resolved proteins were electrophoretically transferred to a polyvinylidene fluoride membrane (Kintner et al, 2004). The blots were incubated in 7.5% non-fat dry milk in Tris-buffered saline overnight at 4°C, and then incubated for 1 h with a primary antibody. The blots were rinsed with Tris-buffered saline and incubated with horseradish peroxidase-conjugated secondary IgG for 1 h. Bound antibody was visualized using the enhanced chemiluminescence assay (Pierce, Rockford, IL, USA). Monoclonal T4 antibody against NKCC1 (1:4,000) and anti-NCX1 monoclonal antibody (1:1,000) were used for detection of NKCC1 and NCX1, respectively. Anti-NCX2 monoclonal antibody (1:500) or anti-NCX3 polyclonal antibody (1:1,000) was also used (Thurneysen et al, 2002).
Intracellular Ca2+ Measurement
Neurons grown on coverslips were incubated with 5 μmol/L fura-2 AM for 45 mins (Lenart et al, 2004; Luo et al, 2005). The cells were washed and the coverslips placed in the open-bath imaging chamber containing HEPES-MEM at 37°C. Using the Nikon TE 300 inverted epifluorescence microscope and a × 40 Super Fluor oil immersion objective lens, neurons were excited every 10 secs at 345 and 385 nm and the emission fluorescence at 510 nm recorded. Images were collected and analyzed with the MetaFluor image-processing software. At the end of each experiment, cells were exposed to 1 mmol/L MnCl2 and 5 μmol/L Br·A23187 in Ca2+-free HEPES-MEM. The Ca2+-insensitive fluorescence was subtracted from each wavelength before calculations (Luo et al, 2005). The MnCl2-corrected 345/385 emission ratios were converted into concentration using the Grynkiewicz equation (Grynkiewicz et al, 1985), as described previously (Lenart et al, 2004).
NCXrev was induced in neurons as described by Hoyt et al (1998). Neurons were exposed to Ca2+-free HEPES buffer for 1 min. NCXrev was initiated by exposing cells to a Na+-free buffer (1.2 mmol/L Ca2+) for 30 to 40 secs, which triggered a rise in [Ca2+]i. Cells quickly regulated Ca2+i to baseline values when they were returned to control HEPES-MEM.
Focal Ischemic Model
Transient MCAO was induced in mice as described previously (Chen et al, 2005). Briefly, the left common carotid artery was exposed and the occipital artery branches of the external carotid artery were isolated and coagulated. After coagulation of the superior thyroid artery, the external carotid artery was dissected and coagulated. The internal carotid artery was isolated, and the extracranial branch of the internal carotid artery was then dissected and ligated. A polyamide resin glue-coated suture (6-0 monofilament nylon) was introduced into the external carotid artery lumen and then advanced ∼9 to 9.5 mm in the internal carotid artery lumen to block the middle cerebral artery blood flow. The suture was withdrawn 30 mins after MCAO and the incision was closed. The core temperature (36.0±0.6°C) was maintained during ischemia and recovery with a heating pad and heating lamp. After recovery, animals were returned to their cages with free access to food and water. At 24 or 72 h of reperfusion, the animals were killed for infarction measurements. Bumetanide (25 mg/kg) (stock was dissolved in 0.5 N NaOH, max 0.09% NaOH) was administered through the left femoral vein immediately before the MCAO induction.
All animal procedures used in this study were conducted in strict compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the University of Wisconsin Center for Health Sciences Research Animal Care Committee.
Infarction Size Measurement
After 24 and 72 h reperfusion, mice were anesthetized with 5% halothane vaporized in N2O and O2 (3:2) and then decapitated. Brains were removed and frozen at −80°C for 5 mins. Two-millimeter coronal slices were made with a rodent brain matrix (Ted Pella Inc., Redding, CA, USA). The sections were stained for 20 mins at 37°C with 2% 2,3,5-triphenyltetrazolium chloride monohydrate (Sigma, St Louis, MO, USA). Infarction volume was calculated with the method reported by Swanson et al (1990) to compensate for brain swelling in the ischemic hemisphere. Briefly, the sections were scanned, and the infarction area in each section was calculated by subtracting the non-infarct area of the ipsilateral side from the area of the contralateral side with NIH image analysis software. Infarction areas on each section were summed and multiplied by section thickness to give the total infarction volume.
In Situ Labeling of DNA Fragmentation
To obtain deep anesthesia for transcardial perfusion of animals, after 30 mins ischemia and 72 h reperfusion, the mice were anesthetized with ketamine (100 mg/kg, intraperitoneally) and xylazine (10 mg/kg, intraperitoneally). The animals were then transcardially perfused with 4% paraformaldehyde in 0.1
Statistical Analysis
Comparisons between groups were made by Student's t-test or analysis of variance using the Bonferroni posttest (SigmaStat, Systat Software, Point Richmond, CA, USA). A P-value of <0.05 was considered as significantly different.
Results
Physiological Parameters and Regional Cerebral Blood Flow in NKCC1+/+, NCX1+/−, and NKCC1+/−/NCX1+/− Mice
Mean arterial blood pressure, pH, PaCO2, PaO2, Na+, and K+ were not significantly different between NCX1+/+/NKCC1+/+, NCX1+/−, or NCX1+/−
Physiological parameters in NCX1+/+/NKCC1+/+, NCX1+/−, and NCX1+/−/NKCC1+/− mice
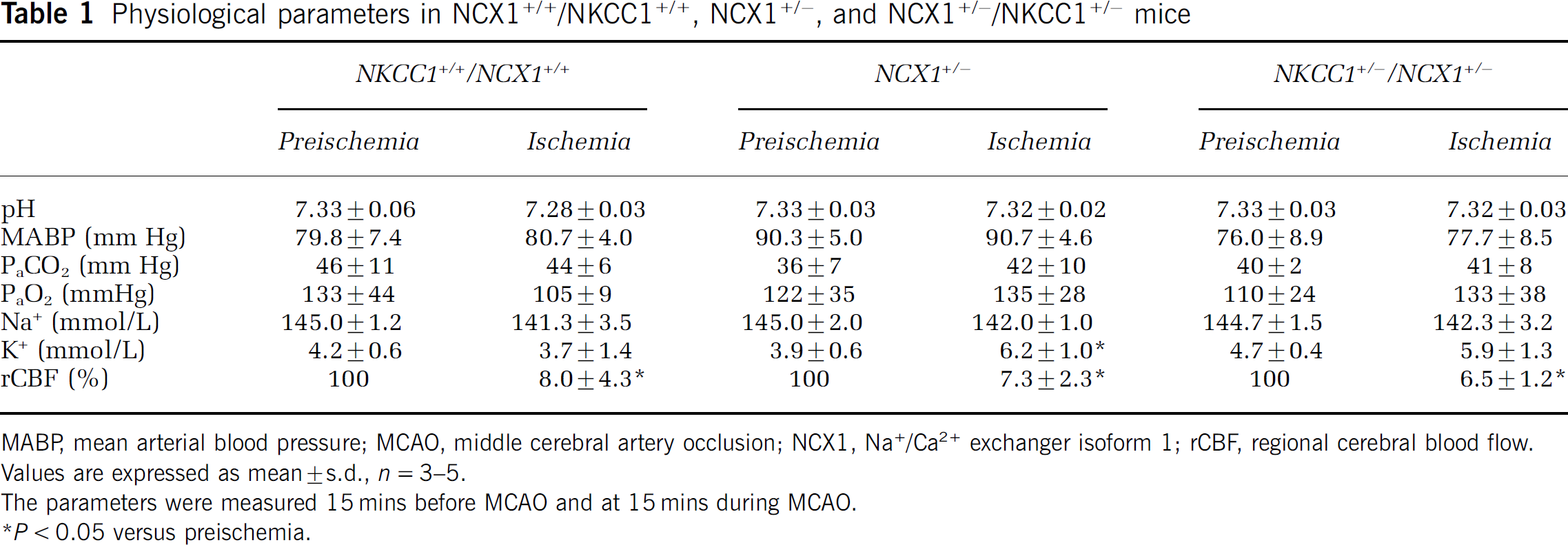
MABP, mean arterial blood pressure; MCAO, middle cerebral artery occlusion; NCX1, Na+/Ca2+ exchanger isoform 1; rCBF, regional cerebral blood flow.
Values are expressed as mean±s.d., n=3–5.
The parameters were measured 15 mins before MCAO and at 15 mins during MCAO.
P<0.05 versus preischemia.
Changes of Na+/Ca2+ Exchanger Isoform 1 Protein Expression in Neuronal Cultures Following Oxygen and Glucose Deprivation/Reoxygenation
Abundant expression of NKCC1 and NCX1 was detected in NCX1+/+ neurons (Figure 1Aa, b) and they were colocalized in most cells (Figure 1Ac). Changes of NCX1 protein expression in neurons were examined at 0, 1, 2, and 21 h REOX after 3 h OGD treatment. Control samples were collected from normoxic NCX1+/+ neuronal cultures. As shown in Figure 1B, 3 h OGD induced an increase in NCX1 full-length protein expression (∼116 kDa). This elevation of the full-length NCX1 protein expression was sustained during 1 to 21 h REOX. The peak value (∼2.4-fold increase in full-length NCX1) occurred at 1 h REOX and it remained elevated at 21 h REOX, although in some samples the level of full-length NCX1 was reduced at 21 h REOX (Figures 1B and 1C). Moreover, NCX1 was cleaved and the ∼70 kDa-cleaved NCX1 was detected at 3 h OGD. Cleaved NCX1 was increased with time and reached ∼10-fold at 21 h REOX (Figures 1B and 1D). However, expression of NCX2 or NCX3 was not altered after either OGD or 1 to 21 h REOX (Figures 1E and 1F). The 60 kDa NCX2 band detected by its specific monoclonal antibody has been characterized previously (Minelli et al, 2007; Thurneysen et al, 2002). Taken together, these findings suggest that NCX1 may play an important role in ischemic neurons.
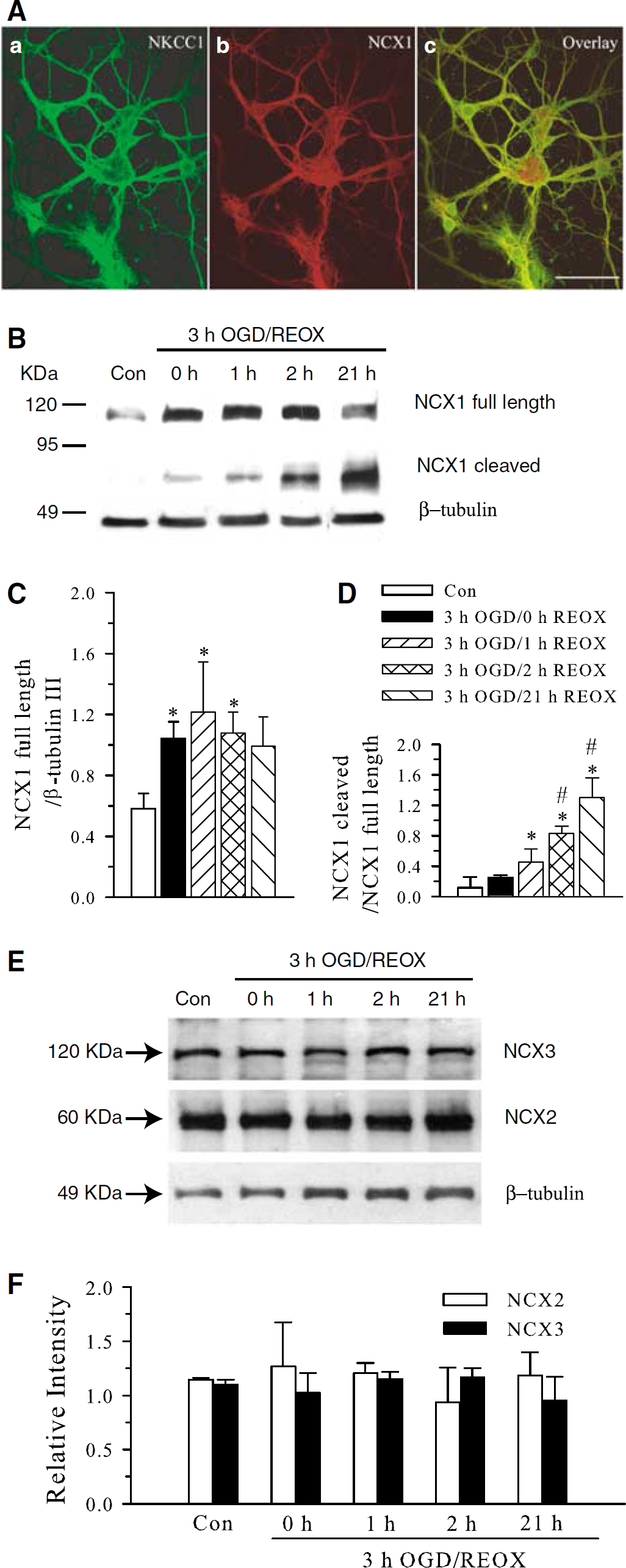
Oxygen and glucose deprivation/reoxygenation-mediated changes in NCX1 expression in NCX1+/+ neurons. (
Reduced Expression of the Full-Length NCX1 and NCX Activity in NCX1+/−/NKCC1+/− Neurons
The full-length NCX1 band (∼116 kDa) was recognized in NCX1+/+ neuronal cultures as before and was decreased by ∼50% in NCX1+/− neuronal cultures (P<0.05, Figures 2A and 2B). In contrast, NKCC1 protein was increased by ∼20% in NCX1+/− neuronal cultures. No significant changes of NCX2 or NCX3 proteins were detected in NCX1+/− neuronal cultures. Screening of NCX1, NCX2, and NCX3 protein in NKCC1−/− neurons or NKCC1−/− astrocytes did not detect any significant changes (data not shown). Therefore, NKCC1+/− neurons or astrocytes were not further tested for such compensatory effects.
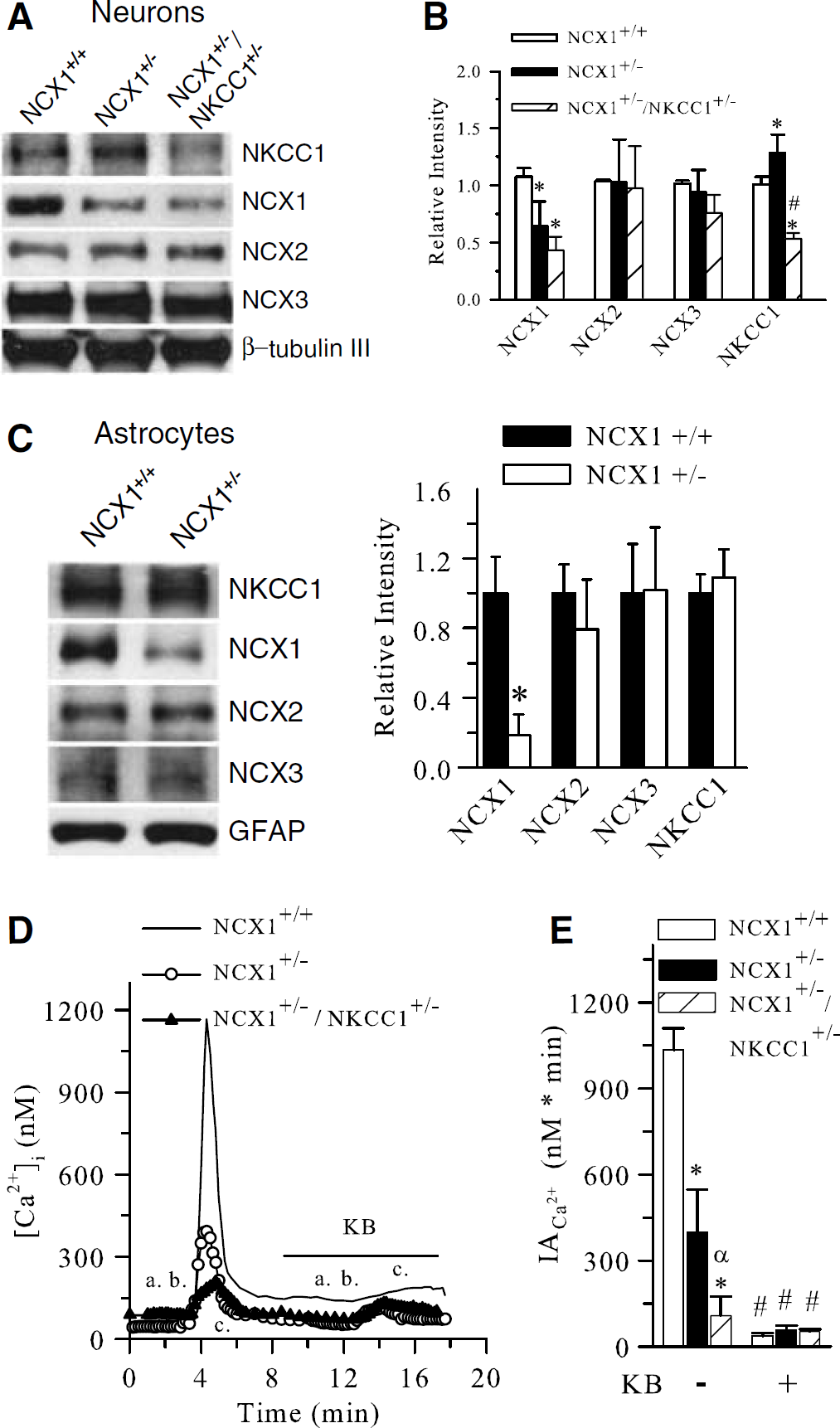
Reduction of NCX1 expression and activity in NCX1+/−/NKCC1+/− neurons. (
Moreover, both NKCC1 and NCX1 proteins were significantly reduced in NCX1+/−/NKCC1+/− neurons. No compensatory responses from NCX2 and NCX3 expression were detected in these cells (Figures 2A and 2B). β III-Tubulin was detected in the same blot of all genotype samples as loading controls and was not changed.
To investigate further that no significant compensation from NCX2 and NCX3 occurs in NCX1+/− brains, we measured expression of NKCC1, NCX2, and NCX3 proteins in astrocytes cultured from either NCX1+/+ or NCX1+/− mice. NCX1 band in astrocytes was as abundant as in NCX1+/+ neuronal cultures (Figure 2C). NCX1+/− astrocyte cultures expressed ∼80% less NCX1 protein (P<0.05). No changes of NKCC1, NCX2, or NCX3 proteins were detected in NCX1+/− astrocyte cultures. A similar level of GFAP was detected in the same blot of all genotype samples.
We then determined whether activity of NCX1 was decreased in NCX1+/− or NCX1+/−/NKCC1+/− neurons by measuring NCXrev. As seen in Figure 2D(a.), after a brief exposure to Ca2+-free buffer, NCXrev in NCX1+/+ neurons was triggered by returning the cells to the Na+-free buffer (1.2 mmol/L Ca2+, Figure 2D(b.)). A large increase in [Ca2+]i was detected in NCX1+/+ neurons (Figure 2D). Ninety percent of the rise in [Ca2+]i was blocked by 10 μmol/L KB-R7943, a nonspecific NCXrev inhibitor. NCX1+/− neurons exhibited significantly less Ca2+ influx triggered by NCX1rev. The integrated area under the transient peak of [Ca2+]i (IACa2+) was 1036±75 nmol/L per min in NCX1+/+ neurons and it was reduced to 399±150 nmol/L min in NCX1+/− neurons (Figures 2D and 2E). The remaining Ca2+ influx in NCX1+/− neurons was further blocked by KB-R7943 (Figures 2D and 2E). Lastly, NCXrev-mediated Ca2+ influx was almost blocked in NCX1+/−/NKCC1+/− neurons (105±64 nmol/L per min).
NCX1+/−/NKCC1+/− Neurons Exhibit Resistance to Ischemia
Because NCXrev-mediated Ca2+ influx was nearly completely suppressed in NCX1+/−/NKCC1+/− neurons, we then investigated whether NCX1+/−/NKCC1+/− neurons have a higher tolerance to OGD/REOX. As shown in Figure 3Aa, d, the basal level of cell death in NCX1+/+ neurons was ∼15±5%. Three-hour OGD and 21-h REOX led to 78±4% cell death in NCX1+/+ neurons (P<0.05, Figures 3Ab, e, and 3B). NCX1+/− neurons did not show neuroprotection (61±8% cell death, P>0.05, Figure 3B). In contrast, NKCC1+/− neurons showed a significant reduction in cell death (42±5%, P<0.05, Figure 3B). The double heterozygous neurons of NCX1+/−/NKCC1+/− exhibited an additional tolerance to ischemic damage (Figure 3Ac, f) and had the lowest cell death rate after 3-h OGD and 21-h REOX (28±5%, P<0.05), which was significantly lower than either NCX1+/− or NKCC1+/− neuron cultures. This finding is consistent with the Ca2+ influx data described above and suggests that NKCC1 in conjunction with NCX1 play a role in ischemic neuronal damage.
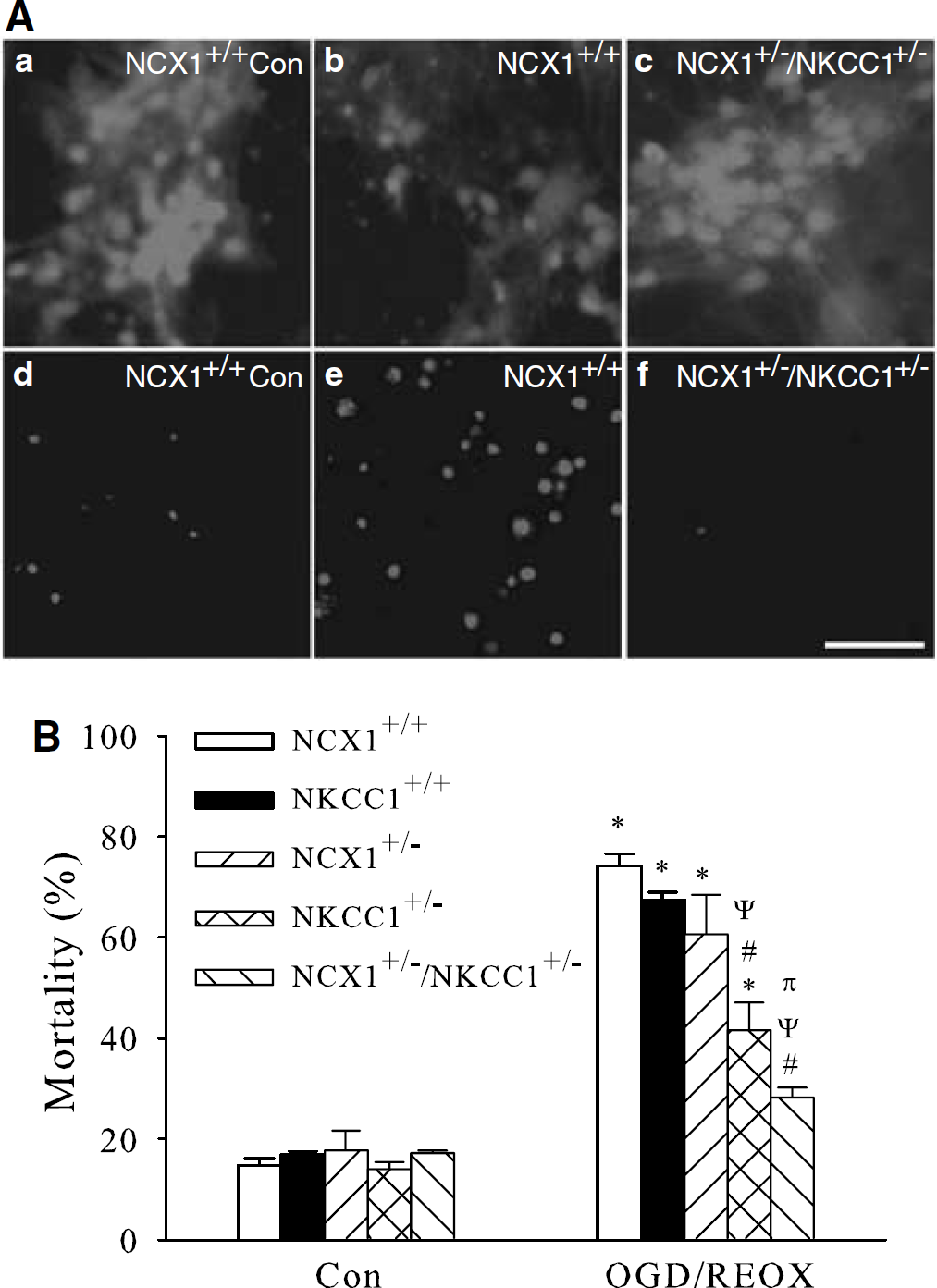
NCX1+/−/NKCC1+/− neurons are resistant to OGD/REOX-mediated damage. (
Reduced Ischemic Cell Death in NCX1+/−/NKCC1+/− Brains after Transient Focal Ischemia
We investigated whether NCX1+/−/NKCC1+/− mice have a higher resistance to cerebral damage in a transient focal ischemia model. NCX1+/+ or NCX1+/− mice exhibited a similar degree of infarction at 24-h reperfusion after 30 mins MCAO (57.3±8.8 and 50.7±3.9 mm3, Figures 4A and 4B). Infarct volume was further developed after 72 h reperfusion in NCX1+/+ or NCX1+/− mice (86.1±10.0 and 100.6±20.0 mm3, P<0.05). In contrast, there was a significant reduction in infarct in NKCC1+/− mice at both 24 and 72 h reperfusion (Figures 4A and 4B). Moreover, NCX1+/−
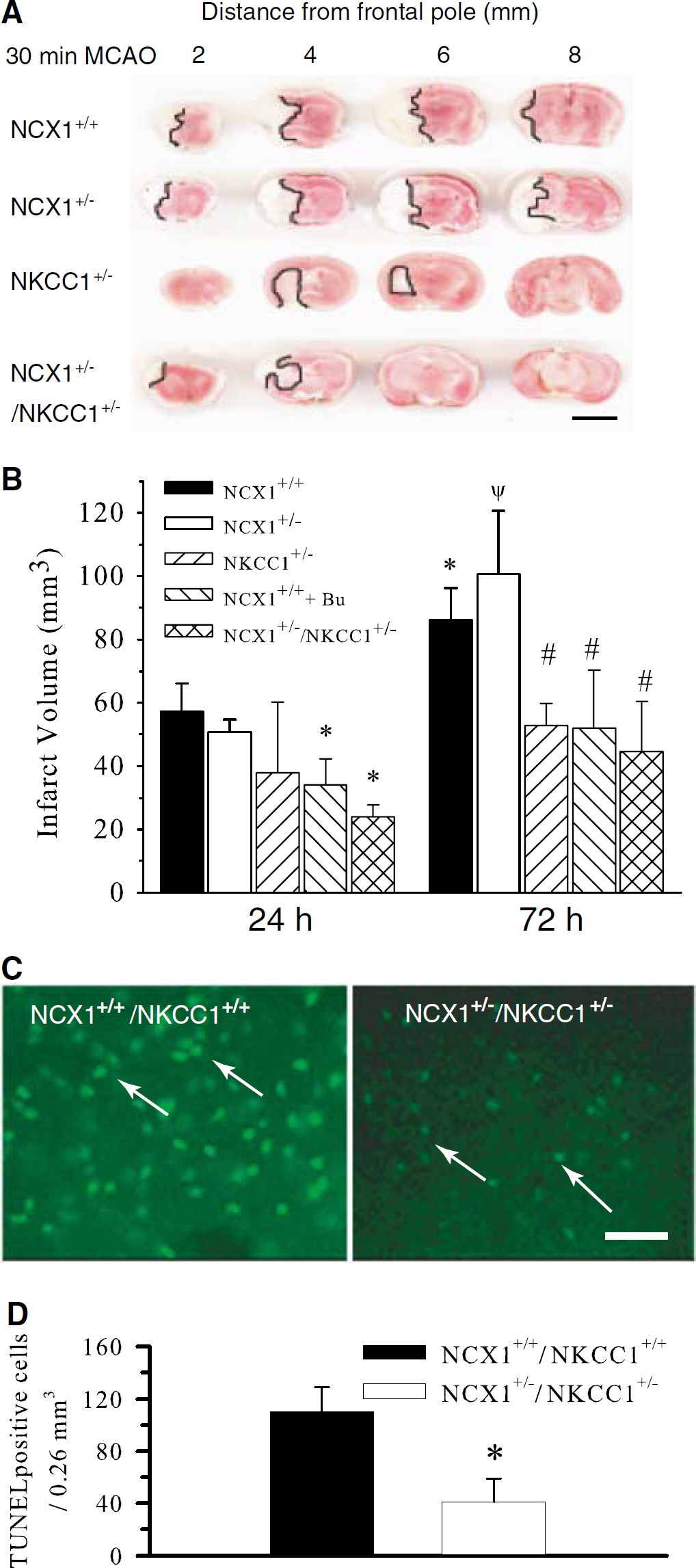
Reduced ischemic brain damage in NCX1+/−/NKCC1+/− mice. (
We hypothesized that activation of NCXrev is in part a result of NKCC1 activation and Na+ overload after ischemia. Therefore, inhibition of NKCC1 activity would prevent NCXrev and should have a similar neuroprotective effect as blocking of NCXrev. As shown in Figure 4B, when a potent NKCC1 inhibitor bumetanide was given (25 mg/kg body weight, intravenously) immediately before MCAO, infarct volume was reduced significantly either at 24 h reperfusion (vehicle versus Bu: 57.3±8.8 versus 34.1±8.2 mm3, P<0.05) or at 72 h reperfusion (vehicle versus Bu: 86.1±9.9 versus 52.0±18.5 mm3, P<0.05). These results are consistent with the data from NKCC1+/− mice.
Furthermore, apoptosis was determined at 72 h reperfusion in the ipsilateral hemispheres of the wild-type brains and NCX1+/−
Because NCX1 protein expression was changed in neurons after in vitro ischemia as described above, we also examined NCX1 protein level in contralateral and ipsilateral hemispheres in NCX1+/+ brains at 0, 6, 24, or 72 h reperfusion. As shown in Figure 5, there were no statistically different changes in expression of full-length NCX1 proteins in ischemic brains at the different times of reperfusion. Some cleavage of NCX1 occurred at 24 and 72 h reperfusion.
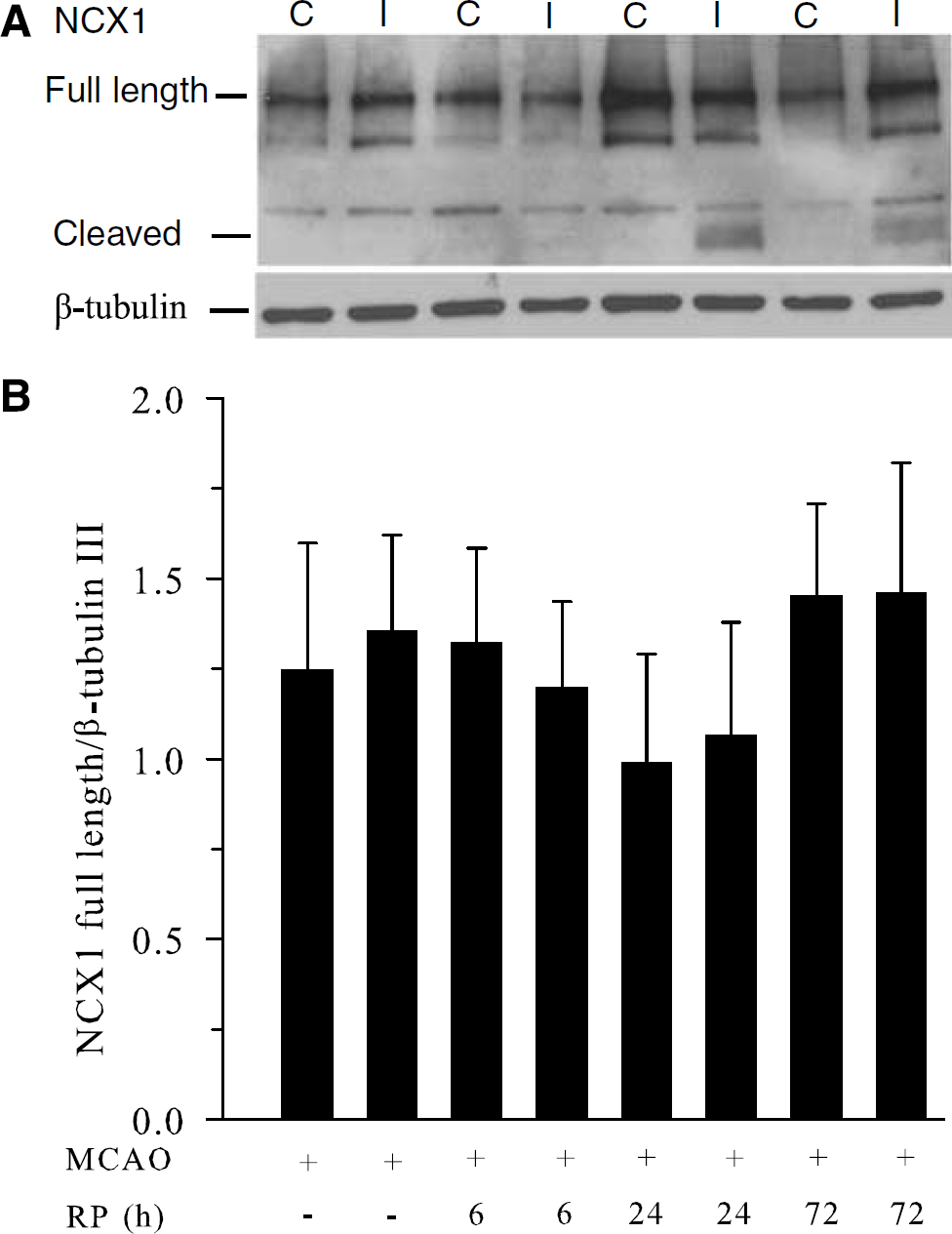
NCX1 expression in NCX1+/+ brains after focal ischemia. (
Discussion
NCX in Ischemic Damage
NCX1–3 isoforms are widely expressed in brain tissues (Annunziato et al, 2004). It was recently reported that all NCX isoforms are predominantly expressed in dendrites and dendritic spines contacted by asymmetric axon terminals (Minelli et al, 2007). In addition, NCX1-3 were also detected in astrocytes in distal processes ensheathing excitatory synapses and perivascular astrocytic endfeet (Minelli et al, 2007). The perisynaptic localizations of NCX1–3 imply that these isoforms may function in buffering intracellular Ca2+ in excitatory postsynaptic sites and in shaping astrocytic intracellular Ca2+ transients after synaptic activity. Therefore, alteration of NCX isoform expression or function after cerebral ischemia will affect Ca2+ handling and contribute to cell damage.
Transient downregulation of NCX1 mRNA occurs in the ischemic core region after 6 to 24 h pMCAO in rats (Boscia et al, 2006). However, in nonischemic regions of the ipsilateral brains, NCX1 transcript is upregulated (Boscia et al, 2006). Transient global ischemia in gerbils induces transient downregulation of NCX1 protein expression in CA1 pyramidal cells and sustained elevation of NCX1 protein level in hippocampal astrocytes (Hwang et al, 2006). Downregulation of NCX2 and NCX3 through small RNA interference exacerbates glutamate-mediated Ca2+ accumulation and subsequent cell death in cerebellar granule neurons (Bano et al, 2005). Antisense oligonucleotide-mediated reduction in NCX1 and NCX3 mRNA and protein expression causes increased cell death in rat brains after pMCAO (Pignataro et al, 2004a). These studies imply that the detrimental effects of the reduction of NCX function may result from inhibition of forward-mode operation of NCX and Ca2+ extrusion in ischemic brains or neurons. This finding is further supported by the recent report on selectively increased resistance to hypoxia in cells overexpressing NCX3 but not NCX1 or NCX2 (Secondo et al, 2007).
Changes of NCX1 Expression after Ischemia
Calpain-mediated selective cleavage of NCX1 and NCX3 has been found in cortex and striatum after tMCAO in rats (Bano et al, 2005). Such cleavage may proteolytically activate the reverse mode of NCX and lead to ischemic cell damage (Omelchenko, personnel communication). In this study, a time-dependent upregulation of NCX1 protein was found in cultured NCX1+/+ neurons during 0 to 21 h REOX. A concurrent increase in NCX1 cleavage occurred during REOX. In contrast, 0 to 72 h reperfusion did not significantly change expression of the full-length NCX1 protein after tMCAO, but cleaved NCX1 occurred at 24 and 72 h reperfusion. These findings suggest that an abundant level of NCX1 (both full-length and cleaved) remains expressed in ischemic brains after transient focal ischemia and could contribute to ischemic damage if functioning in the reverse mode. The discrepancy in upregulation of NCX1 between ischemic neuronal cultures and brains may result from many factors. It could reflect different responses in the two models. However, we may fail to detect a significant change in NCX1 expression in the affected ipsilateral brains by diluting the signals during sampling (mixed cell types and mixed ischemic and nonischemic brain regions).
NKCC1 and NCXrev in Ischemic Damage
NCXrev can occur when intracellular Na+ is overloaded and plasma membrane potential depolarized after ischemia. Inhibition of NCX with KB-R7943, a nonselective inhibitor for NCXrev, is neuroprotective in glucose-deprived/depolarized primary neuron cultures (Kiedrowski et al, 2004). However, when the cells are energized with glucose, Na+/K+-ATPase partially regenerates the Na+ and K+ concentration gradients, which prevents NCXrev (Czyz and Kiedrowski, 2002). This implies that Na+ overload is essential in induction of NCXrev. In cultured astrocytes and neurons, we showed that activation of NKCC1 activity contributes to sustained elevation of [Na+]i, specifically during 60 mins REOX after 2 or 3 h OGD. This Na+ overload triggers NCXrev and intracellular Ca2+ accumulation (Beck et al, 2003; Kintner et al, 2007; Lenart et al, 2004). Moreover, inhibition of NKCC1 activity with bumetanide is neuroprotective against OGD/REOX-mediated cell death (Beck et al, 2003). Ablation of NKCC1 in NKCC1−/− mice reduces cerebral infarction after tMCAO (Chen et al, 2005).
In this study, we investigated further the concerted role of NKCC1 and NCXrev in ischemic damage by examining whether reducing NCX1 activity alone or decreasing activity of NKCC1 and NCX1 is neuroprotective. NCX1+/− neurons expressed ∼50% less protein and ∼60% less NCX activity. However, neither NCX1+/− neurons nor NCX1+/− brains exhibited increased resistance to ischemic damage. This suggests that reduction of NCX1 protein by 50% is not sufficient to affect NCX-mediated Ca2+ extrusion or NCXrev-driven Ca2+ influx after ischemia. Interestingly, 20% more NKCC1 protein was detected in NCX1+/− neurons, which may promote NCXrev after ischemia and enhance ischemic damage.
In contrast, when both NKCC1 and NCX1 were reduced in NCX1+/−/NKCC1+/− neurons, the least cell damage was found after either in vitro or in vivo ischemia. NCX1+/−/NKCC1+/− brains showed a higher tolerance to delayed ischemic damage, reflected by the smallest infarct volume and a fewer TUNEL-positive cells at 72 h reperfusion after tMCAO. These findings imply that NKCC1 is important in ischemic damage, which presumably results from Na+ overload and induction of NCXrev. This speculation is further supported by the increased resistance in NKCC1+/− brains or bumetanide-mediated protection in NKCC1+/+ brains. An alternative interpretation for our results could be that the reduction of NKCC1 activity alone is neuroprotective independent of NCXrev. Thus, there is similar neuroprotection after in vivo ischemia in either bumetanide-treated, NKCC1 knockdown, or double heterozygous-deficient NCX1+/−/NKCC1+/− mice. However, this interpretation fails to account for the higher tolerance to in vitro ischemia in NCX1+/−/NKCC1+/− neurons, compared to either NKCC1+/− or NCX1+/− neurons. Moreover, it cannot explain our previous studies that showed there was a reduction in intracellular Na+ accumulation and organelle Ca2+ accumulation after in vitro ischemia when we inhibited NKCC1 pharmacologically or by genetic ablation in astrocytes. Ca2+ accumulation in organelles could also be significantly reduced by inhibition of NCXrev (Kintner et al, 2007). Additionally, we have shown that NCXrev activity is increased in ischemic astrocytes (Lenart et al, 2004).
In agreement with our view on NCXrev, the protective effects of inhibiting NCXrev were found in other models, such as myocardial and renal ischemia and reperfusion injury (Lee et al, 2005). It was reported that cardiac-specific ablation of NCX1 abolishes Ca2+ influx via NCXrev (Maloyan et al, 2005). Moreover, cardiac-specific NCX1−/− hearts exhibit significantly less necrosis after 20 mins ischemia and 120 mins reperfusion and have improved postischemic cardiac function (Maloyan et al, 2005). However, overexpression of cardiac NCX1 increases susceptibility to ischemia/reperfusion injury in male mice (Cross et al, 1998). Taken together, our results are consistent with the findings in myocardium ischemia and reperfusion injury.
In summary, this study shows that abundant NCX1 protein was expressed in neurons. In vitro ischemia induced upregulation of NCX1 protein expression. Although cleavage of NCX1 protein occurred during REOX, the dominant form was full-length NCX1. Attenuation of NCX1 activity alone was not sufficient to produce neuroprotection after in vitro or in vivo ischemia. Reduction of both NCX1 and NKCC1 activity resulted in the least ischemic damage after the ischemic insult in both models. Taken together, these findings further stress the important role of NKCC1, in conjunction with NCX1, in ischemic damage.
Footnotes
Acknowledgements
We thank Dr Lucio Annuziato for participating in many discussions and for reading the manuscript. We thank Dr Hartmut Porzig for providing NCX2 antibodies.