
Abstract
Aquaporin-4 (AQP4) has been shown to be important in the evolution of stroke-associated cerebral edema. However, the role of AQP4 in stroke-associated cerebral edema as it pertains to sex has not been previously studied. The perivascular pool of AQP4 is important in the influx and efflux of water during focal cerebral ischemia. We used mice with targeted disruption of the gene encoding α-syntrophin (α-Syn−/−) that lack the perivascular AQP4 pool but retain the endothelial pool of this protein. Infarct volume at 72 h after transient focal ischemia (90 mins) in isoflurane-anesthetized mice was attenuated in both sexes with α-Syn deletion as compared with their wild-type (WT) counterparts. There were no sex differences in hemispheric water content in WT and α-Syn−/− mice or regional AQP4 expression in WT mice. In neither sex did α-Syn deletion lead to alterations in end-ischemic regional cerebral blood flow (rCBF). These data suggest that after experimental stroke: (1) there is no difference in stroke-associated cerebral edema based on sex, (2) AQP4 does not involve in sex-based differences in stroke volume, and (3) perivascular pool of AQP4 has no significant role in end-ischemic rCBF.
Introduction
Cerebral edema is an invariable accompaniment of ischemic stroke and is a major cause of morbidity and mortality after large hemispheric infarctions (Hacke et al, 1996). Although there have been numerous experimental studies showing that female sex is relatively protected for sustaining brain injury as compared with men after experimental stroke (McCullough and Hurn, 2003), there is a paucity of studies that have investigated the influence of sex on stroke-associated cerebral edema. In an isolated clinical observational study (Maramattom et al, 2004), female patients were shown to worsen clinically to a greater extent than male patients (72% versus 20%) during their hospital course after large middle cerebral artery territory infarction. However, it remains unclear whether this deterioration was attributable to stroke-associated cerebral edema.
The mechanisms of edema formation after cerebral ischemia are complex and have not been completely elucidated. Historically, poststroke edema has been categorized into a cytotoxic component secondary to energy failure and a delayed vasogenic component secondary to breakdown of the blood-brain barrier with consequent leakage of plasma constituents (Klatzo, 1967). Other secondary mechanisms that have been shown to be important in accentuating ischemia-evoked cerebral edema include impedance of cerebral venous return from cerebral swelling, intrahemispheric diaschisis (Abe et al, 2000), inflammation accentuating blood—brain barrier disruption (Abbott, 2000), neurohormonal responses (Bemana and Nagao, 1999; Chang et al, 2006), and modulating effects of growth factors (van Bruggen et al, 1999). Of recent, aquaporin-4 (AQP4), the most abundant water channel in brain, has been implicated in the pathogenesis of cerebral edema in a variety of brain injury paradigms including ischemic stroke (King and Agre, 1996; Manley et al, 2000; Badaut et al, 2002; Vajda et al, 2002; Amiry-Moghaddam et al, 2003a, 2004; Papadopoulos et al, 2004; Bloch et al, 2005; Ribeiro et al, 2006). AQP4 is strongly enriched at the brain-blood interface (Frigeri et al, 1995; Nielsen et al, 1997). Semiquantitative analyses using immunogold electron microscopy have revealed two AQP4 pools at this site: a major perivascular pool, localized to the luminal membranes of astrocytic end-feet, and a minor endothelial pool, localized to the luminal as well as abluminal membranes of endothelial cells (Amiry-Moghaddam et al, 2004). The end-foot pool of AQP4 is anchored by the dystrophin-associated protein complex (Neely et al, 2001) and is deleted in mdx mice (which lack the dystrophin-associated protein complex) and in mice with targeted disruption of the gene encoding α-syntrophin (α-Syn−/−), a member of the dystrophin-associated protein complex. Both types of mice show a reduced extent of cerebral edema induced by hyponatremia or stroke (Vajda et al, 2002;
The purpose of the present study was not to study the effects of sex steroids on outcomes but to address whether there is a difference in evolution of edema after experimental stroke in adult male and randomly cycling female animals and if AQP4 is involved in this process. Using Syn−/− mice and their wild-type (WT) counterparts of both sexes, the present study was designed to test the hypotheses that: (1) there are significant sex differences in stroke-associated cerebral edema, (2) there are sex differences in AQP4 expression after transient focal ischemia, (3) the perivascular pool of AQP4 is an important determinant of sex-based differences in stroke-associated histological injury as well as cerebral edema, and (4) the perivascular pool of AQP4 modulates regional cerebral blood flow (rCBF) after focal ischemia.
Materials and methods
General Preparation and Animal Surgery
The experimental protocol was approved by the Institutional Animal Care and Use Committee and conformed to the National Institutes of Health guidelines for the care and use of animals in research. All techniques were conducted as previously described (Sawada et al, 2000) with modifications (Zeynalov et al, 2006). All experiments were conducted with male and female mice homozygous for targeted disruption of the gene encoding α-Syn−/−. WT C57Bl/6 mice of both sexes were used as controls. α-Syn−/− mice were bred on a C57Bl/6 background more than 10 generations to avoid effects of differing genetic strains (Adams et al, 2000). Although WT and α-Syn−/− male mice were age matched, female α-Syn−/− mice were approximately 1 to 2 months older on average than their WT counterparts.
Focal Ischemia
Male and female WT and α-Syn−/− mice with body weight of 20 to 28 g were anesthetized with 1.5% to 1.8% isoflurane in oxygen-enriched air, and rectal temperature was maintained at 37°C±0.5°C with heating lamps during the entire surgical procedure until emergence from anesthesia. Rectal temperature was maintained throughout surgical procedures, during ischemia, and until emergence from anesthesia.
Transient focal ischemia (90 mins) was produced by middle cerebral artery occlusion (MCAO; Yang et al, 1994; Hara et al, 1996) with modifications (Sampei et al, 2000; Sawada et al, 2000; Zeynalov et al, 2006) using an intraluminal suture technique in combination with laser-Doppler flowmetry (LDF; Moor Instruments Ltd, Model MBF3D, UK) over the ipsilateral parietal cortex. Briefly, the common carotid artery was temporarily occluded and a 6-0 silicon-coated nylon monofilament was inserted via an external carotid artery stump distal to the internal carotid artery/pterygopalatine bifurcation until LDF signal decreased to < 20% of preischemic baseline values. Middle cerebral artery occlusion was verified by allowing the animal to emerge from anesthesia and carrying out a neurological deficit scoring (NDS; Manley et al, 2000; Sampei et al, 2000) that was as follows: 0 = normal motor function, 1 = flexion of torso and of contralateral forelimb on tail lift, 2 = circling to the contralateral side but normal posture at rest, 3 = leaning to contralateral side at rest, and 4 = no spontaneous motor activity. Animals that did not show reduction in LDF signal or that had an NDS ≤ 1 on emergence from anesthesia were excluded from the study. Mice with clear neurological deficits (score ≥ 2) were reanesthetized for withdrawal of the suture and reperfusion after 90 mins of MCAO. Surgical shams were subjected to similar anesthetic exposure and surgical manipulations except for advancement of intraluminal suture. Animals were housed in separate cages at room temperature and weighed daily until the end of the experiment. After completion of treatments post-MCAO, the brains were harvested for assessment of hemispheric water content, injury volume, AQP4 expression, and rCBF measurements.
Assessment of Infarct Volume
At the end of the experiment (72 h post-MCAO), mice were killed by decapitation under deep isoflurane (5%) anesthesia. The forebrain was sliced into five 2-mm thick coronal sections, which were stained with 1.2% triphenyltetrazolium chloride, as described previously (Sampei et al, 2000; Sawada et al, 2000; Zeynalov et al, 2006). Infarct volume was measured with digital imaging. Infarct volumes were numerically integrated across each section and over the ipsilateral hemisphere. Infarct volumes were measured separately in the cerebral cortex and caudoputamen (CP) complex and expressed as percent of contralateral structure, as previously described (Sawada et al, 2000; Zeynalov et al, 2006).
Assessment of Brain Edema
After decapitation as described above, the brain was quickly removed and dissected along the interhemispheric fissure into the ischemic and nonischemic cerebral hemispheres. Brain edema was assessed by comparing wet-to-dry ratios (WDR) as described previously (Lin et al, 1993; Chang et al, 2006; Chen et al, 2006; Toung et al, 2007). Tissues were weighed with a scale to within 0.1 mg. Dry weight of the brain was determined after heating the tissue for 3 days at 100°C in a drying oven. Tissue water content was then calculated as °%H2O = (1–dry wt/wet wt) × 100% (Lin et al, 1993).
Immunoblotting for AQP4
We modified a previously described Western blot protocol for AQP4 measurements (Kako et al, 1998; Ouyang et al, 1999; Chen et al, 2007). The cerebral cortex was subdissected from WT male and female mice at 48 h of reperfusion after 90 mins MCAO. Tissues were homogenized in cold homogenization buffer A (250 mmol/L sucrose, 60 mmol/L KCl, 15 mmol/L Tris-HCl (pH 7.9), 15 mmol/L NaCl, 5 mmol/L EDTA, 1 mmol/L EGTA), 1 mmol/L DL-dithiothreitol, 0.5 mmol/L phenylmethane-sulfonyl fluoride containing protease (Complete Mini EDTA-Free Protease Inhibitor Cocktail, Roche Applied Science Inc., Basel, Switzerland), and phosphatase inhibitors (Sigma-Aldrich, Saint Louis, MO, USA). Nuclear and membrane fractions pellet was isolated by centrifugation 2,000g for 10 mins at 4°C, and then dispersed with new buffer (Buffer A, 1% Triton 100, 0.1% SDS), followed by 4,000g centrifugation for 10 mins at 4°C. Protein concentrations in the supernatant were determined by BCA protein assay kit (Piece Biotechnology, Rockford, IL, USA), and 5 %μg membrane proteins per lane were loaded into 12% NuPAGE Bis-Tris gel (Invitrogen Corp., Carlsbad, CA, USA) after warming at 37°C for 10 mins. Proteins were then transferred to a polyvinylidene difluoride membrane, which was then blocked (1 h at room temperature) with 5% nonfat dry milk/1 × phosphate-buffered saline/0.1% Tween-20 solution, and probed with a polyclonal anti-AQP4 antibody (1:5000 dilution, Alpha Diagnostics, San Antonio, TX, USA) in blocking solution overnight at 4°C. Blots were then incubated with a secondary horseradish peroxidase-linked goat anti-rabbit antibody (1:2000 dilution) for 1 h at room temperature, and signals were visualized by enhanced chemiluminescence and intensity measured by Quantity One image analysis software (Bio-Rad Laboratories, Hercules, CA, USA). β-Actin was used as a loading control. Band intensities from the ipsilateral hemispheres of ischemic animals were normalized to levels in corresponding contralateral hemispheres.
Regional Cerebral Blood Flow Measurements
End-ischemic rCBF was measured in nonsurvival cohort of C57BL/6 and α-Syn−/− male and female mice (
Assessment of Plasma Osmolality
At the end of the experiment, a sample of blood was drawn by cardiac aspiration to determine serum osmolality (mOsm/L) with an automated freezing point depression micro-osmometer (Advanced Instruments Inc., Norwood, MA, USA) (Chang et al, 2006; Chen et al, 2006, 2007; Toung et al, 2007).
Experimental Groups
All experiments were carried out in a randomized manner by a single investigator. In the first series of experiments in WT and α-Syn−/− male (M) and female (F) mice (
Statistical Analysis
All values are expressed as mean±s.e.m. Physiologic parameters, plasma osmolality, mean LDF measurements, daily and mean body weight loss among groups were subjected to repeated-measures analysis of variance. Infarct volume and differences in hemispheric brain water content were determined by two-way analysis of variance with
Results
In the first series of experiments conducted to determine whether α-Syn deletion attenuates infarct volume in both sexes, NDS, weight loss, and serum osmolality were similar in all treatment groups (data not shown). Premature mortality (before 72 h post-MCAO) was 1/14 in WT-M, 0/13 in WT-F, 1/13 in α-Syn−/−-M and 1/14 in α-Syn−/−-F mice. Three mice (1 WT-M, 1 WT-F, and 1 α-Syn−/−-F) sustained a secondary intracerebral hemorrhage (ICH) and hence were excluded from the final infarct size analysis. Thus, 12 mice in each experimental group were included in the final analysis. Infarct volume was significantly attenuated in the cortex and CP complex in α-Syn−/−-M (cortex: 31%±7%; CP: 75%±7%; percent of contralateral structure) and α-Syn−/−-F (cortex: 25%±5%; CP: 70%±5%) as compared with their WT counterparts (WT-M, cortex: 60%±2%; CP: 98%±1%; WT-F, cortex: 51%±5%; CP: 89%±4%) (Figure 1A and 1B).
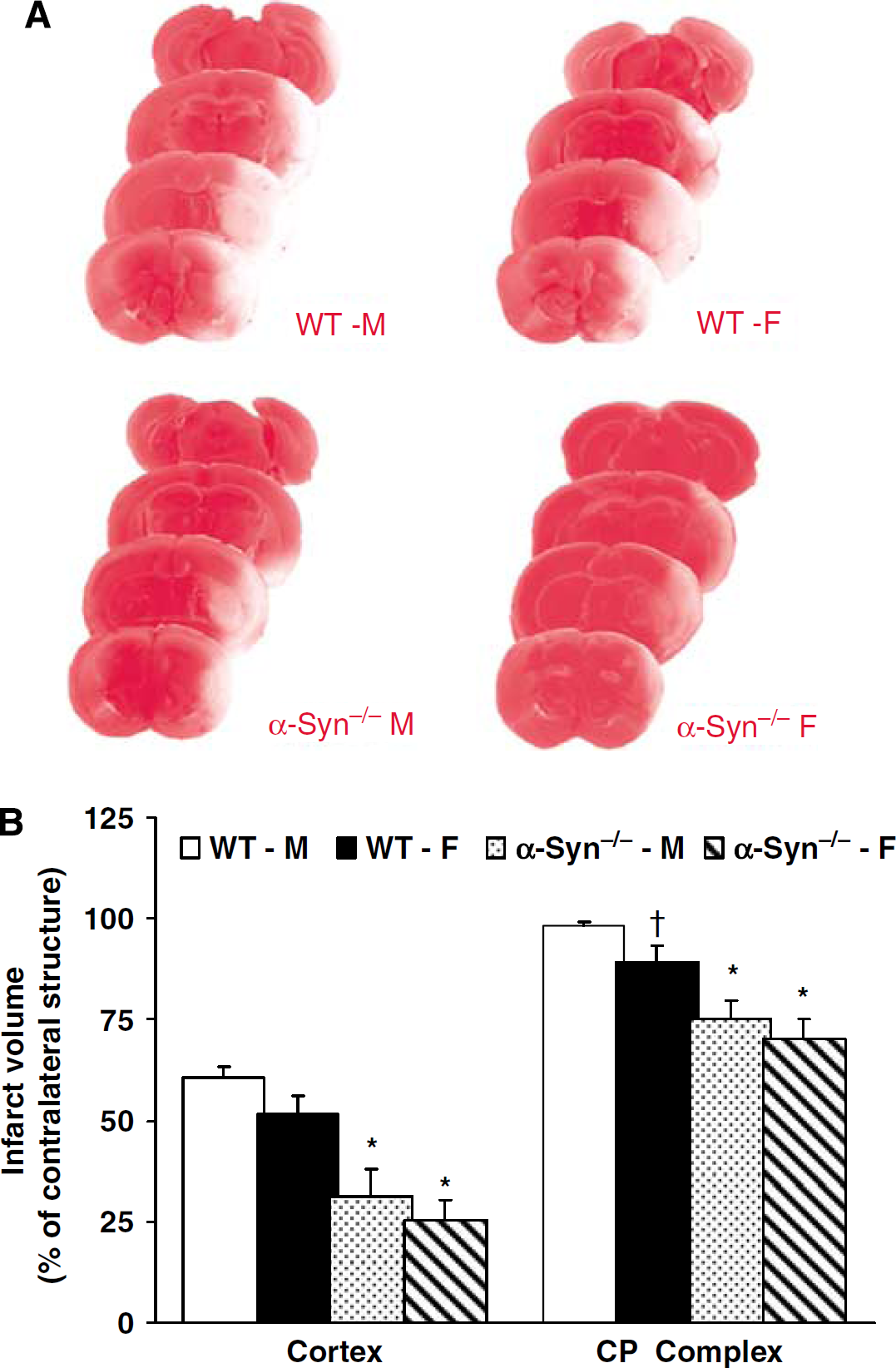
(
In the second set of experiments conducted to determine whether α-Syn deletion attenuates cerebral edema associated with cerebral ischemia in both sexes, NDS and daily and overall weight loss were also similar in all treatment groups (data not shown). Premature mortality (before 48 h post-MCAO) was 0/10 in WT-M, 1/12 in WT-F, 1/13 in α-Syn−/−-M, and 2/13 in α-Syn−/−-F. Four mice (one WT-F, two α-Syn−/−-M, and one α-Syn−/−-F) sustained a secondary intracerebral hemorrhage and were excluded from the final analysis. Thus, 10 mice in each experimental group were included in the final analysis. Ipsilateral hemispheric water content was significantly attenuated in α-Syn−/−-M (82.2% ±0.7%) and α-Syn−/−-F (81.6%±0.7%) as compared with their WT counterparts (WT-M: 84.4%±0.5%; WT-F: 83.9%±0.6%) (Figure 2).
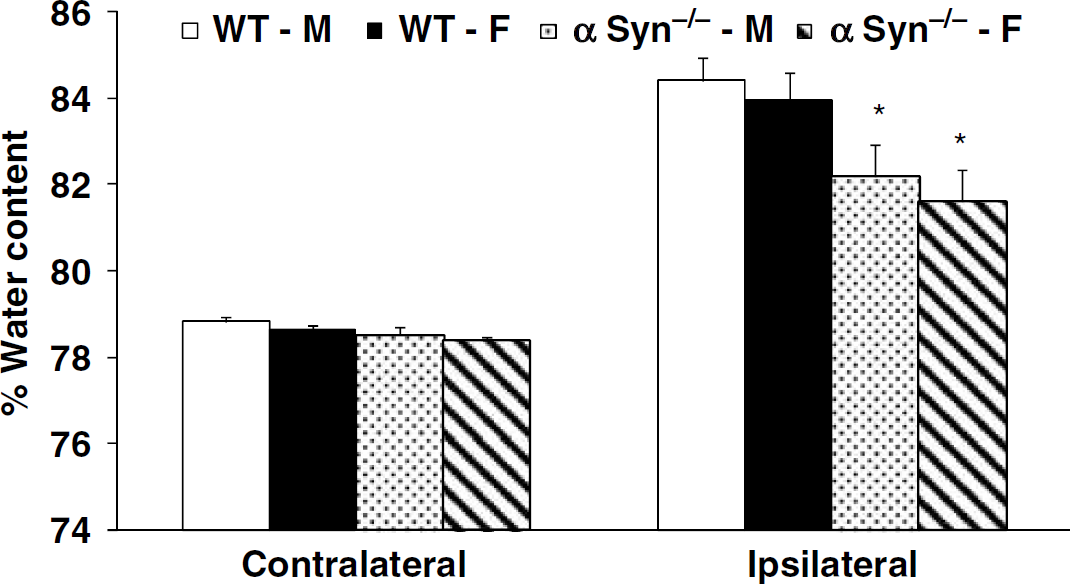
Cerebral hemispheric (ipsilateral and contralateral) water content at 48 h of reperfusion after 90 mins of MCAO in WT-M, WT-F, α-Syn−/−-M, α-Syn−/−-F (
In the third set of experiments conducted to determine if there are sex differences in AQP4 protein expression after cerebral ischemia, there was one death before 48 h post-MCAO in WT-F group. Thus, animals included in the final analysis were WT-M (
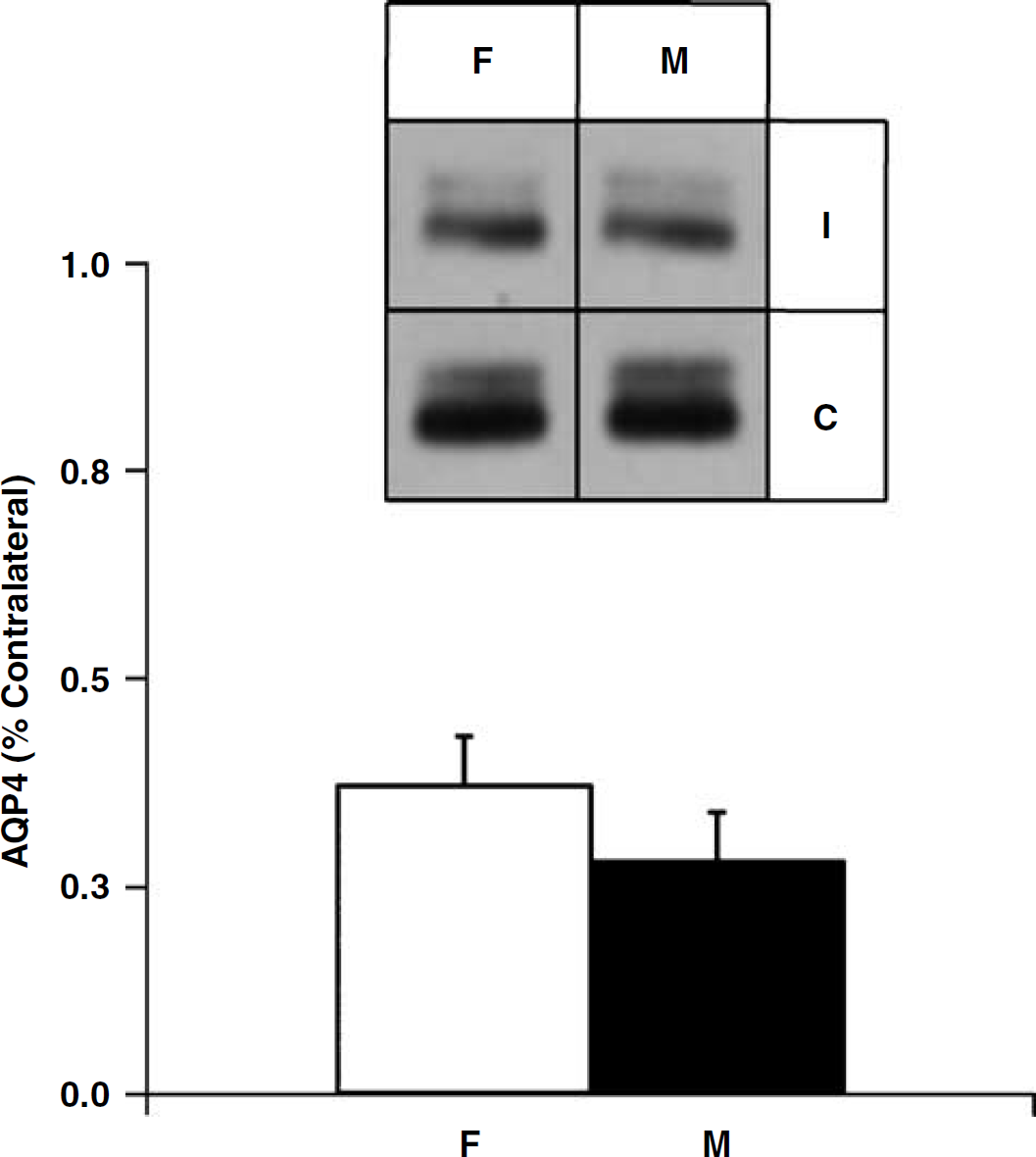
Immunoblot of membrane protein isolated from the cortex of ischemic (I) and contralateral (C) hemispheres in WT-M (
In the fourth set of experiments conducted to determine if α-Syn deletion alters end-ischemic rCBF in both sexes, there was no premature mortality before completion of the experimental protocol. The final analysis included WT-M (
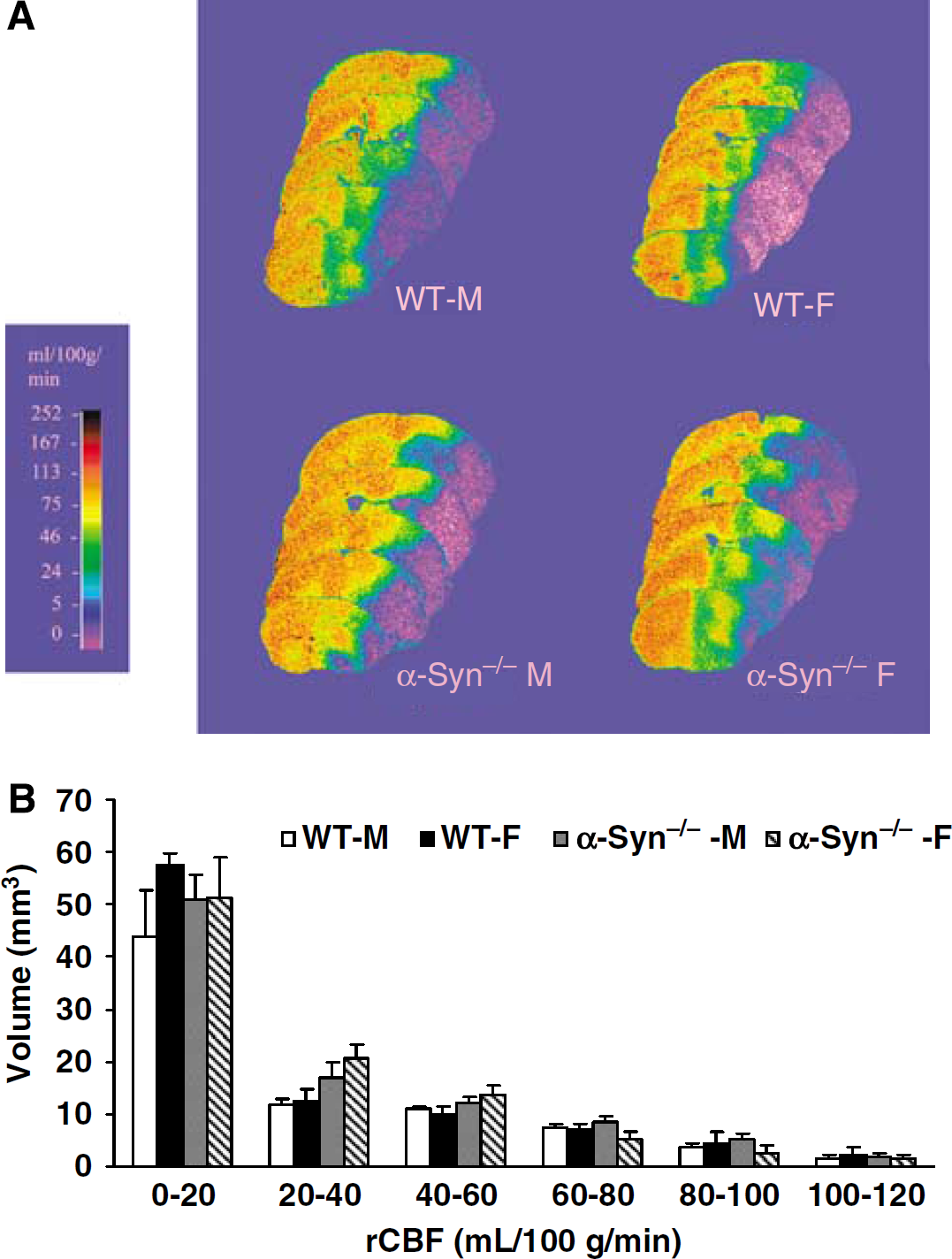
(A) Cumulative distribution of CBF rates within the ischemic hemisphere at end of 90 mins MCAO in WT-M, WT-F, α-Syn−/−-M, α-Syn−/−-F (
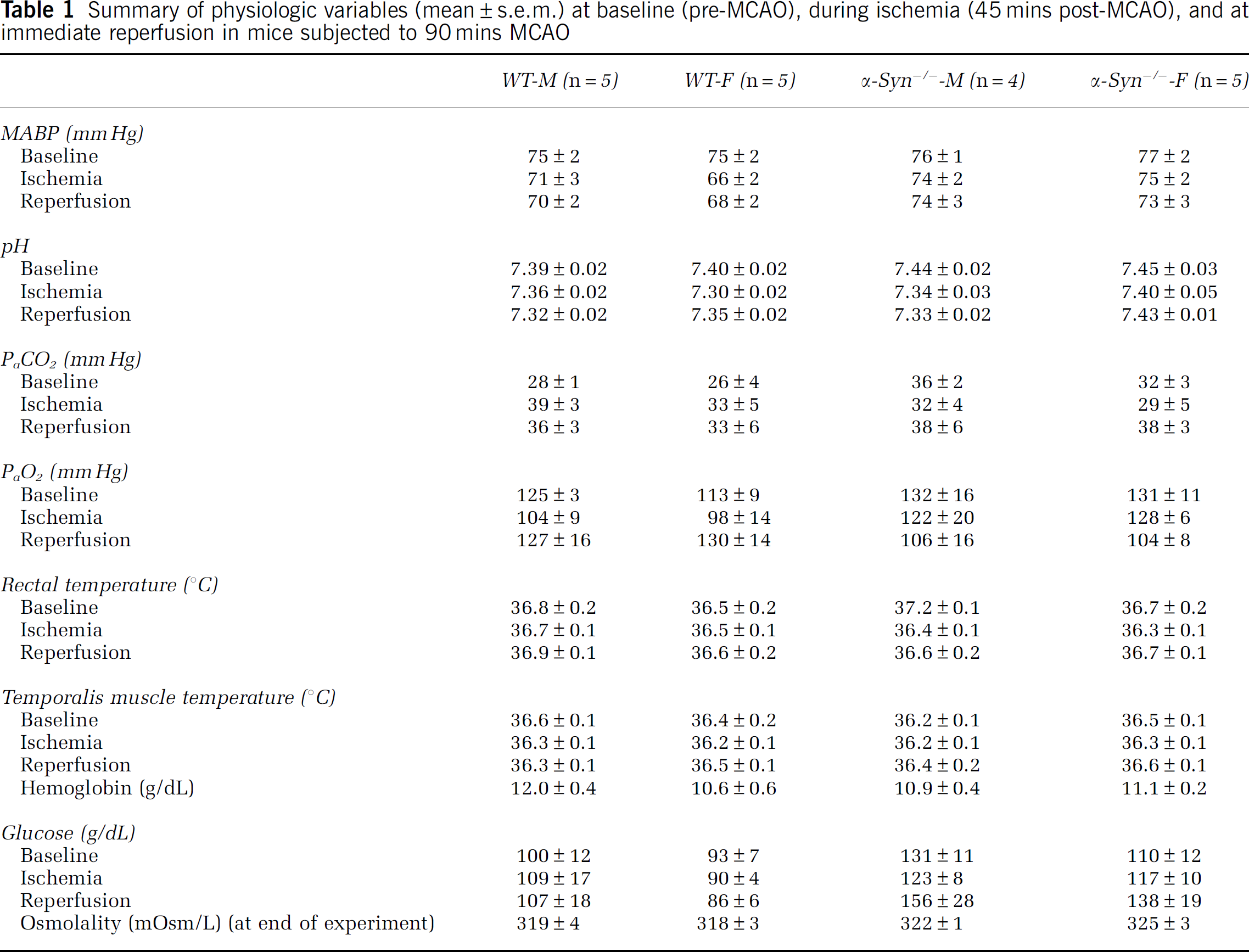
In the fifth set of experiments conducted to determine if α-Syn deletion in both sexes alters physiologic parameters during cerebral ischemia (WT-M:
Summary of physiologic variables (mean±s.e.m.) at baseline (pre-MCAO), during ischemia (45 mins post-MCAO), and at immediate reperfusion in mice subjected to 90 mins MCAO
Discussion
This study shows several important and novel findings. First, both male and female mice that lack the perivascular pool of AQP4 (α-Syn-null mice) have lesser degree of stroke-associated cerebral edema compared with their WT counterparts. However, there are no differences in stroke-associated cerebral edema based on sex. Second, AQP4 does not appear to be involved in sex-based differences in stroke volume and finally, the perivascular pool of AQP4 does not alter end-ischemic rCBF after ischemic stroke.
Sex-based differences in histological and functional outcome after ischemic stroke have received much attention recently. Numerous experimental studies using well-characterized models of ischemic stroke have shown the relative protection from brain injury in the female sex as compared with male sex (Alkayed et al, 1998; McCullough et al, 2001). A variety of mechanisms have been pursued including neuroprotection conferred by endogenous sex steroids (Rusa et al, 1999) that may cause beneficial redistribution of rCBF (McCullough et al, 2001) and modulation of ischemia-evoked release of excito-toxic neurotransmitters (Joh et al, 2006). However, to our knowledge sex-based differences in the magnitude of stroke-associated cerebral edema have not been studied previously.
AQP4 has been implicated in the pathogenesis of cerebral edema associated with ischemic stroke (King and Agre 1996; Manley et al, 2000; Vajda et al, 2002; Amiry-Moghaddam et al, 2003a, Ribeiro et al, 2006). In addition, AQP4 has been shown to facilitate resorption of excess fluid in vasogenic cerebral edema associated with brain tumor and contusive injury (Bloch et al, 2005) and bacterial abscess (Papadopoulos et al, 2004). However, the specific role and function of the perivascular AQP4 pool remains to be fully elucidated. Analyses of gene-targeted or mutant animals that lack perivascular AQP4 have indicated that this AQP4 pool is rate limiting for the rapid water exchange that occurs between blood and brain in the accumulation and resolution phases of brain edema (Vajda et al, 2002; Amiry-Moghaddam et al, 2003a, 2004; Amiry-Moghaddam and Ottersen, 2003c). Specifically, a selective deletion of the perivascular AQP4 pool by targeted disruption of α-syn causes a pronounced decrease in the extent of brain edema after a transient ischemic insult (Amiry-Moghaddam et al, 2003a). α-Syn contributes to the organization of the dystrophin complex in astrocytes (Bragg et al, 2006) and is essential for anchoring of AQP4 to the perivascular end-foot membranes (Neely et al, 2001) where it is densely packed in distinctive rafts orthogonal arrays of proteins (Neely et al, 2001; Rash et al, 2004). We have previously shown that the blood-brain barrier remains intact after α-syn deletion (Amiry-Moghaddam et al, 2004).
The present study takes advantage of the transgenic animal model and provides new insight in the physiological and pathophysiological roles of perivascular AQP4 as it relates sex differences in ischemic stroke. Animals with α-Syn deletion resulted in less injury volume in both sexes as compared with WT counterparts at 72 h post-MCAO. This time point was chosen to ascertain injury volume after the edema had passed its peak. These results are in keeping with our previous studies (Amiry-Moghaddam et al, 2003a) where infarct volume was also significantly attenuated in α-syn−/− mice compared with WT at an earlier time point (24 h post-MCAO). The explanation for the reduction in injury remains elusive at present, but we postulated that it reflects an alleviation of the secondary effects of edema and its beneficial effects on rCBF. No animals were excluded from this part of the study and there was no premature mortality before completion of the experimental protocol. Role of pericytes (Peppiatt et al, 2006) and the possible contribution of volume changes in end-feet and endothelial cells may be important in the regulation of brain rCBF. Both types of volume changes would be expected to affect rCBF through the microvessels by impinging on and occluding their lumen. Our data show that these differences are not secondary to alterations in end-ischemic rCBF, as we observed no differences in male and female animals with α-syn deletion as compared with their WT counterparts. Hence we infer from this data that the perivascular pool of AQP4 does not alter end-ischemic rCBF after focal cerebral ischemia. However, it is plausible that there are differences in rCBF at later time points, although we have previously shown marked heterogeneity in rCBF during reperfusion after focal ischemic stroke (Bhardwaj et al, 2000).
We did not observe any differences in stroke-associated cerebral edema between male and female animals in the present study. Cerebral edema was determined at 48 h after transient focal ischemia, the time point of maximal edema in our animal model (data not shown). Serum osmolality at this time point was comparable in all experimental groups. Serum osmolality is a critical determinant of cerebral edema in variety of brain injury paradigms including ischemic stroke because a hyperosmolar state causes egress of brain water into the vascular compartment (Toung et al, 2007) and can be important confounder in the magnitude of cerebral edema observed in our experimental paradigm. As in our previous studies (Chang et al, 2006; Chen et al, 2006, 2007; Toung et al, 2007), wet-to-dry ratios comparisons were used as a simple and reproducible assessment of brain water in both ischemic and nonischemic hemispheres.
Immunoblotting studies to determine total AQP4 expression (astrocytic and endothelial pools) did not show any appreciable differences in AQP4 expression based on sex. Densitometric analysis shown no sex differences in AQP4 expression in the ischemic cortex (representing ischemic core and penumbral regions) or the CP complex (representing ischemic core predominantly). AQP4 expression was decreased in the ischemic cortex but not in the CP complex as compared with the non-ischemic contralateral hemisphere. AQP4 expression did not differ between naive male and female α-Syn mice (data not shown). It is to be noted, however, that α-syn-null mice show loss of AQP4 from perivascular and subpial membranes but no decrease in other membrane domains in the cerebral cortex, as we shown previously using quantitative immunogold electron microscopy (Amiry-Moghaddam et al, 2004). Furthermore using similar techniques, we have also shown that there is a temporary loss of AQP4 from perivascular end-foot membranes after transient MCAO (Frydenlund et al, 2006).
It must be emphasized that the water flux across the brain-blood interface is not mediated exclusively by AQP4 (Amiry-Moghaddam and Ottersen, 2003c). In addition to a slow diffusion through the plasma membrane, water is subjected to cotransport with ions and organic molecules (monocarboxylate, glucose, and potassium/chloride transporters). We have examined the known water-transporting molecules at the brain-blood interface previously and none of these (except perivascular AQP4) appears to be affected by disruption of the α-syn gene (Amiry-Moghaddam and Ottersen, 2003c). The effects presently observed after α-syn deletion can be attributed to the loss of perivascular AQP4. Whether α-syn deletion and ischemia have additive effects on AQP4 distribution has not been tested but is unlikely given that both interventions alone lead to a complete or near-complete elimination of the polarized AQP4 distribution. It is very likely that all of the downstream effects of α-syn deletion addressed in the present study can be attributed to AQP4, as (1) α-syn deletion has been shown to reduce the AQP4 pool in perivascular membranes by < 90%, and (2) AQP4 is the only water channel expressed in significant amounts at the brain—blood interface. We cannot rule out that α-syn serves to anchor an unknown cotransporter (i.e., a cotransporter not included above). In this unlikely event, the loss of such a cotransporter could have contributed to the changes observed.
Our study has caveats and limitations. We used young animals in our study. On the average α-syn−/− female mice were 1 to 2 months older than their WT counterparts in our study to ensure that animals were weight matched. Body weight is the critical determinant of consistency of injury volume in our model of ischemic stroke because vascular anatomy varies with body weight. Our study cannot comment on the effect of age on AQP4 expression after cerebral ischemia. This study was not designed to investigate the role of sex steroids in the observed outcomes. Although androgens have been shown to have deleterious effects in experimental stroke outcome (Cheng et al, 2007), estrogen has been shown to confer significant neuroprotection and conversely estrogen withdrawal after ovariectomy accentuates injury following experimental ischemic stroke (McCullough and Hurn, 2003). Progesterone has a significant antiedema effect in a variety of brain injury paradigms including traumatic brain injury and ischemic stroke (Guo et al, 2006). We used weight-matched WT and α-syn−/− male and female animals that were randomly cycling during the estrous cycle. We assessed cerebral edema at its peak (48 h post-MCAO) based on our previous work in our model in male animals and it is plausible that the rate and magnitude of stroke-associated cerebral edema is different in male and female animals. Further studies should incorporate aged female animals as well as those after overiectomy to address the interaction of sex steroid hormones and AQP4. Although we observed no alterations in end-ischemic rCBF in male and female mice with α-syn deletion, the study cannot comment on any alterations that may occur during the delayed reperfusion phase. Our study does not provide an explanation for attenuation of injury volume with α-Syn deletion. However, it noteworthy that α-Syn deletion causes other compensatory changes in a variety of proteins including a modest reduction in perivascular Kir4.1 as previously shown (Amiry-Moghaddam et al, 2003b) that may account for attenuation of injury volume. However, other molecules engaged in transport processes across the blood-brain interface (e.g., monocarboxylate transporter 1, glucose transporter 1, excitatory amino-acid transporter 2, and the Na-K-2Cl cotransporter) are not affected by α-Syn deletion. Lastly, we did not observe robust improvements in functional neurologic deficits in α-syn−/− mice as compared with their WT counterparts in our study. Large injury volume with 90-mins MCAO may account for the lack of robust differences in functional outcome in our model.
Conclusions
To our knowledge, this is the first study to investigate the sex-based differences in stroke-associated cerebral edema as it pertains to AQP4 in a well-characterized murine animal model. There appears to be no appreciable sex differences in AQP4 expression after experimental ischemic stroke. However, further studies are needed to investigate the interaction of sex steroid hormones with AQP4 in this injury paradigm. The perivascular pool of AQP4 does not play a significant role in end-ischemic rCBF.
Footnotes
Acknowledgements
We thank Stepahnie J. Murphy, DVM, PhD, for maintaining the colony for transgenic mice, Susan Parker, and Jennifer Young for their technical help and Tzipora Sofare, for her editorial assistance in preparing this paper.