
Abstract
Thrombin and plasmin are serine proteases involved in blood coagulation and fibrinolysis, whose precursors are circulating in blood stream. These blood-derived proteases might play important roles in the pathogenesis of intracerebral hemorrhage by acting on brain parenchymal cells. We previously reported that thrombin induced delayed neuronal injury through extracellular signal-regulated kinase (ERK)-dependent pathways. Here, we investigated potential cytotoxic actions of plasminogen, a precursor protein of plasmin, using slice cultures prepared from neonatal rat brain and intracortical microinjection model in adult rats. Although plasminogen alone did not evoke prominent neuronal injury, plasminogen caused significant neuronal injury when combined with a moderate concentration of thrombin (30 U/mL) in the cerebral cortex of slice cultures. The cortical injury was prevented by tranexamic acid and aprotinin. The combined neurotoxicity of thrombin and plasminogen was also prevented by PD98059, an inhibitor of ERK pathway, as well as by other agents that have been shown to prevent cortical injury induced by a higher concentration (100 U/mL) of thrombin alone. Extracellular signal-regulated kinase phosphorylation after plasminogen exposure was localized in cortical astrocytes. Moreover, microinjection of plasminogen in vivo potentiated thrombin-induced cortical injury, and inhibition of plasmin ameliorated hemorrhage-induced neuronal loss in the cerebral cortex. These results suggest that plasminogen/plasmin system augmenting thrombin neurotoxicity participates in hemorrhagic cortical injury.
Introduction
Intracerebral hemorrhage represents an acute stroke characterized by extravasation of blood into brain parenchyma and formation of hematoma. Although intracerebral hemorrhage (ICH) accounts for 15% of all strokes, no satisfactory medical treatments have been developed against this disorder, and poor prognosis and high mortality have been reported in patients. After hemorrhagic ictus, hematoma may cause brain injury via cytotoxicity of blood-derived molecules as well as via mass effects (Xi et al, 2006). Thrombin is one of the major blood-derived serine proteases released into brain parenchyma when cerebral vessels are ruptured. Besides its important role in blood coagulation, thrombin can induce neuronal apoptotic death and microglial activation in various experimental models (Donovan et al, 1997; Fujimoto et al, 2006, 2007).
Plasmin is another serine protease derived from a circulating precursor protein, plasminogen. Cleavage between Arg-560 and Val-561 of plasminogen molecule, which is catalyzed by plasminogen activators such as urokinase and tissue-type plasminogen activators, generates plasmin (Robbins et al, 1967). Plasmin is well known as a fibrinolytic serine protease that interacts with polymerized fibrin clots to release free fibrin molecule and dissolve clots (Castellino and Ploplis, 2005). The catalytic domain of plasmin is localized in the light chain derived from the C terminus of plasminogen, whereas five kringle domains are within the heavy chain from the N terminus of plasminogen (Patthy, 1985). The kringle domain of plasmin(ogen) possesses lysine-binding activity, and plasminogen binds its substrates such as fibrin through lysine-binding sites, which results in the enhancement of plasmin generation (Hajjar et al, 1986; Wu et al, 1990).
Plasmin(ogen) may have physiologic/pathophysiologic functions in the central nervous system, because expression of plasminogen has been detected in central neurons (Tsirka et al, 1997b). In a pathologic context, neuroxicity of plasminogen in the striatum has been shown in an in vivo experimental model (Xue and Del Bigio, 2001, 2005). In addition, plasmin-mediated degradation of the extracellular matrix enhances vulnerability of hippocampal neurons to excitotoxins (Chen and Strickland, 1997; Tsirka et al, 1997a). A detrimental role of plasminogen/plasmin system in ischemic brain injury has also been proposed (Takahashi et al, 2005).
When ICH occurs, circulating plasminogen and plasminogen activators are released into the cerebral parenchyma, and plasmin generated at the hemorrhagic sites may contribute to the brain injury after ICH. In this study, we investigated the pathogenic roles of plasminogen with relation to ICH. We particularly focused on the interaction of plasminogen with the neurotoxic actions of thrombin.
Materials and methods
Drugs and Chemicals
Drugs and chemicals were obtained from Nacalai Tesque (Kyoto, Japan), unless otherwise indicated. Thrombin from bovine plasma (cat. no. T4648), bovine serum albumin (cat. no. A2153), aprotinin from bovine lung, and clodronate were obtained from Sigma (St Louis, MO, USA). Tranexamic acid was from Aldrich (St Louis, MO, USA). Plasminogen from human plasma (cat. no. 528175), plasmin from human plasma (cat. no. 527621), PD98059, SB203580, 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2), and bisindolylmaleimide (BIM) were from Calbiochem (San Diego, CA, USA). SP600125 was from Tocris Cookson (Bristol, UK). Argatroban was from Sawai Pharmaceutical (Osaka, Japan). Cycloheximide was from Wako Pure Chemical (Osaka, Japan).
Preparation of Slice Cultures
All experimental procedures were approved by our institutional animal experimentation committee, and animals were treated in accordance with the guidelines of the NIH regarding the care and use of animals for experimental procedures. Organotypic slice cultures were prepared essentially according to the methods described previously (Fujimoto et al, 2006). Coronal brain slices of 300 μm thickness were prepared from Wistar rats at postnatal days 3 to 4 (Nihon SLC, Shizuoka, Japan), and six cortico-striatal tissue slices were transferred onto a 30-mm Millicell-CM insert membrane (Millipore, Bedford, MA, USA) in six-well plates. Culture medium, consisting of 50% minimum essential medium/HEPES (Gibco, Invitrogen, Tokyo, Japan), 25% Hanks' balanced salt solution (Gibco), and 25% heat-inactivated horse serum (Gibco) supplemented with 6.5 mg/mL D-glucose, 2 mmol/ L L-glutamine, 100 U/mL penicillin G potassium, and 100 μg/mL streptomycin sulfate (Gibco), was supplied at 0.75 mL/well so that the slices were maintained at the liquid/air interface. Culture medium was replaced with fresh mediun on the next day of culture preparation, and thereafter, every 2 days. Slices were cultured in a humidified atmosphere of 5% CO2 and 95% air at 34°C.
Drug Treatment and Cell Death Assessment
Cultured slices at 9 to 11 days in vitro were incubated for 24 to 48 h in serum-free medium, where minimum essential medium/HEPES substituted for horse serum. Then slices were exposed to drugs and chemicals dissolved in serum-free medium for indicated periods. To assess cell injury, propidium iodide (PI; 5 μg/mL; Wako Pure Chemical) was added to serum-free medium for drug treatment. After 72 h, images of PI fluorescence of each slice were captured through a monochrome chilled CCD camera (C5985; Hamamatsu Photonics, Hamamatsu, Japan) and stored as image files. The average signal intensity in an area of 180 × 180 μm within the parietal cortex was obtained as the fluorescence value of each slice, with the use of NIH Image 1.63 software. In each experiment, slice cultures treated with 100 μmol/L N-methyl-
Immunohistochemistry for Slice Cultures
After drug treatment, slice cultures were fixed with 0.1 mol/L phosphate buffer containing 4% paraformaldehyde and 4% sucrose for 2 h. After rinsing with phosphate-buffered saline (PBS), they were permeabilized and blocked with 0.2% Triton X-100 in PBS containing 1.5% goat serum followed by incubation with primary antibodies overnight at 4°C. Primary antibodies were mouse anti-NeuN (1:200; Chemicon International, Temecula, CA, USA), mouse anti-OX42 (1:300; Dainippon Pharmaceutical, Osaka, Japan), mouse anti-glial fibrillary acidic protein (1:500; Sigma), and rabbit anti-phospho-p44/42 mitogen-activated protein kinase (MAPK) (T202/Y204) (1:250; Cell Signaling Technology, Beverly, MA, USA). After being rinsed with PBS, cultures were incubated with secondary antibodies for 1 h at room temperature. Alexa Fluor 488-labeled goat anti-mouse IgG (1:200; Molecular Probes, Eugene, OR, USA), Alexa Fluor 594-labeled goat anti-mouse IgG (1:200; Molecular Probes), and Alexa Fluor 488-labeled goat anti-rabbit IgG (1:200; Molecular Probes) were used as secondary antibodies. Then cultures were rinsed with PBS and specimens were dehydrated through a graded ethanol series and mounted on slide glasses with glycerol. Fluorescence signals were observed with a laser-scanning confocal microscopic system (MRC1024; Bio-Rad, Hercules, CA, USA).
Plasmin Activity Quantification
Protease activity of plasmin was measured by the rate of substrate cleavage according to a previous report (Schneider and Nesheim, 2004) with modification. D-Val-Leu-Lys p-nitroanilide (S2251 reagent; Sigma), a chromogenic substrate of plasmin, was dissolved in 50 mmol/L Tris-buffered saline at a concentration of 2 mmol/L. Serum-free media with or without drugs and culture supernatants were reacted with an equal amount of S2251 solution at room temperature. Absorbance of p-nitroaniline generated by the cleavage of S2251 reagent was measured at 450 nm on a microplate reader from 3 to 180 mins after initiation of the reaction.
Western Blot Analysis
After treatment with drugs for indicated periods, slice cultures were harvested and homogenized in ice-cold lysis buffer containing 20 mmol/L Tris—HCl (pH 7.0), 25 mmol/L β-glycerophosphate (Sigma), 2 mmol/L EGTA · 2Na, 1% Triton X-100, 1 mmol/L vanadate, 1% aprotinin (Sigma), 1 mmol/L phenylmethylsulfonyl fluoride, and 2 mmol/L dithiothreitol. Samples were mixed with sample buffer composed of 124 mmol/L Tris—HCl (pH 6.8), 4% sodium dodecyl sulfate, 10% glycerol, 0.02% bromophenol blue, and 4% 2-mercaptoethanol. After being boiled for 5 mins, samples were subjected to 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis for 70 mins, followed by transfer to polyvinylidene difluoride membrane (Millipore) for 70 mins. Membranes were blocked with 5% nonfat milk and subsequently probed overnight with mouse anti-phospho-p44/42 MAPK (T202/Y204) (1:2,000; Cell Signaling Technology) and anti-p44/42 MAPK (1:1,000; Cell Signaling Technology). The membranes were rinsed and incubated with horseradish peroxidase-conjugated goat anti-mouse IgG (1:10,000; Jackson Immunoresearch Laboratories, West Grove, PA, USA) or goat anti-rabbit IgG (1:10,000; Jackson Immunoresearch Laboratories). After incubation with secondary antibodies, membranes were rinsed and bound antibodies were detected with the enhanced chemiluminescence kit (Amersham Biosciences, Buckinghamshire, UK) according to the manufacturer's instructions. The band intensities were analyzed with NIH image 1.63.
Surgical Procedures for Microinjection and Drug Administration
Male Sprague—Dawley rats initially weighing 220 to 280 g were used. Animals were kept at constant ambient temperature (23±2°C) under a 12-h light and dark cycle with free access to food and water. Drug administration was performed according to a previous report (Fujimoto et al, 2007) with modification. Briefly, rats were anesthetized with pentobarbital (50 mg/kg, intraperitoneally; Dainippon Sumitomo Pharmaceutical, Osaka, Japan) and placed in a stereotaxic frame (Narishige, Tokyo, Japan). After scalp incision, a hole was drilled on the skull. Thrombin, plasminogen, and collagenase were administered via a stainless steel injection cannula (outer diameter (o.d.) 0.35 mm) into the parietal cortex at 0.2 mm anterior, 3.0 mm lateral, 2.5 mm ventral from bregma. The solution of these drugs in a volume of 2 μL/rat was infused at a constant rate of 0.5 μL/min. The injection cannula was left in place at least for additional 5 mins to prevent backflow of drugs. Aprotinin and tranexamic acid were administered intraperitoneally. Aprotinin was administered at a dose of 2 mg/kg at 30 mins after intracortical injection of collagenase, followed by administration of 1 mg/kg from the next day twice daily. Tranexamic acid was administered at a dose of 300 mg/kg at 30 mins after intracortical injection of collagenase, followed by administration of 150 mg/kg from the next day twice daily. Control rats received intraperitoneal injection of the equivalent volume of saline.
Histologic Examinations After Microinjection
After indicated periods from intracortical injection, rats were anesthetized again and perfused through the heart with PBS followed by 4% paraformaldehyde. Brains were removed from the skull, and postfixed in 4% paraformaldehyde and dehydrated with 15% sucrose solution overnight at 4°C. After freezing, coronal brain sections (16 μm) containing the injection site were prepared and mounted onto slides. For immunohistochemitry, specimens were autoclaved (121°C for 15 mins) for epitope retrieval. They were permeabilized and blocked with 0.5% Triton X-100 in PBS containing 1.5% horse serum for 1 h at room temperature. Specimens were then incubated with mouse anti-NeuN (1:200) overnight at 4°C, and after being rinsed with PBS, they were incubated with biotinylated horse anti-mouse IgG (1:200; Vector Laboratories, Burlingame, CA, USA) for 1 h at room temperature. After being rinsed, specimens were treated with the avidin—biotinylated horseradish peroxidase complex (Vectastain Elite ABC kit, Vector Laboratories) and then peroxidase was visualized with diaminobenzidine and H2O2. Bright-field images were captured through a monochrome chilled CCD camera. For injury induced by thrombin with or without plasminogen, we defined the region where no NeuN-positive cells were observed as the injured region, and measured the injured area with NIH Image 1.63. For injury resulting from collagenase-induced hemorrhage, we counted the number of NeuN-positive cells in an area of 230 × 320 μm at the center of hematoma formed in the cortex.
Statistics
Data are expressed as means±s.e.m. Statistical significance of difference was evaluated with paired t-test, Mann—Whitney U-test, or one-way analysis of variance followed by Student—Newman—Keuls test. Probability values less than 5% were considered significant.
Results
We applied plasminogen (0.1 to 10 U/mL) for 72 h to cortico-striatal slice cultures. Propidium iodide fluorescence was used as a measure of cell injury in the cortical region. Plasminogen up to 10 U/mL did not induce cortical cell injury (Figure 1F). Moreover, although we have previously shown that thrombin induced shrinkage of the striatal tissue in slice culture (Fujimoto et al, 2006), we did not observe any changes in the striatal area after application of plasminogen (data not shown). Notably, when plasminogen at 3 U/mL, but not at 1 U/mL, was applied to slice cultures concomitantly with a moderate concentration (30 U/mL) of thrombin that did not induce prominent cortical injury by itself, a significant degree of cortical injury was evident at 72 h (Figures 1A to 1D, 1G). When plasminogen was applied from 24 h before application of 30 U/mL thrombin, significant cortical injury was produced also at 1 U/mL and in a concentration-dependent manner bewteen 1 and 3 U/mL (data not shown). Propidium iodide fluorescence in the cortical region of slice cultures treated with a combination of thrombin and plasminogen was colocalized with immunoreactivity against NeuN, a neuronal marker (Figure 1E), suggesting that neurons were injured by this treatment. However, plasminogen did not potentiate NMDA-induced cortical neuron injury, even when slices received 24-h pretreatment with plasminogen (Figure 1H). In addition, plasminogen did not affect the degree of striatal shrinkage induced by thrombin (Figure 1I). Plasminogen did not show any toxic effects when combined with bovine serum albumin at a protein concentration corresponding to that of 30 U/mL thrombin (data not shown). These results suggest that plasminogen specifically evokes cortical injury by a combination with thrombin.
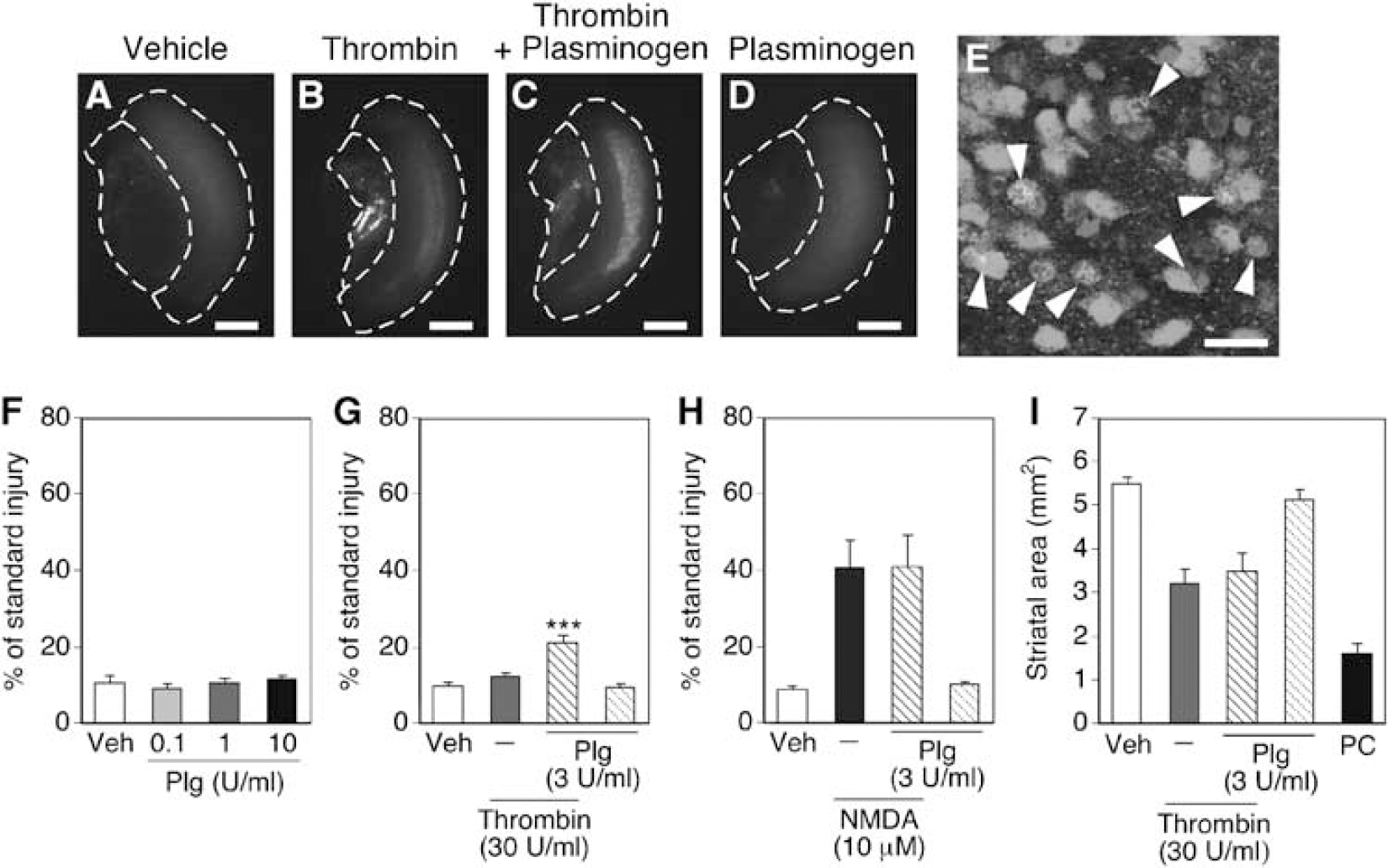
Injury induced by thrombin and plasminogen in cortico-striatal slice cultures. (
Plasminogen can be converted into a serine protease plasmin by plasminogen activators. We then examined the involvement of plasmin protease activity on cortical injury induced by thrombin and plasminogen. To measure plasmin activity, we incubated serum-free medium containing plasmin (0.1 U/mL) or plasminogen (3 U/mL) with a chromogenic substrate, S2251, at room temperature. Analysis of the absorbance of the degradation product indicated that plasmin cleaved S2251 time-dependently, whereas medium alone or plasminogen was without effect (Figure 2A), confirming that S2251 was a suitable substrate of plasmin but not of plasminogen. On the basis of these observations, we examined whether plasmin was generated after application of plasminogen to slice cultures. The supernatants of slice cultures treated with 3 U/mL plasminogen for 72 h cleaved S2251 gradually, whereas those of slice cultures treated with vehicle showed no effect (Figure 2B). These results suggest that a fraction of plasminogen was converted into plasmin during incubation with slice cultures. Moreover, cortical injury induced by thrombin plus plasminogen was prevented by aprotinin (20 μg/mL), a plasmin inhibitor, and tranexamic acid (3 mmol/L), a lysine analog that inhibits plasminogen activation (Figures 2C and 2D). In this set of experiments, we used aprotinin and tranexamic acid at concentrations that were sufficient to inhibit plasmin activity but did not inhibit thrombin activity (Engles, 2005). Indeed, aprotinin and tranexamic acid did not ameliorate cortical injury induced by a high concentration (100 U/mL) of thrombin (Figures 2E and 2F), suggesting that the effects of these drugs were mediated by inhibition of plasmin activity.
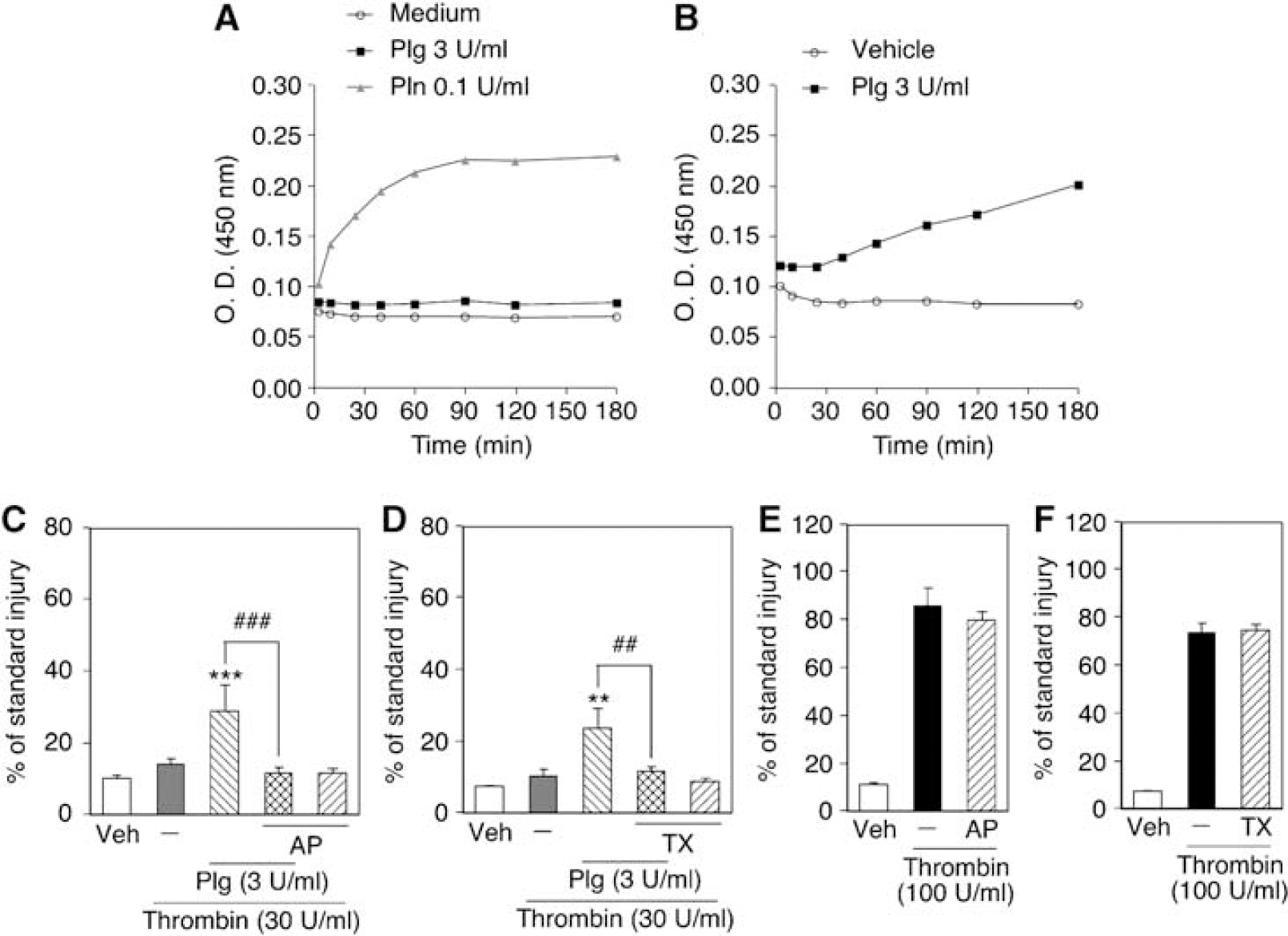
Involvement of plasmin activity in cortical injury induced by a combination of thrombin and plasminogen. (
We previously demonstrated that thrombin-induced cortical injury was prevented by inhibition of the extracellular signal-regulated kinase (ERK) pathway, Src family tyrosine kinase, protein kinase C, and de novo protein synthesis as well as by an inhibitor of thrombin protease activity. In contrast, inhibition of p38 MAPK and depletion of microglia had no effect, and inhibition of c-Jun N-terminal kinase exacerbated thrombin-induced cortical injury (Fujimoto et al, 2006). Then, we examined the involvement of these pathways in neurotoxicity of thrombin plus plasminogen. Selective inhibitors for various signaling pathways were used at the same concentrations as those used in our previous study (Fujimoto et al, 2006). As shown in Figures 3A to 3E, the combined neurotoxicity of thrombin and plasminogen in the cortical region was prevented by inhibitors of the ERK pathway (PD98059, 100 μmol/ L), Src family tyrosine kinase (PP2, 100 μmol/L), protein kinase C (BIM, 3 μmol/L), de novo protein synthesis (cycloheximide, 1 μg/mL), and thrombin protease activity (argatroban, 300 μmol/L). However, an inhibitor of p38 MAPK (SB203580, 100 μmol/L) and depletion of microglia by clodronate (100 μg/mL) did not prevent cortical injury, and an inhibitor of c-Jun N-terminal kinase (SP600125, 100 μmol/L) exacerbated the injury (Figures 3F to 3H). Overall, the effects of various agents on neurotoxicity of thrombin plus plasminogen were similar to those on neurotoxicity of a high concentration of thrombin alone (Fujimoto et al, 2006).
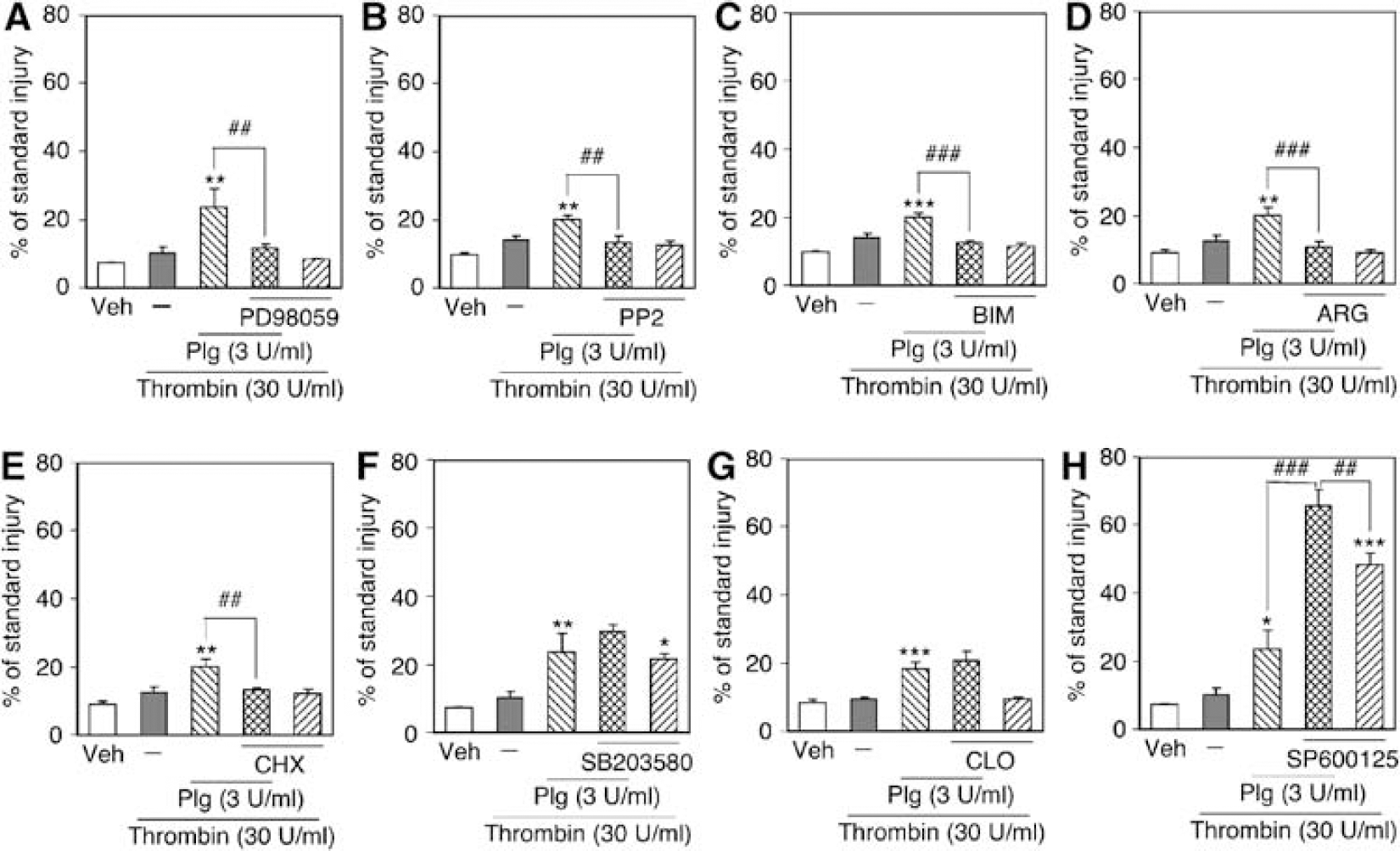
Effects of (
We further investigated the involvement of ERK in combined neurotoxicity of thrombin and plasminogen. Levels of ERK phosphorylation in slice cultures were examined by Western blot analysis after 3 or 24 h of incubation with or without thrombin and plasminogen. At 3 h, plasminogen (3 U/mL) increased phosphorylated ERK level to 267±55% of vehicle treatment (n = 8, P = 0.019, evaluated by paired t-test). The increased level of ERK phosphorylation was sustained even after 24 h from the onset of plasminogen treatment (Figure 4A). Consistent with our previous study (Fujimoto et al, 2006), thrombin also caused ERK phosphorylation at 3 h, which decreased thereafter (Figure 4A). To determine cell types exhibiting ERK phosphorylation after 3 h of plasminogen treatment, we performed immunofluorescence staining with a combination of antibodies against phosphorylated ERK and cell type-specific marker proteins. The majority of phosphorylated ERK immunofluorescence in the cortical region was colocalized with immunofluorescence of an astrocyte marker, glial fibrillary acidic protein (Figure 4B), but not with a neuronal marker, NeuN (Figure 4C), or a microglial marker, OX42 (data not shown). These results suggest that plasminogen evokes persistent ERK phosphorylation in cortical astrocytes.
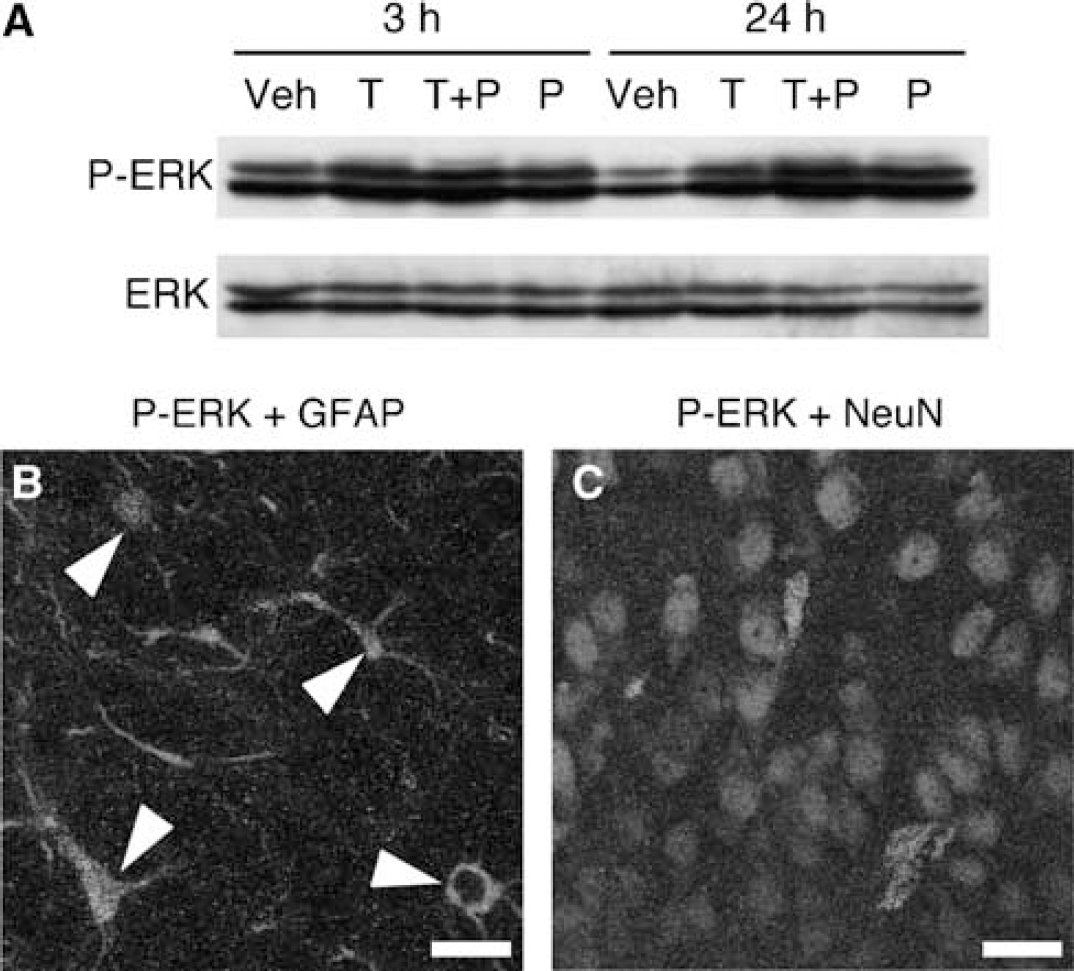
Phosphorylation of ERK induced by thrombin and plasminogen. (
On the basis of these in vitro observations, we next examined whether plasminogen could potentiate thrombin neurotoxicity in vivo. For this purpose, we injected thrombin with or without plasminogen into the rat cortex. The dose of thrombin was set to 1 U, because in preliminary experiments 10 U thrombin alone caused severe cortical injury. After 72 h, several coronal brain sections were prepared from each rat and immunostained with anti-NeuN antibodies. As shown in Figure 5A, thrombin injection into the cerebral cortex produced a distinct region with decreased NeuN immunoreactivity around the injection site, as with the case of thrombin injection into the striatum in our previous study (Fujimoto et al, 2007). Because there were very few NeuN-positive cells within this region and we could easily distinguish the border between the injured region and the intact region, we assessed the degree of neurotoxicity by the injured area defined as the region with scarce NeuN immunoreactivity around the injection site. As expected, the injured area of the section containing the injection site, as revealed by a cannula track, was the largest among several coronal brain sections examined. This means that the cortical injury expanded concentrically from the injection site. In this series of experiments, plasminogen (0.3 U) again augmented thrombin-induced cortical injury at 72 h (Figures 5B and 5C).
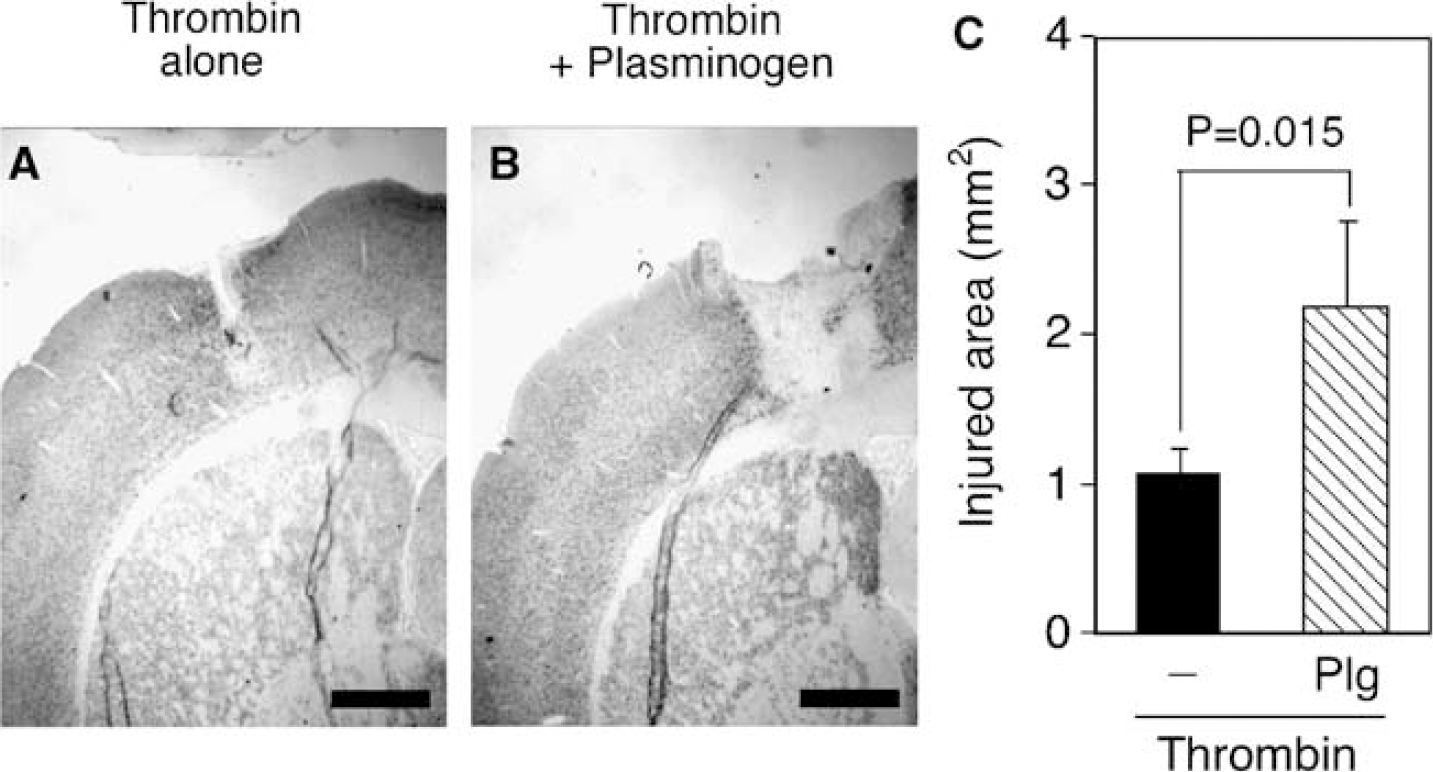
Plasminogen potentiates thrombin-induced cortical neuron loss in vivo. (
Finally, we examined possible involvement of plasminogen in hemorrhage-induced brain injury in vivo. Injection of 0.1 U collagenase into the rat cortex led to hematoma formation and a substantial decrease of NeuN-positive cells inside the hematoma. Unlike in the case of thrombin injection, however, a moderate number of NeuN-positive cells remained viable even in the center of the collagenase injection site (Figures 6A, 6B, 6E, and 6F). Therefore, we evaluated neuroprotective effects of drugs by the number of NeuN-positive cells at the hematoma center 72 h after collagenase injection. Aprotinin (2 mg/kg per day) intraperitoneally administered after collagenase injection partially but significantly increased the number of NeuN-positive cells at the hematoma center, compared with saline-administered control (Figures 6C, 6G, and 6I). Tranexamic acid (300 mg/kg per day) also tended to increase the number of surviving NeuN-positive cells, although the effect did not reach statistical significance (Figures 6D, 6H, and 6I). The dose of aprotinin in this set of experiments was almost the maximal one that could be prepared from the commercial product (Sigma, A6279), and the dose of tranexamic acid was chosen according to a previous report (O'Brien et al, 2000). We also compared the hematoma size of each treatment group. When the area of hematoma was defined as the cortical region with decreased NeuN immunoreactivity in the coronal brain section containing the injection site, hematoma size of saline-administered control was 2.12±0.28 mm2 (n = 10). Hematoma sizes of rats treated with aprotinin and tranexamic acid were 2.63±0.30 mm2 (n = 9) and 2.70±0.41 mm2 (n = 7), respectively, both of which were not significantly different from that of control group. These results suggest that the neuroprotective effect afforded by regulation of plasmin activity is independent of inhibition of fibrinolytic activity of plasmin.
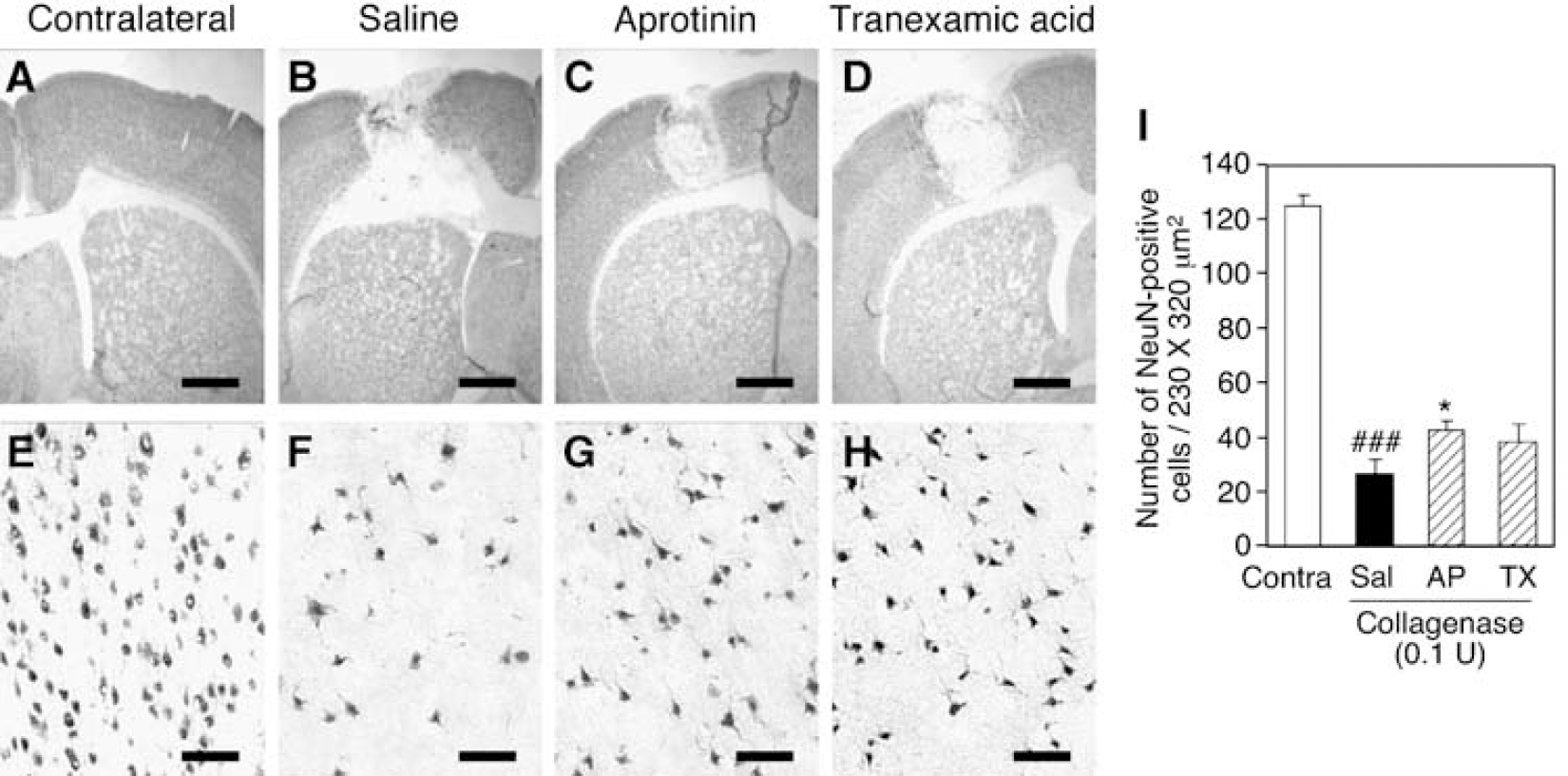
Involvement of plasmin in hemorrhage-induced cortical injury. (
Discussion
Plasminogen is a circulating zymogen of fibrinolytic serine protease plasmin, and plasma concentration of plasminogen is estimated to be ~2U/mL (Xue and Del Bigio, 2005). Although neurotoxicity of plasminogen in the striatum has been reported by in vivo experimental model (Xue and Del Bigio, 2001, 2005), we did not observe neurotoxicity of plasminogen in cortico-striatal slice cultures even at 10 U/mL, a higher concentration than that in plasma. Instead, we found that a prominent effect of plasminogen was potentiation of neurotoxicity of thrombin (30 U/mL) in the cerebral cortex. As previously shown, thrombin alone can produce cortical injury at a higher concentration of 100 U/mL (Fujimoto et al, 2006). Therefore, the action of plasminogen can be viewed as lowering of the threshold of neurotoxic actions of thrombin. The plasma concentration of prothrombin, the precursor of thrombin, is estimated to be 200 U/mL (Lee et al, 1996), which means that the concentration of thrombin used in this study (30 U/mL) is probably attainable in the case of hemorrhagic events. Plasminogen had no effect on thrombin toxicity in the striatum, which might reflect the fact that the cellular mechanisms of thrombin neurotoxicity are different between the cerebral cortex and the striatum (Fujimoto et al, 2006).
We also suggest that the potentiating effect of plasminogen on thrombin neurotoxicity was mediated by plasmin generated from plasminogen processing. This view was supported by the finding that the supernatant of slice cultures treated with plasminogen exhibited plasmin activity. Moreover, cortical injury induced by a combination of thrombin and plasminogen was abolished by aprotinin, a plasmin inhibitor. Although aprotinin is a protease inhibitor with a broad spectrum, the concentration of aprotinin applied to slice cultures (20 μg/mL), corresponding to ~0.6 μmol/L, was much lower than the concentration that inhibited thrombin (30 μmol/L; Engles, 2005). Moreover, aprotinin did not abolish cortical injury and striatal shrinkage induced by a higher concentration of thrombin. Therefore, the effect of aprotinin on neurotoxicity of thrombin plus plasminogen was likely attributable to inhibition of plasmin activity.
Similar to aprotinin, tranexamic acid abolished cortical injury induced by combined application of thrombin and plasminogen, without any effect on injury induced by a high concentration of thrombin. These results indicate that the protective effect of tranexamic acid was also mediated by inhibition of plasmin(ogen) activity. There are two plausible mechanisms for the effect of tranexamic acid. Firstly, tranexamic acid may inhibit the conversion of plasminogen to plasmin. Plasminogen binds to fibrin and cell surface through lysine-binding sites located in the kringle domain (Hajjar et al, 1986; Wu et al, 1990), which promotes the conversion of plasminogen by tissue-type plasminogen activator (Miles et al, 2005; Plow et al, 1995). Lysine analogs such as tranexamic acid reversibly bind to the kringle domain (Prentice, 1980), decreasing plasmin production (Ellis et al, 1991). Secondly, interference of association of plasmin(ogen) with cell surface may be responsible for the effect of tranexamic acid. For example, G-protein-coupled receptors sensitive to pertussis toxin have been shown to function as a plasmin receptor in monocytes and endothelial cells (Chang et al, 1993; Syrovets et al, 1997). Because the responses of these cells to plasmin were inhibited by tranexamic acid, the actions of plasmin may be mediated not only by its protease activity but also by its binding to unidentified cell surface receptors through the kringle domain (Chang et al, 1993; Syrovets et al, 1997). Another report in this context has shown that α-enolase is expressed on the surface of neurons as a plasminogen receptor, and that binding of plasminogen to neurons is abolished by tranexamic acid (Nakajima et al, 1994). Possible involvement of protease activity and receptors of plasmin(nogen) in potentiation of thrombin neurotoxicity remains to be addressed.
Argatroban, a thrombin inhibitor, abolished cortical injury induced by a combination of thrombin and plasminogen, confirming that protease activity of thrombin was crucial for the combined neurotoxicity. Moreover, inhibitors of the ERK pathway, Src family tyrosine kinase, protein kinase C, and de novo protein synthesis prevented the combined neurotoxicity, whereas an inhibitor of p38 MAPK was without effect and an inhibitor of c-Jun N-terminal kinase exacerbated the injury. These pharmacologic profiles totally mimic those of neurotoxicity of a higher concentration of thrombin (Fujimoto et al, 2006). Thus, plasminogen may potentiate thrombin neurotoxicity by allowing low concentrations of thrombin to recruit effectively neurotoxic signaling mechanisms. Surprisingly, persistent ERK phosphorylation in response to plasminogen was exclusively observed in astrocytes, although thrombin-induced ERK phosphorylation has been observed mainly in neurons (Fujimoto et al, 2006). These results imply that some cell-to-cell interactions between astrocytes and neurons should be involved in potentiation of thrombin neurotoxicity by plasminogen. Extracellular signal-regulated kinase phosphorylation in astrocytes is known to trigger astrogliosis accompanied by proliferation and expression of cyclooxygenase (Brambilla et al, 2002; Mandell and VandenBerg, 1999), and astrogliosis is observed in many neurodegenerative conditions (Pekny and Nilsson, 2005). Because reactive astrocytes release various inflammation-related molecules including cytokines (Hu et al, 1997; Suzumura et al, 2006), activation of astrocytes by plasminogen may lead to enhanced release of cytokines that make neurons vulnerable to thrombin. This possibility should also be addressed in future investigations. Finally, we cannot formally exclude the possibility that the protease activity of thrombin itself is somehow enhanced by plasminogen/ plasmin.
Potentiation of thrombin neurotoxicity by plasminogen in the cerebral cortex was also observed in a rat brain microinjection model, suggesting that the same cellular mechanisms of synergistic cytotoxicity of thrombin and plasminogen as those in vitro also operate in vivo. Moreover, involvement of plasminogen in hemorrhagic brain injury was ascertained by the protective effect of aprotinin against cortical injury in a collagenase-induced hemorrhage model. As demonstrated in our recent study of intrastriatal hemorrhage (Ohnishi et al, 2007), the hematoma region contained remaining viable neurons as revealed by NeuN immunoreactivity, and we observed a significant protective effect of aprotinin as the increase in the number of NeuN-positive cells. Aprotinin and tranexamic acid in this study did not reduce the hematoma size in the cortex. Although these drugs have been clinically used to prevent blood loss (Engles, 2005; Mahdy and Webster, 2004), prior administration of high doses of aprotinin and tranexamic acid is required to prevent uncontrolled plasma extravasation in vivo (O'Brien et al, 2000). Therefore, relatively weak neuroprotective effects of aprotinin and tranexamic acid systemically administered in this study might be attributable to insufficient inhibition of plasmin activity by these drugs. Recently, molecules that promote blood coagulation and prevent hematoma growth, such as recombinant factor VII, attract considerable interests as a therapeutic strategy against ICH (Mayer and Rincon, 2005; Mayer et al, 2005). Because inhibition of plasmin is expected to reduce hemorrhage and also to prevent neuronal injury mediated by protease activity, regulation of plasminogen/plasmin system could be an attractive therapeutic intervention.
In conclusion, we demonstrated here that plasminogen potentiated thrombin neurotoxicity in the cerebral cortex both in vitro and in vivo. These effects of plasminogen appeared to be mediated by plasmin protease activity and ERK phosphorylation in astrocytes. Recently, exacerbation of thrombin-induced neuronal death by a combination with matrix metalloprotease-9 has been reported (Xue et al, 2006). Elucidation of interactions of various protease systems with thrombin-mediated cytotoxic pathways may lead to discovery of novel therapeutic strategies for hemorrhagic brain injury.