
Abstract
Secondary insults such as hypotension or hemorrhagic shock (HS) can greatly worsen outcome after traumatic brain injury (TBI). We recently developed a mouse combined injury model of TBI and HS using a controlled cortical impact (CCI) model and showed that 90 minutes of HS can exacerbate neuronal death in hippocampus beneath the contusion. This combined injury model has three clinically relevant phases, a shock, pre hospital, and definitive care phases. Mice were randomly assigned to four groups, shams as well as a CCI only, an HS only, and a CCI + HS groups. The CCI and HS reduced cerebral blood flow (CBF) in multiple regions of interest (ROIs) in the hemisphere ipsilateral and contralateral to injury. Hemorrhagic shock to a level of ~30 mm Hg exacerbated the CCI-induced CBF reductions in multiple ROIs ipsilateral to injury (hemisphere and thalamus) and in the hemisphere contralateral to injury (hemisphere, thalamus, hippocampus, and cortex, all P < 0.05 versus CCI only, HS only or both). An important effect of HS duration was also seen after CCI with maximal CBF reduction seen at 90 minutes (P < 0.0001 group-time effect in ipsilateral hippocampus). Given that neuronal death in hippocampus is exacerbated by 90 minutes of HS in this model, our data suggest an important role for exacerbation of posttraumatic ischemia in mediating the secondary injury in CCI plus HS. In conclusion, the serial, non invasive assessment of CBF using ASL-MRI (magnetic resonance imaging with arterial spin labeling) is feasible in mice even in the complex setting of combined CCI + HS. The impact of resuscitation therapies and various mutant mouse strains on CBF and other outcomes merits investigation in this model.
INTRODUCTION
Secondary insults such as hemorrhagic hypotension or hemorrhagic shock (HS) can greatly worsen outcome after traumatic brain injury (TBI). 1 This combination has long been known to double mortality in civilian TBI, and has taken on greater significance recently in combat casualty care as a result of blast injuries resulting from improvised explosive devices. In blast injury, polytrauma such as traumatic amputation or HS from shrapnel contained in the improvised explosive devices are observed and similarly increase mortality and morbidity. 2 Specifically, hypotension after severe blast TBI is associated with extremely high mortality rates. 3
Autoregulatory mechanisms can defend the uninjured brain against hemorrhagic hypotension or HS; however, after TBI, blood pressure autoregulation of cerebral blood flow (CBF) is often compromised, and CBF is reduced particularly early after the injury.4,5 Thus, blood pressure reductions that would normally be tolerated can produce CBF reductions that contribute to secondary ischemia. This can be true even in the setting of mild TBI. 6 Similarly, HS and administration of subsequent resuscitation fluids result in hemodilution with reduced oxygen carrying capacity of blood that can further compromise oxygen delivery to the injured brain. 7 In addition, TBI reduces the capacity of the organism to maintain blood pressure during hemorrhage. 8 Thus, hemorrhage represents a particularly compromising insult to the brain after TBI.
Much of the investigation of the effect of hemorrhagic hypotension or HS on experimental TBI has focused on the study of acute hemodynamic effects (i.e., intracranial pressure, cerebral perfusion pressure, and brain tissue oxygen) or evaluation of various resuscitation fluids (i.e., crystalloids, colloids, hypertonic saline, or blood substitutes).9–13 Most of this work has been performed in large animal models, such as pigs, to facilitate intracranial pressure monitoring. Few studies have been performed in rat14–20 or mouse models. Recently, we developed and published a series of reports using a new mouse model of combined TBI plus HS, using controlled cortical impact (CCI).17–19 In addition, we previously reported on the ability to use magnetic resonance imaging (MRI) with arterial spin labeling (ASL) to serially and non invasively study CBF in mice, 21 including after CCI. 22 We now report the application of this ASL-MRI method to serially map CBF in mice subjected to HS, CCI, the combination of CCI plus HS, and shams.
MATERIALS AND METHODS
Animal Model
All experiments were approved by the Institutional Animal Care and Use Committees of Carnegie Mellon University, the University of Pittsburgh, and the United States Army. The protocol used and details of this report are also in accordance with ARRIVE guidelines. Male C57BL/6J mice aged between 11 and 15 weeks were used in this study (Charles Rivers Laboratories, Wilmington, MA, USA). The mice were fed with laboratory chow and water ad libitum and maintained under temperature controlled conditions with 12 hours light/dark cycles. Animals were randomly assigned to four experimental groups, sham (n = 6), CCI only (n = 7), HS only (n = 7), and CCI + HS (n = 5).
Anesthesia was induced in the mice as previously described. 21 Mice were intubated and mechanically ventilated with 2% isoflurane in a 1:1 N2O/O2 gas mixture during surgery. A femoral artery catheter was surgically inserted for continuous blood pressure monitoring and arterial blood sampling, while a femoral venous catheter was inserted for blood withdrawal and fluid replacement.
Experimental Protocol
The experimental protocol is shown in Figure 1. After catheterization, anesthesia was reduced to 1% isoflurane in room air for a 10-minute equilibration period before injury protocols. The CCI was performed on mice according to group assignment. In the combined insult group, hemorrhage was started 5 minutes after CCI. The amount of hemorrhage volume required to reduce mean arterial blood pressure (MABP) to the target was determined in a previous study. 17 For mice subjected to HS alone, a volume between 2.5 and 2.7 mL/100 g was needed to achieve the target MABP of ~35 to 40 mm Hg. After CCI, a smaller volume of 2.0 mL/100 g was required to reach the target MABP. Hemorrhagic shock was induced over 15 minutes in a decelerating manner, with 50% of the total volume removed in the first 5 minutes and the remaining 50% in the final 10 minutes.
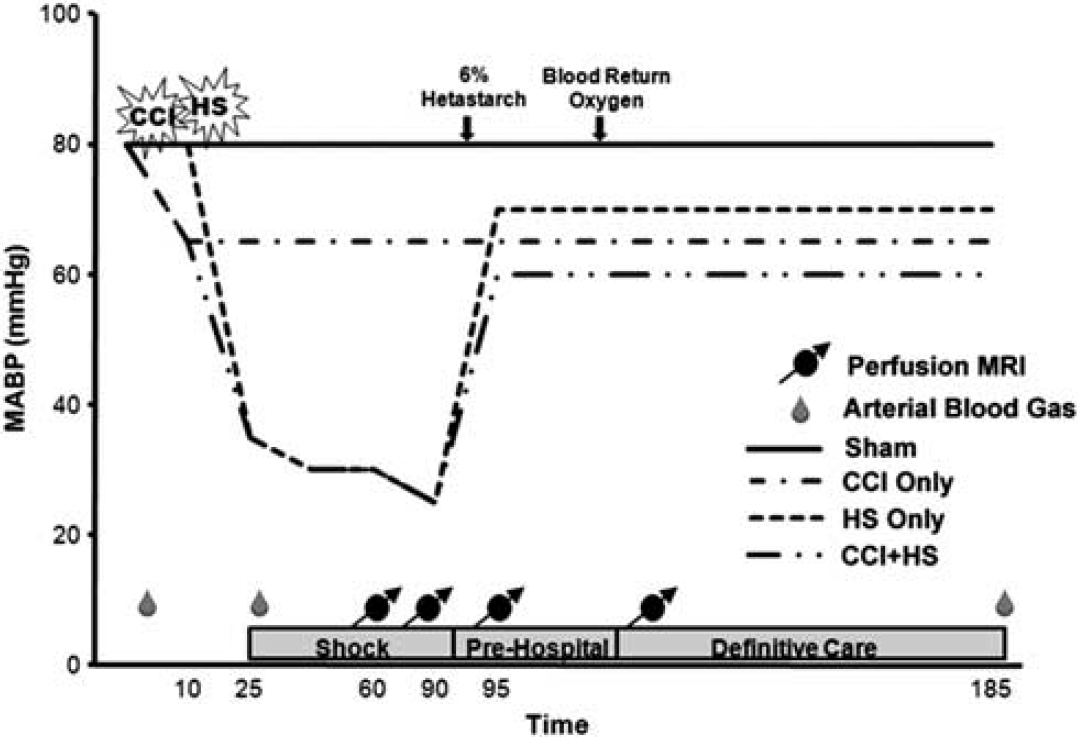
Experimental design. Experimental timing was the same for sham mice but without insult or resuscitation measures. Animals were only given resuscitation fluid if mean arterial blood pressure (MABP) was <50 mm Hg. Blood was withdrawn and later returned to hemorrhagic shock (HS) only and controlled cortical impact (CCI) + HS mice. MRI, magnetic resonance imaging.
Mice remained unresuscitated during this period referred to as the ‘shock’ phase, for a total of 90 minutes. During this period, mice were positioned on the cradle and placed within the MRI scanner. After completion of the ‘shock’ phase, a 30-minute ‘pre hospital’ period began and 6% hetastarch (Hextend, Hospira Inc., Lake Forest, IL, USA) was rapidly infused in 0.1 mL aliquots until MABP reached ≥50 mm Hg, pressure was maintained at this level with subsequent 0.1 mL injections of 6% hetastarch if necessary. At the end of this 30-minute period, all animals were switched from room air to 100% O2. Shed blood was returned over 6 minutes to mice in the appropriate groups. This period of ‘definitive care’ was continued for an hour, during this time MABP was maintained at ≥50 mm Hg using 6% hetastarch.
Controlled Cortical Impact
The mouse CCI model was used as previously described 23 with minor modifications.24,25 Mice that underwent CCI were placed in a stereotaxic holder and a temperature probe was inserted through a burr hole into the left frontal cortex and a 4-mm craniotomy was performed over the left parietal bone and the bone flap was removed for trauma. Once brain temperature reached 37 ± 0.5°C and was maintained at this temperature for 5 minutes, a vertically directed CCI was delivered (5.0 m/s at a depth of 1.0 mm). After injury, the bone flap was replaced, sealed with dental cement and the incision closed.
Magnetic Resonance Imaging Image Acquisition
The magnetic resonance studies were performed on a 4.7-Tesla, 40 cm bore Bruker AVANCE AV system (Billerica, MA, USA), equipped with a 12-cm diameter shielded gradient insert and a home-built radio frequency coil. Image acquisition parameters were identical to those reported in a prior study. 21 Briefly, pilot spin-echo images were used to verify the position of the center of the contusion. Perfusion images were acquired in the ‘Shock’ period at 60 and 90 minutes, during the ‘Pre Hospital’ phase after the return of MABP to target levels, and during the ‘Definitive Care’ period after the return of shed blood. Perfusion studies were performed using a continuous ASL26,27 imaging technique (spin echo, 64 × 40 matrix interpolated to 64 × 64, repetition time (TR) = 2,000 ms, summation of 3 echoes, TE (echo time) = 10, 20, and 30 ms and two averages). The inversion plane for the labeling radio frequency was positioned ± 2 cm from the perfusion detection plane. The spin-lattice relaxation time of tissue water (T1obs) 28 was measured from a series of spin-echo images (TR = 8,000, 4,300, 2,300, 1,200, 650, 350, 185, and 100 ms, TE = 9 ms, 2 averages, and a 64 × 40 matrix interpolated to 64 × 64). Spin-labeling efficiency 29 was determined from intensities within the carotid arteries (gradient echo, 45° flip angle, 8 averages, TR/TE = 100/9.6 ms, 256 × 256 matrix and spin-labeling applied at ± 6 mm).
Image Analysis
All image processing was performed with the Bruker ParaVision 3.0.2 image analysis software (Bruker, Billerica, MA, USA). Regions of interest (ROIs) defined the left (ipsilateral) and right (contralateral) hemisphere, as well as within each hemisphere, the cortex, hippocampus, thalamus, and the amygdala/piriform cortex, guided by assignments from a mouse brain atlas.
30
Cortical ROIs were drawn to comprise the entire cortex including herniated tissue within each hemisphere (cortex), or an area beginning 1 mm from midline to the midpoint of the arc of the cortex (cortex-contusion rich). After CCI, the cortex-contusion rich region contains an area that has a large contribution from the contusion. Maps of (MC – ML) · MC−1 were generated from the perfusion data. MC is the signal intensity from the control image and ML is the signal intensity with ASL. The T1obs maps were generated from the series of variable TR images by a three parameter non linear fit to:
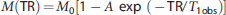
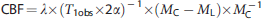
Statistical Analysis
Results are reported as the mean ± s.d. A repeated-measures ANOVA was used to compare CBF values and physiology between different groups. Post hoc testing used the Bonferroni correction for multiple comparisons. For the physiology parameters, post hoc comparisons were made across treatment groups. For CBF, post hoc comparisons included treatment and time and for this parameter, based on the number of comparisons a P-value of <0.0083 was required to achieve statistical significance.
RESULTS
Physiology
Arterial blood gases were assessed immediately before and after the blood withdrawal, or HS, and at the conclusion of the MRI measurements. Mean PaCO2 and pH did not differ between pre shock and post shock values for all groups (Table 1). Pa
Arterial blood gasesa
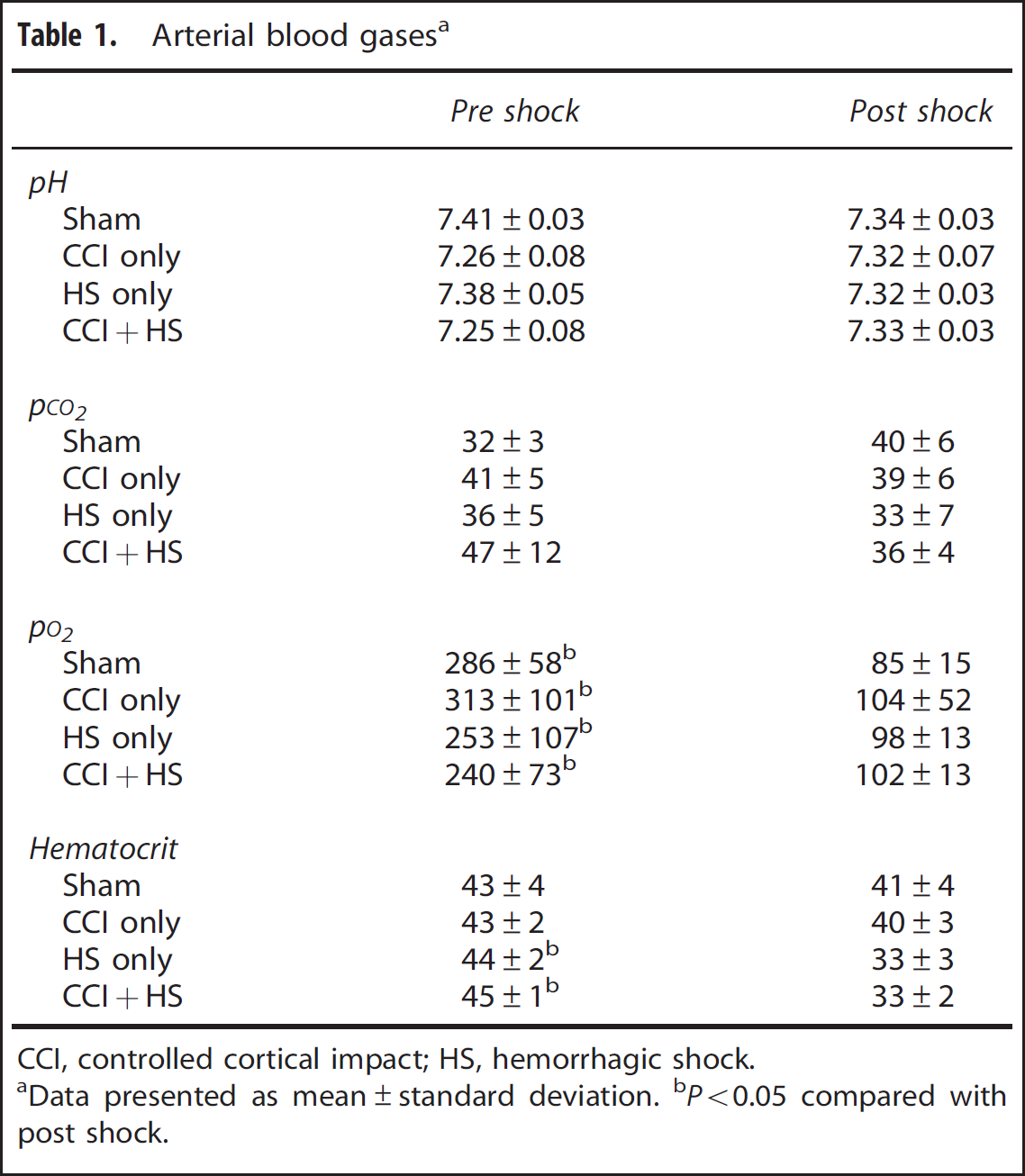
CCI, controlled cortical impact; HS, hemorrhagic shock.
Data presented as mean±standard deviation.
P<0.05 compared with post shock.
As designed, MABP was markedly reduced at 60 and 90 minutes of shock for the HS and CCI + HS groups (to a level of ~28 mm Hg) versus the sham and CCI groups (P < 0.05, Table 2). The CCI group was also significantly different than the sham group at the 90-minute shock time point (Table 2)—although this reduction in MABP was much more modest (~46 mm Hg) to that seen in either group subjected to hemorrhage. The mean heart rate of the sham and HS groups during the definitive care period was significantly (albeit modestly) higher than during the 60-minute shock phase (Table 2).
Physiologic dataa
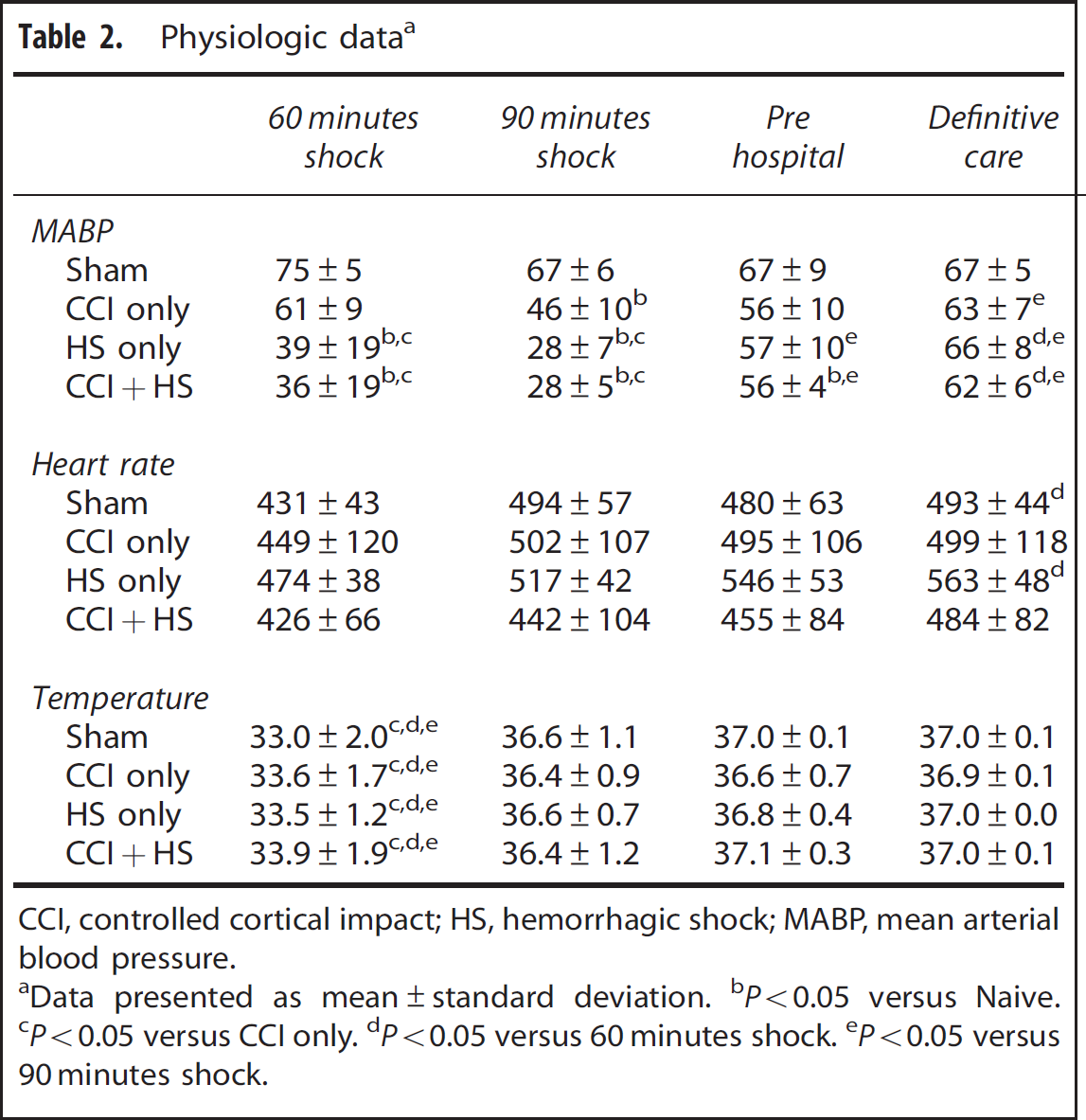
CCI, controlled cortical impact; HS, hemorrhagic shock; MABP, mean arterial blood pressure.
Data presented as mean±standard deviation.
P<0.05 versus Naive.
P<0.05 versus CCI only.
P<0.05 versus 60 minutes shock.
P<0.05 versus 90 minutes shock.
After resuscitation, MABP in all three groups improved toward sham levels, although the MABP of the CCI + HS group during the pre hospital phase was still significantly lower than sham (Table 2).
Rectal temperature in all groups was significantly lower after 60 minutes of shock versus later time points (Table 2). Mice were positioned within the bore of the spectrometer only minutes before the 60-minute time point and thus there was insufficient time for the warm air heating system to completely normalize temperature. However, there were no differences between any of the groups. At all of the later time points, rectal temperature was within the normal physiologic range in all groups.
Resuscitation Volumes
Total infusion volumes of Hextend required for resuscitation to achieve the target MABP of ≥50 mm Hg during both the pre hospital and definitive care periods were as follows: sham mice did not required any resuscitation fluid, CCI only mice received 0.28 ± 0.15 mL, HS only mice received 0.54 ± 0.27 mL and CCI + HS mice received 0.59 ± 0.09 mL. The CCI, HS, and CCI + HS groups differed significantly from the sham group (P < 0.05) and the CCI + HS group was significantly different than the CCI only group (P < 0.05). These volumes represent ~11, 22, and 24 mL/kg of Hextend in the CCI, HS, and CCI + HS groups, respectively.
Regional Cerebral Blood Flow
Representative CBF maps obtained from sham, CCI, HS, and CCI + HS mice are shown in Figure 2. Mean regional CBF values for the hemisphere ipsilateral to injury are shown in Figure 3. At 60 minutes of shock, there were significant reductions in CBF in CCI, HS, and CCI + HS groups versus sham for the hemisphere, amygdala/piriform cortex, and thalamus (Figure 3). In the ipsilateral hippocampus, CBF was significantly reduced in CCI and CCI + HS versus sham, while in the contusion rich cortex, CBF was reduced in CCI and CCI + HS versus sham and HS (Figure 3).
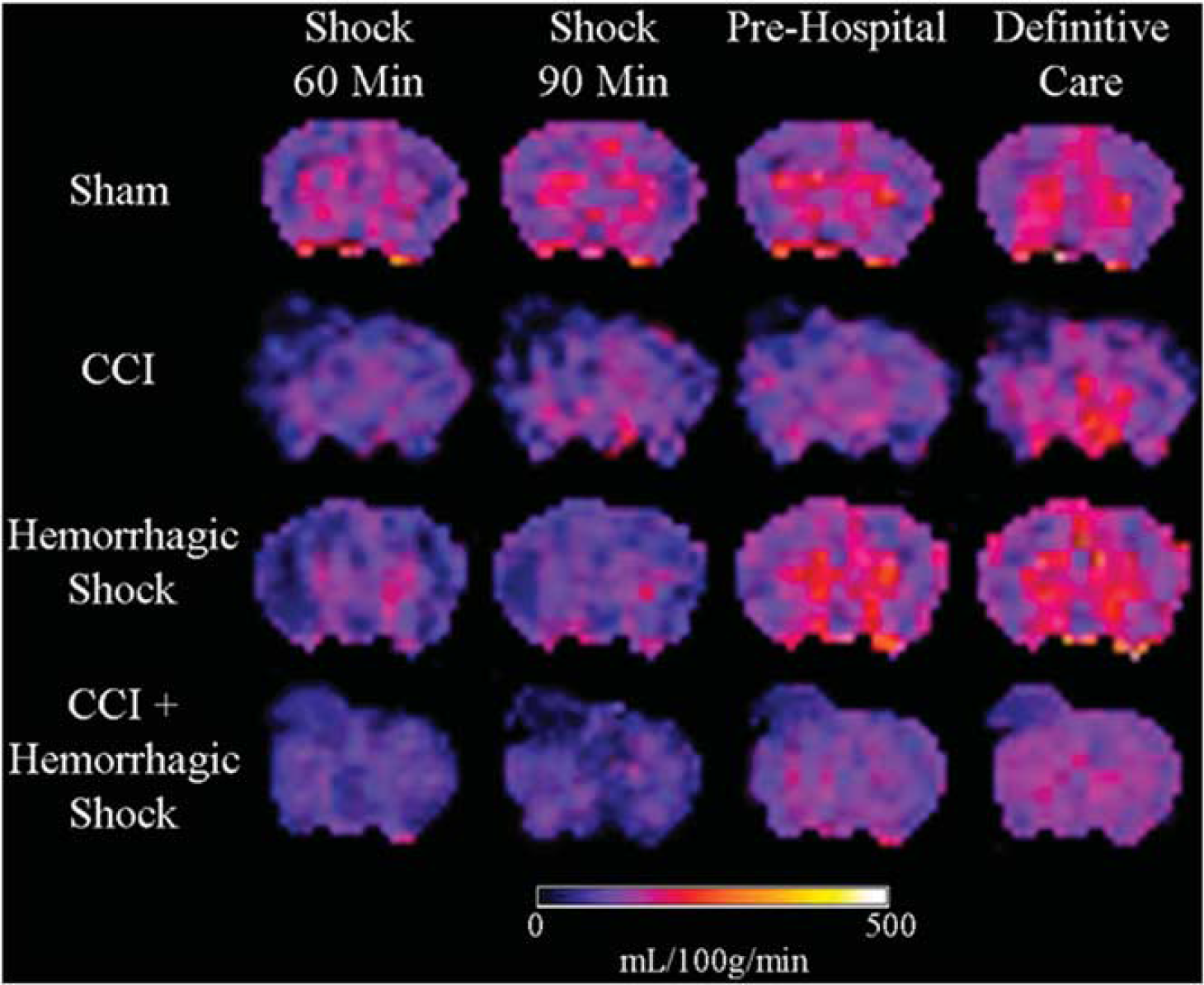
Representative color cerebral blood flow (CBF) maps of mouse brains from the four groups during each period of the experiment. CCI, controlled cortical impact.
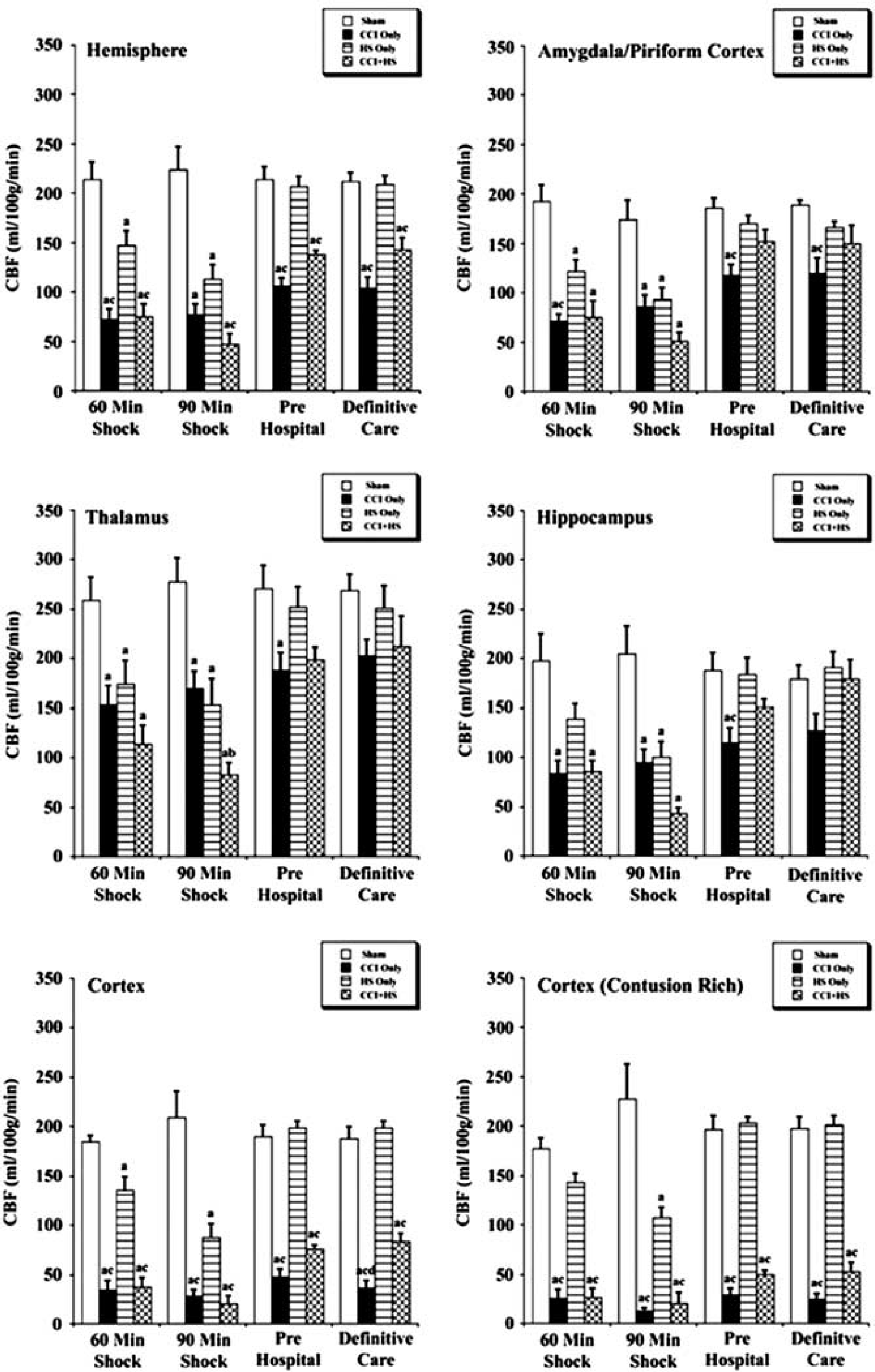
Regional cerebral blood flow (CBF) values (mean ± s.e.) for the ipsilateral hemisphere for sham mice (n = 6), controlled cortical impact (CCI) only (n = 7), hemorrhagic shock (HS) only (n = 7), and CCI + HS mice (n = 5). aP < 0.05 compared with sham, bP < 0.05 versus CCI only, and cP < 0.05 versus HS only.
At the 90 minutes after shock time point, CBF for the CCI, HS, and CCI + HS groups for all brain regions was significantly reduced versus sham (Figure 3). Cerebral blood flow for the CCI + HS group was significantly reduced versus CCI mice in the thalamus, while both the CCI and CCI + HS groups had significantly reduced CBF versus the HS group in the hippocampus, cortex, and contusion rich cortex. A significant effect of the duration of shock on neuronal death in the ipsilateral hippocampus was previously reported in this model 17 with exacerbation of damage when the HS duration was extended from 60 to 90 minutes in mice subjected to CCI + HS. Comparison of CBF in the hippocampus ipsilateral to injury in mice in the CCI + HS group in the current study of CBF revealed a group time effect (P < 0.0001) and a reduction in CBF from 86.20 mL/100 g per minute at 60 minutes to 43.40 mL/100 g per minute at 90 minutes (P < 0.029) (Figure 3).
During the pre hospital phase, mice were resuscitated with Hextend only if MABP was <50 mm Hg. Cerebral blood flow rebounded slightly for the CCI and CCI + HS groups, whereas it recovered to near sham values in the HS group (Figure 3).
Cerebral blood flow in the hemisphere, cortex, and contusion rich cortex ROIs was significantly lower for the CCI and CCI + HS groups versus both sham and HS groups in the pre hospital phase (Figure 3). Cerebral blood flow for the CCI mice was also significantly reduced versus sham and HS groups in the amygdala/piriform cortex and hippocampus and versus sham in the thalamus during the pre hospital phase. Thus, the rebound in CBF during reperfusion in the pre hospital phase was greater in the CCI + HS group than in CCI alone in these brain regions.
At the beginning of the definitive care phase, the shed blood was re-infused in the HS and CCI + HS groups. Cerebral blood flow for the HS only group remained similar to sham (Figure 3). However, CBF remained significantly lower in the ipsilateral hemisphere, cortex, and contusion rich area of the cortex in the CCI and CCI + HS groups versus sham and HS only groups (Figure 3). For the CCI group, CBF was reduced in the amygdala/piriform cortex versus sham and HS mice and also in the cortex compared with CCI + HS (Figure 3).
The CBF values in the hemisphere and cortex contralateral to impact were significantly reduced in the CCI, HS, and CCI + HS groups versus sham (Figure 4). While in the contralateral amygdala/piriform cortex, CBF was significantly lower for CCI and CCI + HS groups versus sham at the 60-minute shock time point and for the CCI, HS, and CCI + HS mice versus sham at the 90-minute shock time point (Figure 4). In the contralateral thalamus, CBF was reduced only in the CCI + HS group at the 60-minute shock time point, but was reduced in CCI, HS, and CCI + HS groups at 90 minutes versus sham (Figure 4).
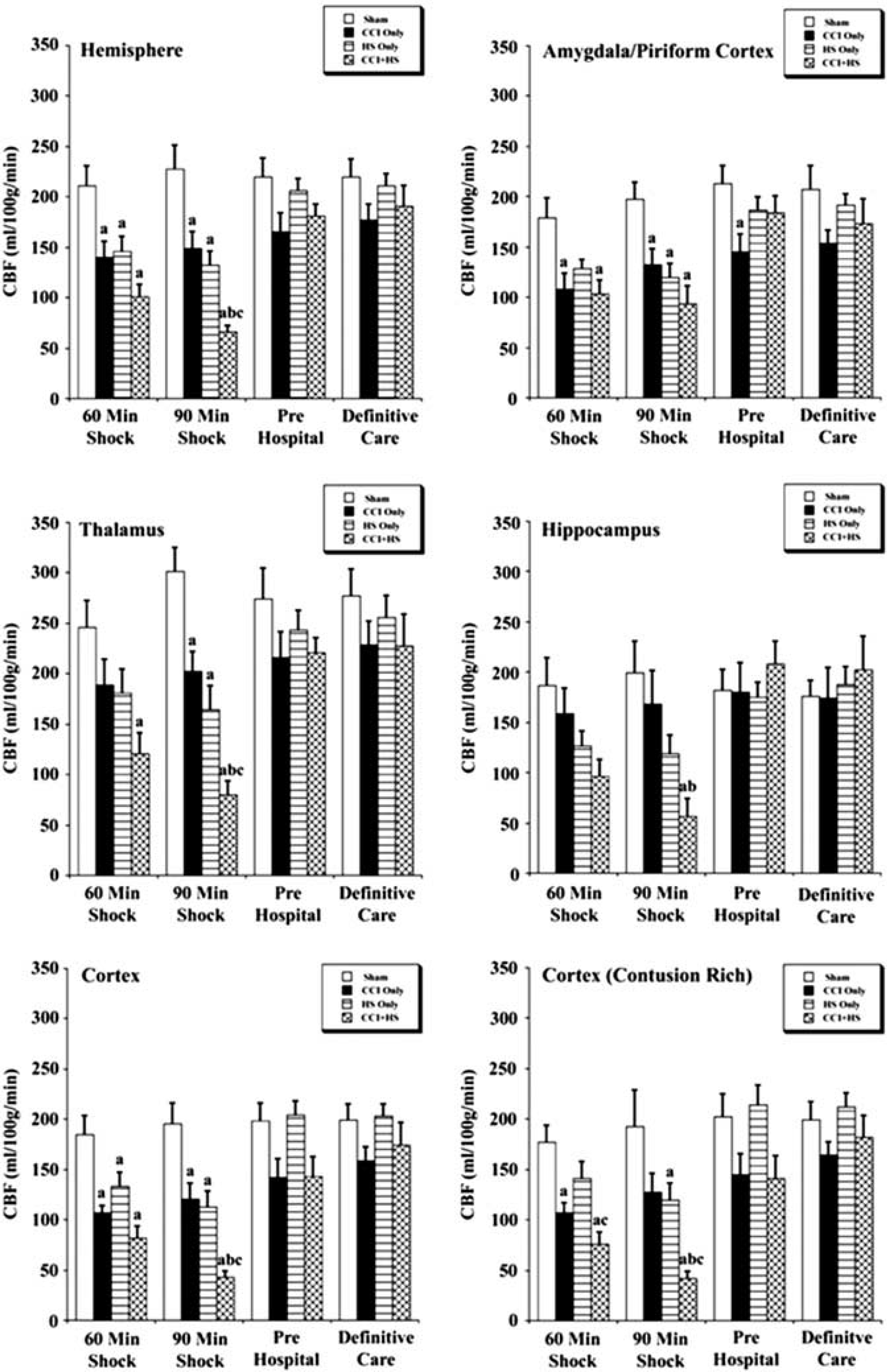
Regional cerebral blood flow (CBF) values (mean ± s.e.) for the contralateral hemisphere for sham mice (n = 6), controlled cortical impact (CCI) only (n = 7), hemorrhagic shock (HS) only (n = 7), and CCI + HS mice (n = 5). aP < 0.05 compared with sham, bP < 0.05 versus CCI only, and cP < 0.05 versus HS only.
The CBF in the CCI + HS group was also significantly reduced in the contralateral hemisphere, thalamus, cortex, and contusion rich cortex versus CCI only and HS only groups at the end of the 90-minute shock period, which suggests that the combined injury exacerbates global flow reductions during shock (Figure 4). After resuscitation, in the pre hospital and definitive care phases, CBF in the contralateral hemispheric ROIs was not different than sham (Figure 4).
DISCUSSION
We have assessed the impact of HS on CBF at 60 and 90 minutes after CCI in a mouse model in which the impact of HS on neuronal death in the hippocampus has been established. 17 Hemorrhagic shock contributed to a global exacerbation of a CCI-induced reduction in CBF most notably at 90 minutes after the onset of HS. The impact on HS on this CBF reduction was greatest in the ipsilateral thalamus and hippocampus, and was also seen in most ROIs in the contralateral hemisphere. We did not, however, observe a significant exacerbation of hypoperfusion in the cortex or contusion rich cortical ROIs that were defined comparing CCI versus CCI + HS at either 60 or 90 minutes of shock. In addition, during the pre hospital and definitive care resuscitation phase, surprisingly, CBF recovery toward baseline was somewhat greater in the CCI + HS group than in CCI alone—although CBF reductions during the resuscitation phase were relatively modest compared with those seen in the shock phase. Although CBF recovered in the pre hosptial phase, it was compromised at 60 minutes after TBI in the CCI and CCI + HS models, and severely compromised at 90 minutes in the CCI + HS model—a time when metabolic demand is increased in hippocampus. 33 Given that we previously showed that CA1 neuronal death is exacerbated in the hippocampus ipsilateral to the insult in this model,17–19 presumably the flow decrement in the initial 90 minutes after CCI + HS is sufficient to ultimately lead to CA1 neuronal death. An initial hyperemic period after ischemia in hippocampus was followed by complete recovery of CBF in the subacute period. This is seen in other models that exhibit CA1 neuronal death such as global brain ischemia and has been shown in experimental cardiac arrest in rats using the same ASL-MRI method. 34 Studies of CBF in hippocampus at more delayed time points after injury after hippocampal neuronal death has occurred are also warranted.
To our knowledge, this is the first report of the assessment of CBF in a model of combined TBI plus HS in mice. Technical challenges to these studies are considerable, given the size of mice and the necessity to closely monitor their physiology and control blood gases for ASL-MRI studies. However, the ability to carry out these studies in a 4.7-T magnet provides the unique opportunity to serially assess both global and regional CBF in the mouse brain and evaluate key ROIs with regard to exacerbation of secondary injury. Given our success in establishing this and other mouse models of TBI + HS17,35 and in light of the emerging importance of secondary insults in exacerbating brain injury, particularly in the setting of blast TBI,2,3,13,36 the availability of a mouse model to study the interaction between TBI and HS on CBF dysregulation could be of considerable importance. Our approach should open up the possibility of studying molecular mechanisms involved in secondary injury after TBI using knockout and other mutant mouse strains.
Few studies have reported on the use of rodents to explore the effect of HS on secondary damage after TBI and conflicting results have been noted. Matsushita et al 14 reported that hemorrhage-induced hypotension to an MABP of 60 mm Hg for 30 minutes after fluid percussion injury in rats produced an ~50% reduction in CBF at 20 minutes and doubled contusion volume at 72 hours. Schütz et al, 15 however, assessed the effect of a similar level of hemorrhage (MABP of 50 to 60 mm Hg) for 30 minutes after fluid-percussion injury in rats but did not report exacerbation of either brain edema or lesion volume, rather noted worsening of cognitive deficits. Recently, Robertson et al 20 reported on a model of mild CCI + HS in rats to test the effect of erythropoietin as a possible therapy. After controlled hemorrhage to an MABP of ~40 mm Hg, they observed an ~60% reduction in cortical CBF. This level of hypotension is similar to that seen in our model at 60 minutes of HS but not as low as what was seen at 90 minutes (~28 mm Hg). In our model, CBF in the cortex and contusion rich cortical ROIs was reduced ~80% in both the CCI and CCI + HS groups. Thus, it is possible that near maximal CBF reductions were already present in cortex even without a secondary insult. In contrast, HS for a duration of 90 minutes in our model exacerbated the CBF reductions in the hippocampus, thalamus, and in multiple ROIs in the contralateral hemisphere. This suggests regions further from the site of impact may represent penumbral zones that are at risk for injury exacerbation by HS, rather than just the pericontusional cortex. Although histology was not performed in these mice, supporting this possibility, in a recent study, we reported substantial hemispheric tissue loss outside of the contusion along with expansion of the contusion at 21 days after injury in a similar murine model of CCI + HS, albeit with a lower MAP and shorter HS phase. 35 Similar to our findings, a comprehensive review by DeWitt and Prough 37 identified five studies in rat models of TBI that combined hemorrhage or ischemia which examined neuropathology.14,38–41 Two of these studies revealed increased hippocampal neuronal death after the second insult. Taken together with our results, these studies suggest that the pattern of exacerbation of hypoperfusion and damage produced by a secondary insult such as HS can vary from model to model and likely depends on the severity of the TBI, proximity of the brain region assessed to the injury site, and likely the type, severity, and duration of the secondary insult. In future studies, it will be important to assess the effect of HS on neuropathology on other brain regions after CCI in our model given the effect of HS on CBF reductions in thalamus and ROIs in the contralateral hemisphere.
The HS alone insult that we used did not produce hippocampal neuronal death in our prior report 17 and a similar level of HS in rats in a study by our group showed that similar levels of HS in rats does not produce either hippocampal neuronal death in rats or cognitive deficits. 42 However, our current study clearly shows that the level of HS used was below the lower limit of blood pressure autoregulation of CBF in mice. Hemorrhagic shock alone at both the 60- and 90-minute time points produced a global (~25% to 30%) reduction in CBF that was seen in most ROIs. However, without TBI, this level of hypoperfusion for even 90 minutes was not associated with hippocampal neuronal death in our prior report 17 and consistent with that finding we noted a complete recovery of CBF in the pre hospital and definitive care phases in all ROIs in the HS only group. Our data are consistent with Lacombe et al 43 who reported that the lower limit of CBF autoregulation in normal C57BL/6 mice is ~60 mm Hg. It is unclear if more modest reductions in MABP would exacerbate hypoperfusion and damage after CCI in our model. However, blood pressure autoregulation of CBF is disturbed after TBI and injured animals can tolerate only small variations in MABP, 4 while maintaining constant CBF. As previously indicated, early post injury hypermetabolism as reported by Hovda et al 33 and others may underlie the enhanced vulnerability to hypoperfusion given the fact that flow reductions in the hippocampus (to a level of ~40 mL/100 g per minute) are not to a level that one would consider, in and of themselves, to be below the ischemic threshold in the absence of TBI. Consistent with enhanced vulnerability of the traumatically injured brain to hemorrhage, Glass et al 44 reported that even mild normotensive hemorrhage exacerbated lesion volume versus TBI alone in pigs and thus factors such as hemodilution with reduced oxygen delivery, stimulation of systemic cytokine production by hemorrhage early after injury, reduced neuroprotective gene expression, 45 or stimulation of peri-contusional depolarizations 46 could also exacerbate damage. Further study is needed to define the depth and duration of hemorrhage needed to exacerbate damage. This could be important given that the recently recognized magnitude of the clinical problem of mild TBI 47 and the fact that even mild TBI can produce loss of blood pressure autoregulation of CBF in patients. 6
It was surprising that CBF recovery in the pre hospital resuscitation phase was somewhat more complete in the CCI plus HS group than in the CCI only group—although CBF values were only modestly reduced in most ROIs in the CCI group. It is possible that some degree of volume depletion was observed in the CCI group since those mice received less fluid than the HS and CCI + HS groups. We did not use maintenance fluids in any of the groups since they are rarely used in studies of CCI in rodent models. However, we use sustained anesthesia in the MRI studies which could have influenced the state of hydration. Alternatively, the ‘recovery’ of CBF during resuscitation may, as previously suggested, represent reperfusion hyperemia in brain regions subjected hypoperfusion followed by resuscitation after HS or CCI + HS.48,49
There are several limitations to this study. First, we used Hextend as the resuscitation fluid. Hextend is the standard of care in resuscitation of blast TBI victims in combat casualty care and is a logical choice for this work. However, in civilian TBI + HS, resuscitation with normal saline or Lactated Ringer's solution would be more likely. Whether use of a crystalloid would produce a different CBF pattern during resuscitation in this model is unclear. However, we previously reported that hippocampal neuronal death was not different in our model in mice resuscitated with either Hextend or Lactated Ringer's. 18 Second, the level of hypotension during HS in this study was somewhat more severe than that observed in our prior reports with this model despite an identical volume controlled degree of hemorrhage and a similar CCI level.17–19 Intubation and mechanical ventilation were required for these studies, unlike the use of this model on the benchtop, and this could affect MABP via effects on venous return to the heart. We used isoflurane anesthesia for these studies. Isoflurane exhibits neuroprotective effects and alters CBF in experimental TBI. 50 The effect of HS on CBF after CCI could have been different if an alternative anesthetic was used. However, isoflurane is the most commonly used anesthetic in TBI models. 51 We selected a treatment threshold of 50 mm Hg for resuscitation in the pre hospital phase. Although this is a reasonable target the optimal MABP for pre hospital care of the TBI victim remains unclear. We used a model of sustained volume controlled HS to superimpose on TBI. Other models such as pressure controlled HS 35 or uncontrolled HS with ongoing bleeding could potentially produce different results. However, this approach was the most technically feasible for use with MRI studies on mice. Finally, we did not assess intracranial pressure or cerebral perfusion pressure in this model. This also was not technically feasible given the limited space constraints of the MRI bore.
In conclusion, the serial, non invasive assessment of CBF using ASL-MRI is feasible in mice even in the complex setting of combined CCI + HS. Hemorrhagic shock to a level of ~30 mm Hg exacerbated the CCI-induced CBF reductions in multiple ROIs including thalamus and hippocampus ipsilateral to injury, and in multiple ROIs in the hemisphere contralateral to injury. The impact of therapeutic strategies on CBF and other outcomes merits investigation in this model. A mouse model of combined TBI + HS also affords the unique opportunity to use knockout strains to study mechanisms of cerebrovascular dysregulation in the unique setting of TBI + HS.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
Footnotes
ACKNOWLEDGEMENTS
The authors thank Marci Provins for assistance with manuscript preparation.