
Abstract
The first few hours and days after subarachnoid hemorrhage (SAH) are characterized by cerebral ischemia, spasms of pial arterioles, and a significant reduction of cerebral microperfusion, however, the mechanisms of this early microcirculatory dysfunction are still unknown. Endothelial nitric oxide production is reduced after SAH and exogenous application of NO reduces post-hemorrhagic microvasospasm. Therefore, we hypothesize that the endothelial NO-synthase (eNOS) may be involved in the formation of microvasospasms, microcirculatory dysfunction, and unfavorable outcome after SAH. SAH was induced in male eNOS deficient (eNOS–/–) mice by endovascular MCA perforation. Three hours later, the cerebral microcirculation was visualized using in vivo 2-photon-microscopy. eNOS–/– mice had more severe SAHs, more severe ischemia, three time more rebleedings, and a massively increased mortality (50 vs. 0%) as compared to wild type (WT) littermate controls. Three hours after SAH eNOS–/– mice had fewer perfused microvessels and 40% more microvasospasms than WT mice. The current study indicates that a proper function of eNOS plays a key role for a favorable outcome after SAH and helps to explain why patients suffering from hypertension or other conditions associated with impaired eNOS function, have a higher risk of unfavorable outcome after SAH.
Introduction
Subarachnoid hemorrhage is a subtype of (hemorrhagic) stroke with high mortality and morbidity 1 and survivors of the disease often remain significantly disabled.2–4 It is increasingly recognized that changes on the level of the cerebral microcirculation occurring within the first few hours and days after aneurysm rupture play an important role for early brain injury (EBI) and unfavorable outcome after SAH.5–9 Microvasospasms of pial arterioles and a subsequent reduction of parenchymal microperfusion which have been shown to occur as early as three and up to at least 72 hours after experimental SAH and two to three days after SAH in patients significantly contribute to post-hemorrhagic ischemia,10–15 however, the molecular mechanisms of this process are largely unknown.
Endothelial nitric oxide (NO) is an important mediator of proper cerebro-vascular function. 16 After SAH, NO concentration decreases rapidly17–20 and replenishing NO bioavailability reduced microvasospasms, improved cerebral perfusion, provided neuroprotection, and improved outcome in experimental models of SAH.15,21–26 Despite the important role of NO after SAH, the molecular mechanisms of NO depletion are still not well defined. Indirect evidence from experimental studies suggests that the endothelial NO-synthase (eNOS) may be dysfunctional after SAH 17 and studies in humans demonstrated that genetic polymorphisms decreasing eNOS activity are linked to increased susceptibility for cerebrovascular disease and an increased risk for unfavorable outcome after SAH.27–29 In the current study we therefore hypothesize that NO produced by eNOS plays a crucial role for post-hemorrhagic microcirculatory dysfunction, early brain injury, and outcome after SAH.
A major obstacle when investigating eNOS is the lack of potent and isoform specific inhibitors and antibodies. 30 Since this is believed to be one of the major causes of heterogeneous results in other brain injury models, such as ischemic stroke or traumatic brain injury, we used a genetic approach, i.e. eNOS deficient mice, to investigate the role of eNOS for outcome and microcirculatory dysfunction after SAH.
Materials and methods
Ethical statement
All procedures performed on animals, group size calculation, and all statistical methods used to analyze in vivo data were reviewed and approved by the Government of Upper Bavaria. The results of the study are reported in accordance with the ARRIVE guidelines. 31 Animal husbandry, health screens and hygiene management checks were performed in accordance with Federation of European Laboratory Animal Science Associations (FELASA) guidelines and recommendations. 32 All surgical procedures and data analysis were performed in a randomized fashion by a researcher blinded to the genotype of the animals. Group allocation was obtained by drawing lots.
Experimental animals
eNOS deficient animals were purchased from Jackson Laboratories (strain #002684, B6.129P2-Nos3tm1Unc/J). In these animals, eNOS knock out is achieved by replacement of 129 base pairs on exon 12 of the eNOS gene by a neomycin cassette which disrupts the calmodulin binding site of the protein and introduces a premature stop codon into the transcripts. 33 Animals were then backcrossed into their C57BL/6J background for more than ten generations. We bred heterozygous mice in order to obtain homozygous and heterozygous offspring as well as littermate controls. Male mice (mean age 8 weeks) with an average bodyweight of 22.5 g were used for experiments.
Anesthesia and monitoring
Anesthesia was induced as previously described 34 under continuous monitoring of end-tidal CO2 partial pressure, oxygen saturation, heart rate (by oximetry at the hind paw, SpO2-MSE, Kent Scientific Corporation, Torrington, CT, USA), and body temperature (by a feedback controlled heating pad, FHC Bowdoinham, Bowdoin, ME, USA). Intracranial pressure was measured contralateral to SAH induction using a piezoelectric probe (Codman ICP Express, DePuy Synthes, Umkirch, Germany) placed in the right temporoparietal epidural space as previously described.35,36 Cerebral blood flow was measured via laser Doppler fluxmetry (Perimed, Järfälla, Sweden) over the ipsilateral MCA territory.35,36 In chronic experiments, arterial blood pressure was measured non-invasively with a tail-cuff (Kent Scientific, Torrington, CT, USA). After surgery and monitoring, anesthesia was antagonized as previously described 34 and animals were kept in a heating chamber at 34 °C and 40% humidity for up to 24 h in order to prevent hypothermia and dehydration. For intravital microscopy, animals were re-anesthetized and re-intubated 2.5 h after SAH induction. For imaging, an open cranial window (4 x 4 mm) was prepared over the left middle cerebral artery territory 1–1.5 mm rostral to the coronal suture and 2 mm lateral of the midline (Figure 1(a), left and upper right panel). Blood pressure was continuously monitored via a femoral artery catheter. The femoral vein was cannulated for application of the plasma marker fluorescein isothiocyanate (FITC)-dextran (Merck, Darmstadt, Germany).
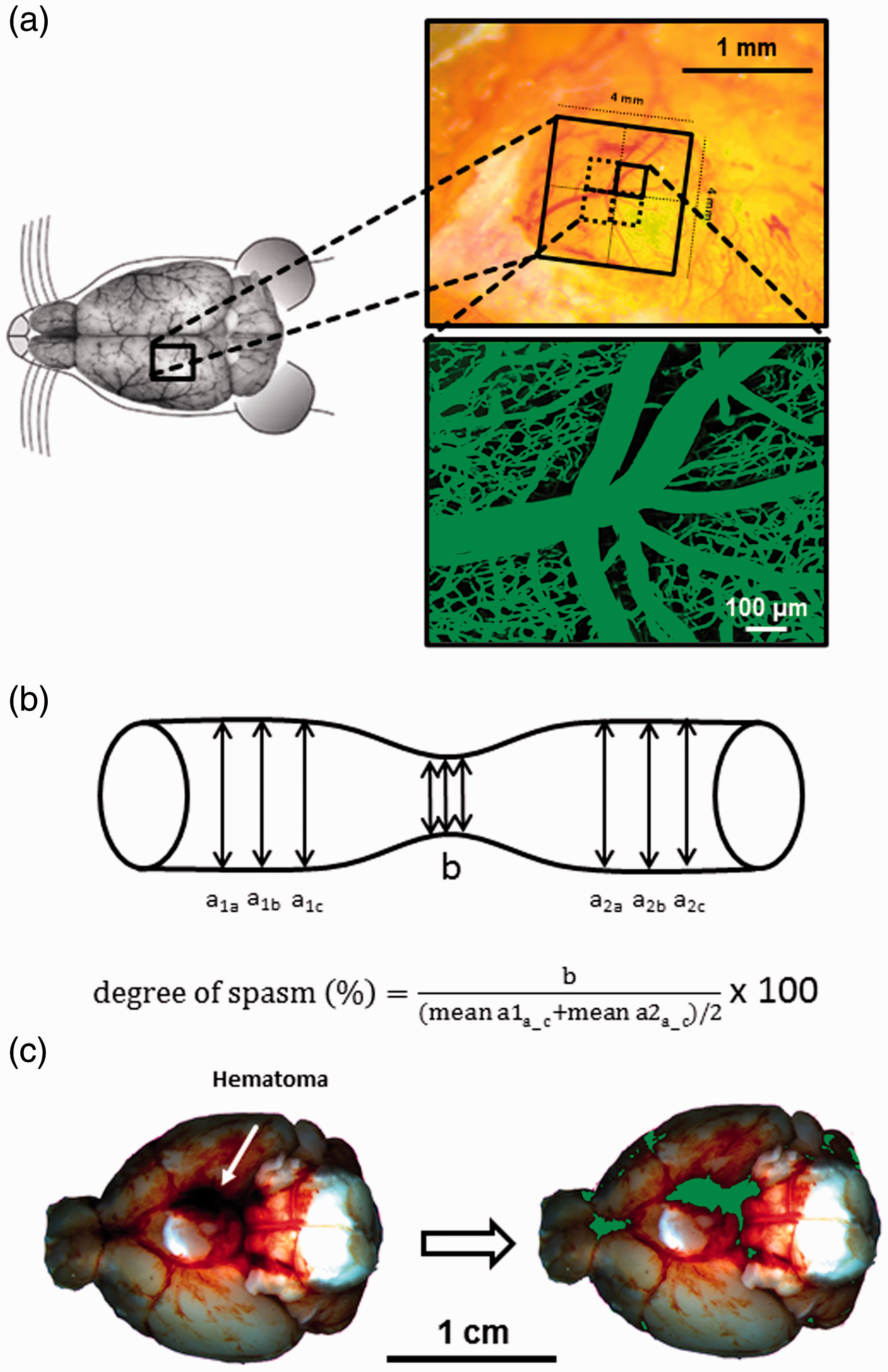
Intravital microscopy - Experimental setup. (a) Schematic overview of cranial window placement for intravital microscopy. (b) Assessment of microvasospams formation and severity. (c) Example of a hematoma at the skull base after SAH (left) and the quantification of hematoma size (green) by an automated algorithm (right).
Induction of subarachnoid hemorrhage
Subarachnoid hemorrhage was induced by the endovascular filament perforation model. 14 ,15,35 Briefly, a 5-0 monofilament was inserted into the left external carotid artery and advanced towards the circle of Willis under continuous control of cerebral perfusion and intracranial pressure until a sharp increase in intracranial pressure above 60 mmHg and a drop in cerebral perfusion below 20% of baseline was observed as a sign of successful vessel perforation. Animals not fulfilling these criteria were excluded from further analysis. A detailed step-by-step video of the procedure including probe placement can be accessed here: doi: 10.3791/50845). Rebleedings after subarachnoid hemorrhage were defined as secondary increases of ICP by 10 mmHg or more within 10 seconds or less; analysis of secondary ICP events/rebleedings was done automatedly using the intraoperatively recorded data. 36
2-Photon microscopy
In vivo 2-photon microscopy was performed three hours after induction of subarachnoid hemorrhage every 10 minutes for 90 min as previously described37–39 using a confocal microscope (LSM 7, Zeiss, Germany) equipped with a Li:Ti laser (Chameleon, Coherent, Santa Clara, CA, USA) and a water immersion objective (20x Plan Apochromat, NA 1.0, Zeiss, Germany) as well as a micromanipulator table for exact placement. Vessels were visualized by i.v. injection of 0.1 ml FITC dextran (0.5% in saline). Three-dimensional images of four adjacent regions of interest (425 x 425 µm each) in the geometrical middle of the exposed area within the craniotomy were acquired up to a depth of 400 µm and then digitally stitched together to form one single image stack of 850 x 850 µm for further analysis (Figure 1(a), lower right panel).
Data analysis
Physiological monitoring was performed at a sample rate of 1 Hz and averaged every 20 seconds. Intravital microscopy data was analyzed using ImageJ version 1.52p. Microvascular constriction and the number of microvasospasms were evaluated as previously described in arterial vessels with diameters ranging from 10 until 80 µm.14,38 A vessel segment was considered spastic when its diameter was 85% or less than non-constricted vessel segments; spasm severity is given as percent constriction of vessel diameter compared to non-constricted vessel segments (Figure 1(b)). Vessel diameter was assessed for all arterial vessels captured in each scan every 10 –15 µm along the course of the vessel which was traced throughout the 3 D dataset; non-spastic vessel diameters were averaged before and after a spastic segment, the degree of vasospasm was then calculated as described in Figure 1(b). For assessment of total perfused vessel volume all FITC-positive pixels of a stack were summed up and expressed as percentage of all pixels within this stack. All data was analyzed in a blinded fashion by a researcher unaware of the experimental series and the genotype of the animals.
Tail bleeding time
Bleeding time was determined as previously described. 15 Briefly, the distal one millimeter of the tail was amputated and the tail was placed perpendicularly in 37 °C warm saline. The time between incision and stop of bleeding was determined visually.
Determination of hematoma size
All animals were perfused with saline, then brains were carefully removed in a prone position so the blood remained at the brain base. Afterwards, the brains were placed in a prone positions under a stereomicroscope (Leica M205, Zoom: 10.6 x, resolution max. 5251p/mm; Meyer Instruments, Houston, USA) and fotographed at high-resolution. The setup for brain positioning (in a custom made white plastic holder), the position of the lights, and the camera settings were not altered during the whole experiments in order to achieve comparable illumination and image quality. The background of the image obtained was manually extracted using GIMP (The GIMP team, GIMP 2.10.6, https://www.gimp.org, 1997–2019). Using a purpose made JAVA code written for ImageJ, each image was then automatically analysed and visually crosschecked by a researcher blinded towards experimental group and genotype. The code differentiates between light red blood in the subarachnoid space and the darker red blood representing the basal blood clot as described below:
Color composition of each pixel was analysed and expressed as red (r), green (g), and blue (b) value (each ranging from 0 to 1). If r + g+b < dark_threshold (0.235), the pixel was counted as dark red/belonging to the blood clot. The threshold was established by analyzing images from previous methodological studies using the MCA perforation model and was found to provide a good differentiation between the light red/dark red blood. Pixels exceeding this threshold were quantified as light red, i.e. representing subarachnoid blood, if they fulfilled r>(g + b)/red_sensitivity. Red sensitivity was assumed to be 0 as we were not interested in quantifiying the subarachnoid hematoma, therefore these pixels were not counted. The total number of pixels fulfilling one of two conditions (r + g+b <= dark_threshold or r>(g + b)/red_sensitivity) were then counted and divided by the total number of pixels comprising the brain base. The results of automated analysis was anecdotally crosschecked by a blinded researcher manually marking pixels of the blood clot; results corresponded to a high degree (Figure 1(c)).
Statistical analysis
Sample size calculations were performed before starting the study with the following parameters: alpha error 0.05, beta error 0.2, calculated standard deviation ranged from 15–20% (depending on the parameter investigated), and biologically relevant difference 30%. This calculation also needs to account for mortality that is approximately 30% in 3 days in this model. When interim analysis showed a relevantly increased mortality in eNOS transgenic animals, their number had to be increased in order to achieve the calculated group numbers. In order to guarantee adequate randomization and blinding an equal number of wildtype animals was randomized thereby increasing n in the WT group. Since the resulting sample size was below 25, all data was assumed to be not normally distributed even when it passed normality testing (Kolmogorov-Smirnov method) in order to avoid overestimating a possible effect; therefore, only non-parametrical statistical tests were used for statistical analysis. Two groups were compared with the Mann Whitney rank sum test, multiple groups with Kruskal-Wallis ANOVA on ranks, and repeated measurements with the Friedman test. When indicated, the Dunnet test was performed post-hoc. All calculations were done using a standard statistical software package (SigmaStat 3.0, Jandel Scientific, Erkrath, Germany). Differences between groups were considered to be significant at p < 0.05.
Results
After induction of SAH wild type mice displayed a sharp increase in intracranial pressure to around 60 mmHg followed by a recovery to about 20–30 mmHg within a few minutes (Figure 2(a), dark grey circles); no animal had to be excluded due to insufficient primary ICP-increase (<60 mmHg). In heterozygous and homozygous eNOS deficient animals peak ICP values were more than 25 mmHg higher than in wild type mice (p < 0.001 vs. WT control, Kruskal-Wallis test, Dunnet´s post hoc test). While in heterozygous animals ICP values recovered over time and reached those of wild type mice at the end of the observation period, ICP in homozygous eNOS mice remained significantly elevated (Figure 2(a), light grey and open circles). Cerebral blood flow dropped significantly in all experimental groups immediately after SAH (Figure 2(b)), however, the drop in CBF was significantly more pronounced in heterozygous and homozygous eNOS deficient mice. While heterozygous animals recovered to control levels within two minutes, homozygous eNOS mice showed a significantly reduced CBF for more than six minutes (Friedman test).
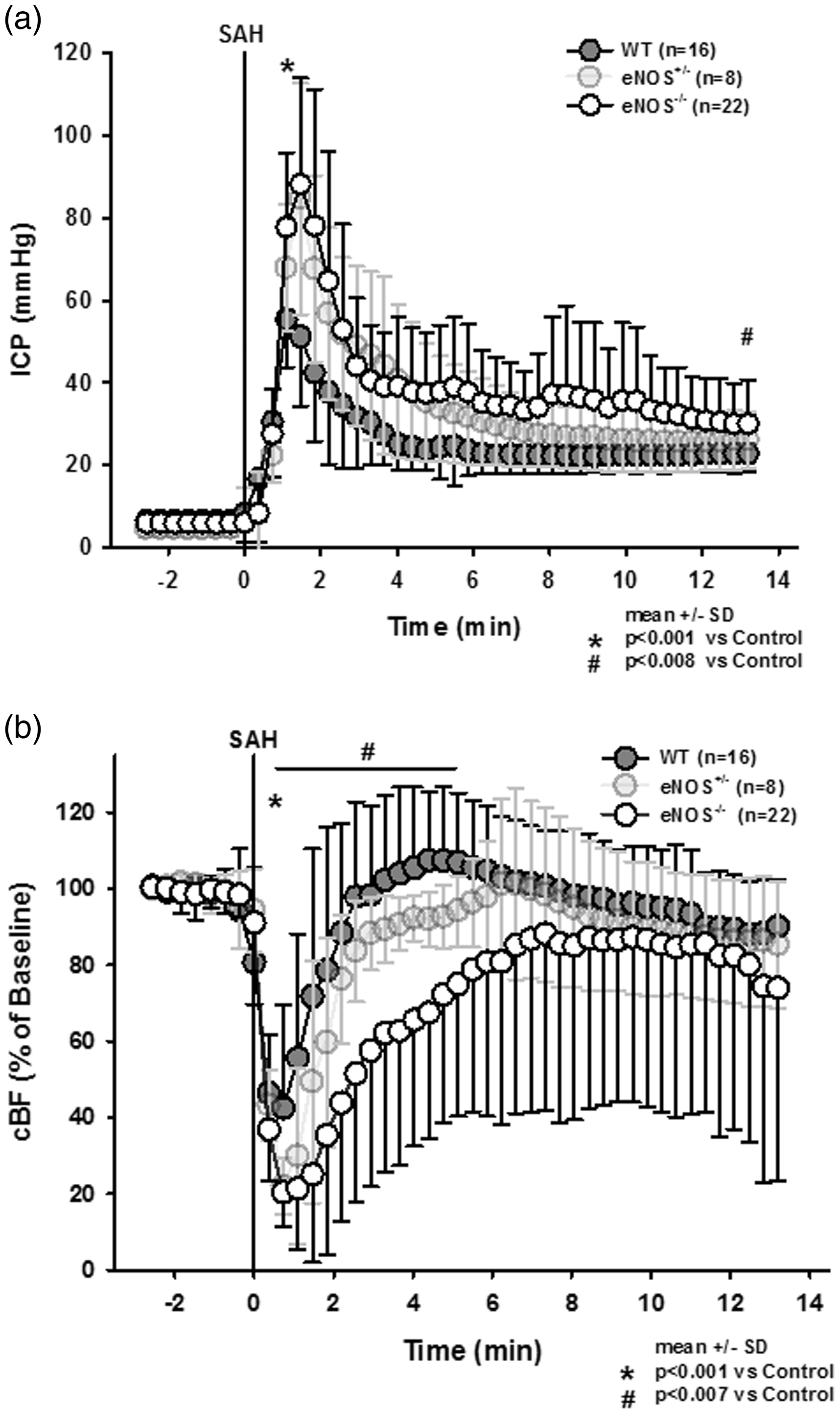
Intracranial pressure and local cerebral blood following subarachnoid hemorrhage. (a) Peak intracranial pressure is significantly and gene-dose-dependently elevated in eNOS deficient mice. After few minutes, ICP drops again but remains significantly elevated in eNOS homozygous animals compared to wild type littermates. (b) Cerebral perfusion drops to lower values in eNOS deficient as compared to wild type mice. While in heterozygous and wild type mice CBF recovers to baseline values within a short time, CBF in homozygous eNOS deficient mice remains significantly impaired.
When ICP recordings from single animals were analyzed, we observed several secondary ICP increases indicating re-bleedings, specifically in eNOS deficient animals. Secondary increases in intracranial pressure accompanied by drops in CBF have been described to be associated with radiologically detectable re-bleeding in CT or angiography 40 and occurred in our previous experimental studies41,42 where they were associated with worse outcome; while brain edema formation and/or hydrocephalus may account for elevation of ICP, the onset of these phenomenona is not as rapid and it is not as readily reversible, therefore occurrence of re-bleeding events are the most feasible explanation for these occurrences. In order to quantify these observations, an additional cohort of eNOS–/– and corresponding control animals were continuously monitored for up to 180 minutes after SAH. Exemplary traces for a wild type and an eNOS–/– mouse (Figure 3(a)) show no re-bleedings in wild type, but multiple re-bleedings in eNOS–/– mice. Quantification of these events showed a three-fold increase in the number of re-bleedings in eNOS–/– mice as compared to wild type controls or heterozygous eNOS animals (Figure 3(b); p = 0.004 vs. WT, p = 0.022 vs. eNOS+/–, Kruskal-Wallis test, Dunnet´s post hoc test).
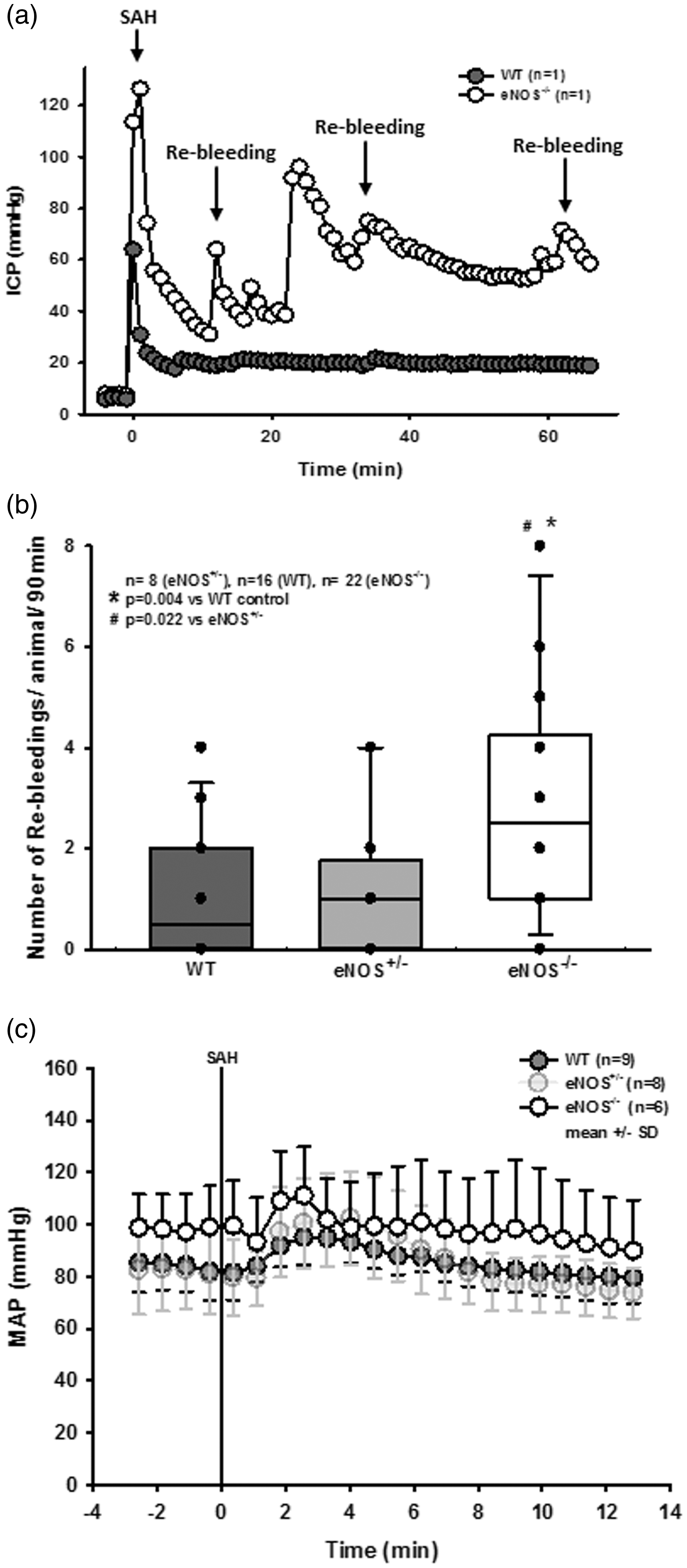
Re-bleeding events. (a) Exemplary ICP traces in single mice show repetitive re-bleedings in an eNOS–/– animal while no such events were observed in a wild type littermate. (b) Quantification reveals a significantly elevated number of re-bleeding events in eNOS homozygous animals compared to wild type and heterozygous mice (p = 0.004 and 0.022, respectively). (c) Invasive arterial blood pressure recordings in homozygous (dark grey symbols) and heterozygous (light grey symbols) eNOS deficient mice and wild type littermates before and after SAH. Already before SAH eNOS–/– mice had a slightly increased arterial blood pressure. Within the first four minutes after SAH all three genotypes showed the typical increase in blood pressure due to increased intracranial pressure, the Cushing response. Thereafter, arterial blood pressure returned to baseline levels in all investigated genotypes.
Re-bleedings may be caused by high blood pressure or by insufficient hemostasis. Since high blood pressure as well as impaired hemostasis were described in eNOS–/– mice,33,43–45 we analyzed both parameters in our experimental series. When investigating systemic arterial blood pressure in wild type and heterozygous and homozygous eNOS deficient mice, we indeed observed a non-significant trend towards a higher blood pressure of about 10 mmHg in homozygous eNOS deficient mice before and after SAH (Figure 3(c)). However, the Cushing response was similar in all investigated animals and no blood pressure peaks were observed in eNOS deficient mice. Hence, we were not convinced that this increase in blood pressure is the main reason for the quite impressive re-bleeding phenotype observed in homozygous eNOS deficient mice. Therefore, we investigated hematoma size as a proxy for hemostasis and found a more than twice as large hematoma area in eNOS–/– mice as compared to wild type controls (Figure 4(a); p < 0.001, Mann Whitney rank sum test). Since larger hematomas point towards an altered bleeding phenotype in eNOS deficient mice, we assessed the tail bleeding time in our animals. Indeed, eNOS deficient mice had an almost three times longer bleeding time than wild type control mice which had a physiological bleeding time of about one minute. (Figure 4(b); p = 0.006, Mann Whitney rank sum test). Bleeding time analysis does not allow for exact differentiation of platelet function and vascular endothelial function as well as the influence of arterial blood pressure. As we did detect a non-significant difference of mean arterial blood pressure between the genotypes, we believe that incomplete hemostasis in combination with (slightly) increased blood pressure is the mechanism behind increased SAH intensity and the re-bleeding phenotype observed in eNOS–/– mice. In order to address the aim of the current study, i.e. the role of eNOS on the cerebral microcirculation after SAH, we subjected wild type and eNOS–/– mice to SAH and observed cerebral blood vessels in vivo by 2-photon microscopy. One wild type and one eNOS mouse had to be excluded due to technical issues during cranial window preparation. Exemplary 2-PM image stacks representing the cerebral microcirculation down to a depth of 400 µm in sham operated mice show a slightly rarefied capillary density in homozygous eNOS mice compared control animals (Figure 5(a), upper panels). This was confirmed by quantification of the perfused vessel volume which showed a significantly decreased vessel density in eNOS–/– mice (Figure 5(b), striped bars; p < 0.009, Mann Whitney rank sum test). Of note, this automated evaluation method quantifies the total cerebral microvasculature, i. e. the venous as well as the arterial side. After SAH, there was a significant vessel rarefication in wild type and eNOS–/– animals as compared to control conditions (Figure 5, lower panels and Figure 5(b), solid bars; p < 0.038, Kruskal-Wallis test, Dunnet´s post hoc test). The decrease in vessel density, however, was more pronounced in eNOS–/– mice. The absolute difference between wild type and eNOS–/– mice was similar to the status before SAH indicating that the loss of endothelial NO production does not aggravate the already existing vascular phenotype in eNOS–/– mice.
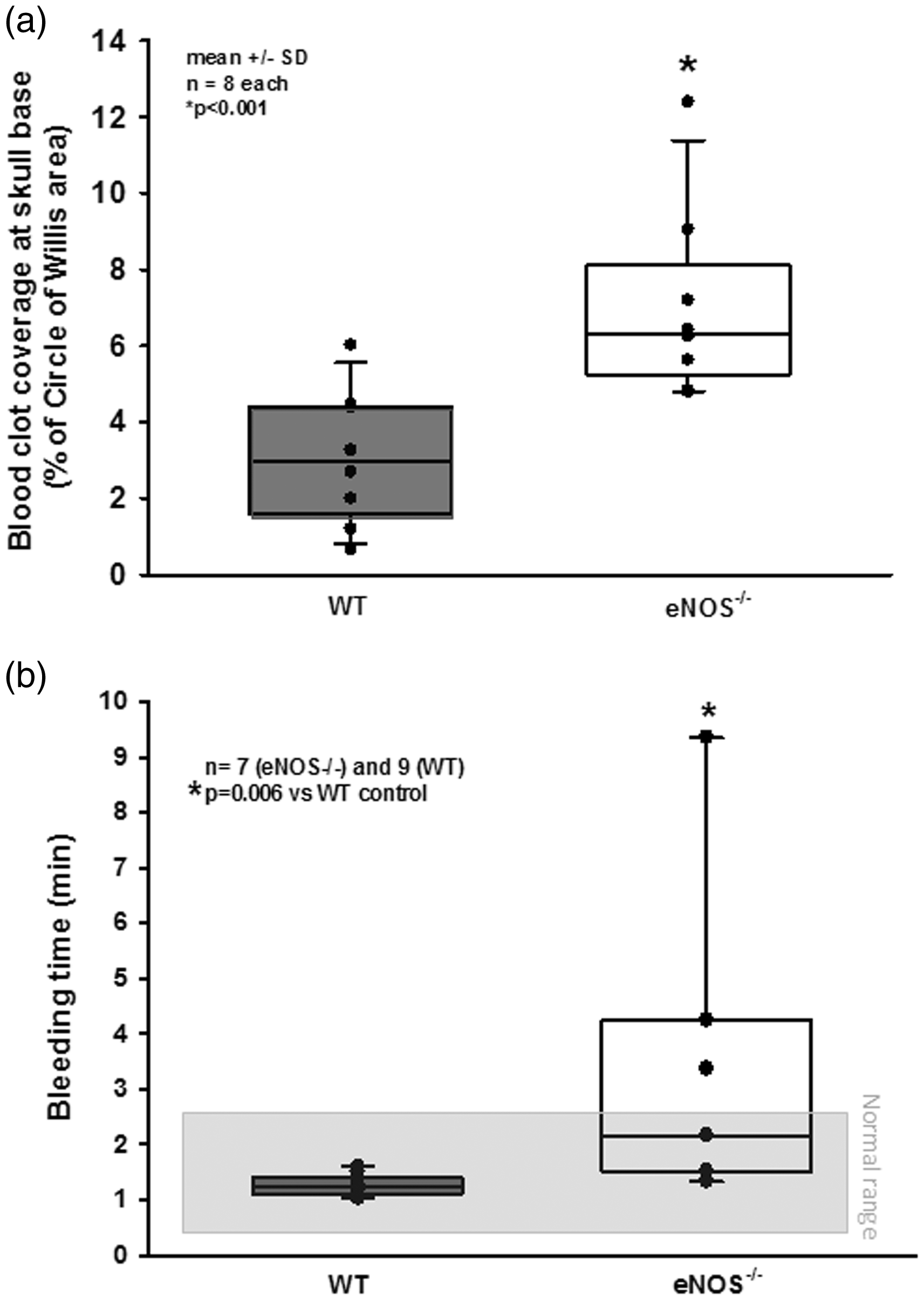
Possible causes for increased SAH severity in eNOS–/– mice. (a) eNOS–/– mice had significantly larger hematomas at the skull base as compared to wild type littermates after SAH. (b) Tail bleeding time was significantly elevated in eNOS deficient animals.
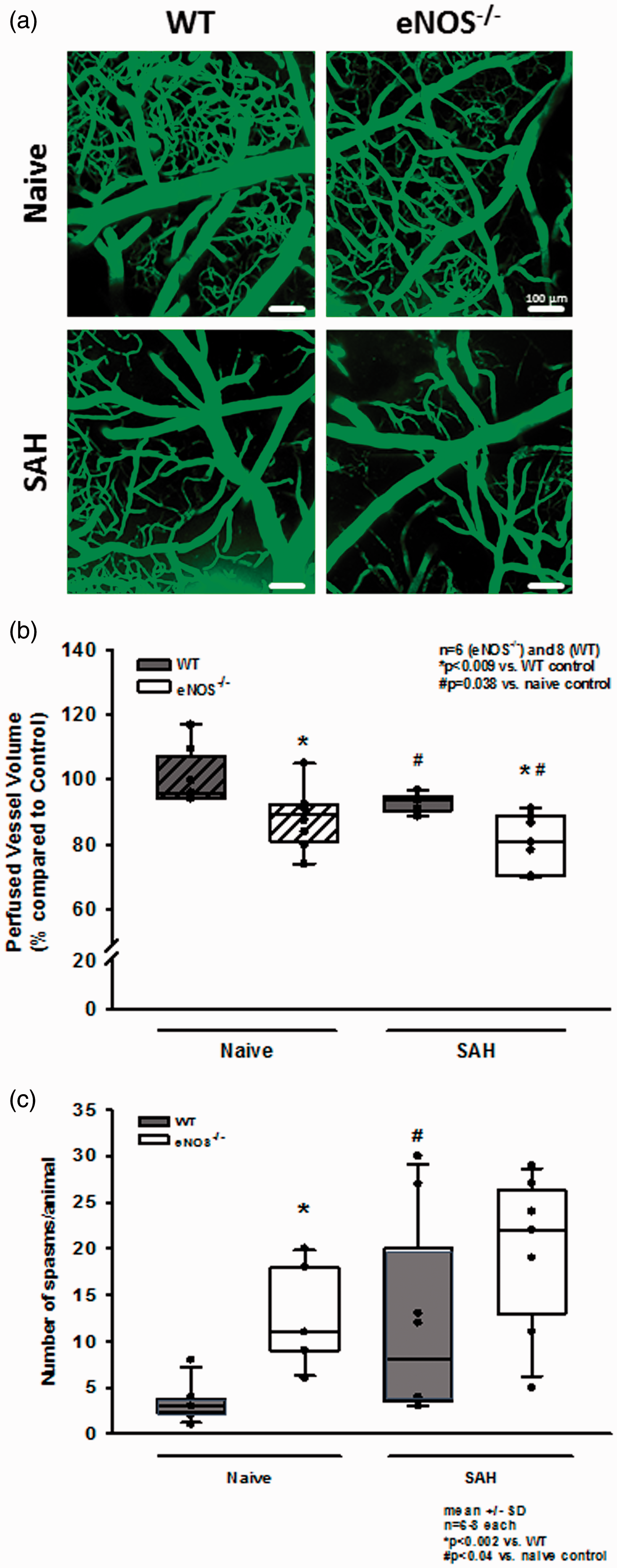
Changes of the cerebral microcirculation after SAH. (a) Exemplary IVM screenshots depicting the cerebral microcirculation in naïve animals (upper panels) and after induction of SAH (lower panels). eNOS deficient mice already show a slight narrowing and rarefication on the capillary level. After SAH, there is (increased) microvascular constriction in both genotypes. (b) Corresponding to the exemplary picture, total perfused vessel volume is slightly but significantly lower in eNOS–/– homozygous animals compared to wild type mice. After SAH, perfused vessel volume is significantly lower in both genotypes, indicating relevant microvascular constriction and – thus – microcirculatory dysfunction. The drop in perfused vessel volume, however, is significantly more pronounced in eNOS–/– than in wt. (c) Microvasospasms occur very rarely under physiological conditions in wild type controls (left side, dark bar). The number of MVS in eNOS–/– is significantly elevated. After SAH, there is significant spasm formation in wt mice (right side of panel). This increase is more pronounced in eNOS–/– mice (striped bars).
For evaluation of micro-arteriolar spasms about 360 vessel segments with an average length of 174 µm were analyzed in wild type and eNOS–/– mice. Following sham surgery very few spasm-like constrictions were observed in wild type mice, while eNOS–/– mice had a more than three-fold higher rate of spontaneous micro-arteriolar spasms (Figure 5(c), striped bars; p = 0.002, Mann Whitney rank sum test). Lack of basal production of the vasodilator NO in endothelial cells may well explain this so far unidentified phenotype. After SAH the number of micro-arteriolar spasms almost tripled in wild type mice, while in eNOS–/– mice the number of spasms increased only by about 40%, though from a higher level (Figure 5(c), solid bars; p = 0.04, Mann Whitney rank sum test). As already determined in earlier studies, 14 , 15 micro-arteriolar spasms after SAH occurred predominantly in microvessels with a diameter below 30 µm (Supplemental Figure 1a and b). These data indicate that eNOS deficiency causes a reduction of cerebro-vascular density and the formation of micro-arteriolar spasms already under physiological conditions. SAH worsens this phenotype and may thus cause additional brain damage. This conclusion is supported by the massively elevated mortality in eNOS deficient animals. While none of the 16 investigated wild type animals died after SAH and heterozygous eNOS transgenic mice showed a moderately increased mortality of 25% (2/8 mice), 48% of investigated eNOS–/– mice (12/25) did not survive the first three hours after hemorrhage (Figure 6; p < 0.001, Log-Rank test).
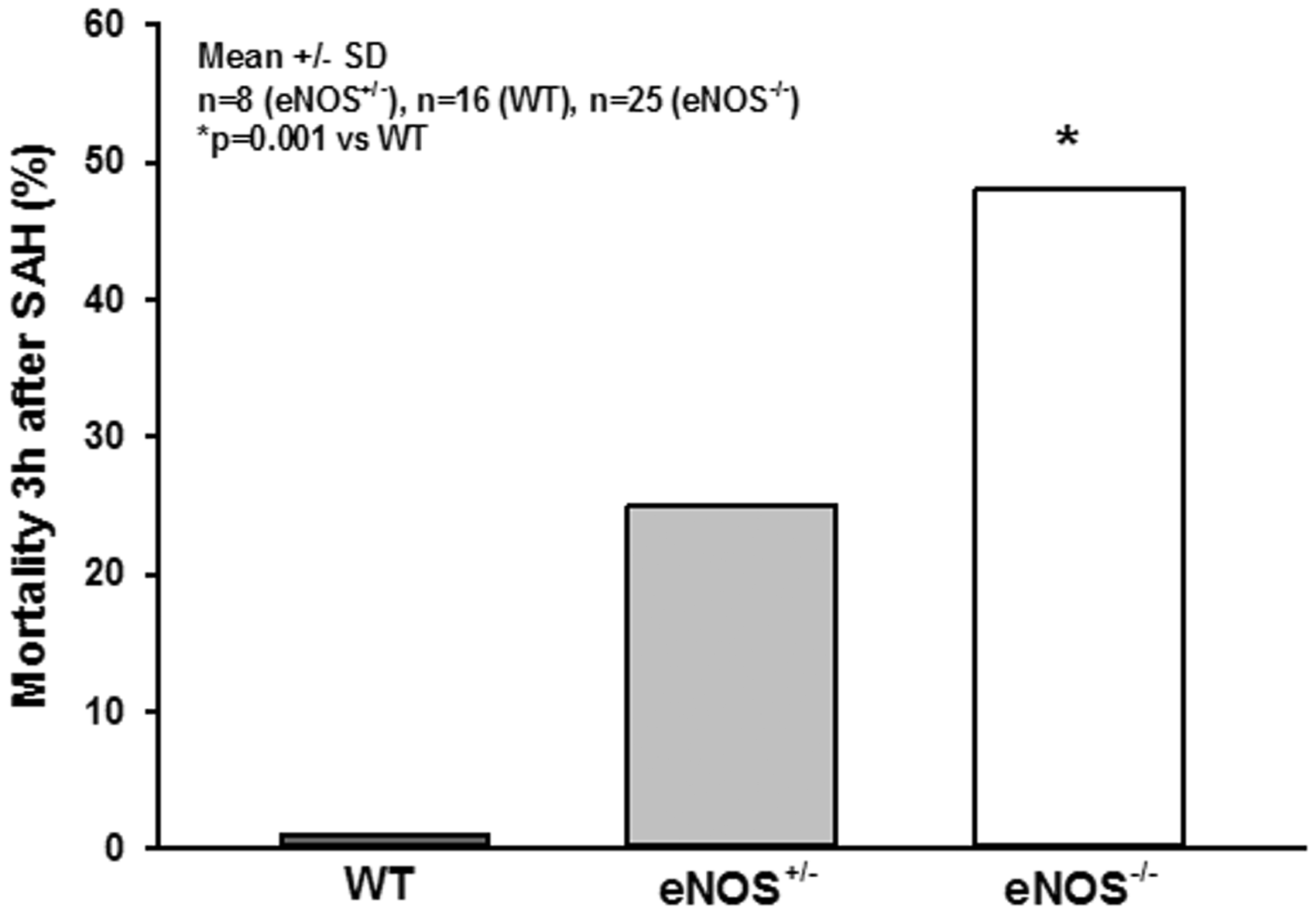
Mortality. While all wild type animals survived the early phase after SAH and heterozygous animals showed a moderately elevated mortality rate of 25%, nearly half of all homozygous eNOS–/– mice died within the first 3 hours after hemorrhage induction.
Discussion
In the present study, we demonstrate that eNOS-deficient mice have significantly increased morbidity and mortality after experimental SAH. Hemorrhage is more severe in eNOS–/– animals as indicated by higher peak ICP values and increased hematoma size; this increased SAH severity is most probably caused by elevated blood pressure and a longer bleeding time found in eNOS transgenic animals. Hence, a more severe SAH is part of the post-hemorrhagic phenotype of eNOS transgenic animals. However, also post-hemorrhagic microcirculatory changes are more pronounced in eNOS–/– mice suggesting an important role of eNOS derived NO for the maintenance of proper microvascular function after SAH.
Interestingly, we also found that eNOS deficient mice have a reduced microvessel density and a significant number of microvasospasms already before SAH. These findings are consistent with the important function of eNOS-derived NO for the maintenance of microvascular stability and vascular tone under physiological conditions. These changes did not result in changes in cerebral blood flow (CBF) since an elevated systemic blood pressure compensated the reduced microperfusion, conclusions consistent with published results of normal CBF in eNOS deficient mice. 46
Furthermore, the present results demonstrate that SAH leads to a marked deterioration of microcirculatory function which - in combination with the increased hemorrhage severity observed in the present study – results in significantly increased post-hemorrhagic mortality. These findings corroborate the importance of eNOS-derived NO and eNOS-dysfunction for the development of early brain injury after subarachnoid hemorrhage.
Early local depletion of nitric oxide has been identified to play an important role in the development of post-ischemic47–51 and post-traumatic52–55 brain damage. While a secondary increase of nitric oxide concentration at later time points – most probably due to activation of inducible NO-synthase has repeatedly been demonstrated to be deleterious in these types of brain injury,56–61 endothelial NO production by eNOS has consistently been considered neuroprotective.19,24,49,62–69
As for subarachnoid hemorrhage, an early decrease of the NO concentration has been observed in experimental studies17,18,65,70,71 and in patients.9,15,72–74 This early NO depletion is linked to microcirculatory dysfunction such as the formation of microvasospasm and microarterial constriction.15,21,75,76 At later time points the lack of NO has been linked to the formation of (angiographic) vasospasm.22,67,77,78 Low nitric oxide concentrations also have been shown to facilitate cortical spreading depolarizations79,80 which may contribute to the development of delayed ischemia after subarachnoid hemorrhage.79,81,82 It has been speculated that eNOS dysfunction may (partially) be responsible for early NO depletion, especially in brain vessels19,83–85 and that this may be a cause for early microcirculatory dysfunction. 19 The role of eNOS for maintaining proper microcirculatory function is also supported by the fact that eNOS does not play a significant role in the formation of macrovasospasm after SAH. 56
The influence of SAH on eNOS activity and expression, however, remained unclear with studies finding no effect of SAH on eNOS activity86–88 or an increased eNOS activity after experimental SAH. 89 It has also been reported that uncoupling of the homodimeric eNOS complex, which occurs under pathological conditions and has also been reported after experimental subarachnoid hemorrhage, can lead to excess reactive oxygen species formation that aggravates post-ischemic and post-hemorrhagic brain damage. 89 Still, this was found to be accompanied by NO depletion. 89 Hence, the biochemical investigation of eNOS after SAH generated heterogeneous results so far.
In order to overcome the limitations of the biochemical approach, eNOS deficient mice were already previously subjected to experimental SAH. 90 In this previous study, wild type and eNOS deficient mice received an injection of blood into the prechiasmatic cistern and following this procedure less vasospasm of the middle cerebral artery, less microthrombosis, and less apoptotic cell death was observed. These findings were quite surprising, since in all other investigated animal models eNOS deficiency univocally resulted in worse outcome in the context of cerebrovascular pathology.64,68,71,91–93 One possible explanation may be that the prechiasmatic blood injection model was developed to be able to specifically investigate the effect of blood on larger cerebral arteries located in the subarachnoid space. Hence, the model lacks the clinically relevant feature of vessel perforation and all the subsequent, pathophysiological equally important sequels, e.g. intracranial hypertension, global cerebral ischemia, and microvascular dysfunction. The fact that all relevant changes observed in eNOS deficient mice in the current study are not part of the prechiasmatic blood injection model (increased re-bleeding rate, larger hematoma volume, and impaired microperfusion) may explain the differences between the two studies. One possible drawback of the perforation model is (additional) endothelial injury to extracranial or circle of Willis vessels during advancement of the filament towards the perforation site which deviates from the human pathology where vessel wall injury occurs only at the rupture site; this, however, may incidentally occur in all experimental groups including sham-operation and should therefore not influence our results. Also, focal hypoperfusion or ischemia in the MCA territory may occur immediately after MCA puncture in our model, but is quickly followed by global ischemia when ICP peaks, thereby inducing global hypoperfusion. Of note, hypoperfusion and subsequent ischemia/ischemic damage distal to the vessel rupture site has been described in human SAH94,95 so this component may be considered a pathophysiological feature which is reproduced by the model. As hemorrhage is more severe in eNOS transgenic animals, the focal ischemia component immediately after rupture may be more pronounced in mutant mice, which may add to the increased SAH pathology observed in eNOS–/– mice.
In previous studies using the same setup as used in the current study,39,96,97 craniotomy or an open cranial window preparation did not per se induce microvascular changes or affect vessel reactivity (neurovascular coupling, CO2 reactivity); also, there is no evidence of cortical spreading depolarization caused by surgical manipulation unless animals had increased sensitivity towards CSD. 98 While eNOS transgenic animals have not been shown to have increased susceptibility to develop CSDs and while we did not detect any indirect signs of neurophysiological changes (e. g. reversible waves of hyperperfusion in CBF recording) we cannot exclude that eNOS transgenic animals may react to cranial window preparation with CSDs which may alter or negatively influence cerebral hemodynamics in eNOS–/– mice. As iatrogenic CSDs are more likely to occur when using a traumatic drilling technique utmost care was taken during window preparation; imaging was started 20 minutes after window preparation to minimize possible artifacts. Also, we performed repetitive measurements over a period of up to 110 minutes after surgical manipulation and averaged results. Therefore, we think it highly unlikely that the results obtained in this study are significantly biased by the technique used. Lastly, we would like to mention that the changes observed in the present study were evidenced via a cranial window placed over the left parietal region, ipsilateral to SAH induction at the skull base. After SAH by MCA perforation, blood distributes evenly over the convexity of both hemispheres along perivascular spaces; 36 although we performed a regional analysis we therefore have no reason to believe that the phenomena observed in the present study are limited to the left parietal region.
The present results suggest an important role of endothelial NO production for the pathophysiology of SAH. Chronic lack of endothelial NO, as also observed in patients suffering from hypertension and atherosclerosis, a risk factor for SAH, seems to reduce neuro-vascular capillary density and to cause arteriolar spasms already under physiological conditions. If SAH occurs on top of this preexisting microvascular pathology, lack of NO causes more severe bleedings due to impaired hemostasis and induces a high rate of re-bleedings, one of the most life-threatening complications of SAH. Hence, the findings of the current study may help to explain why patients who suffer from hypertension or other disorders associated with decreased endothelial NO production have a disproportional high rate of unfavorable outcomes after SAH. 99 Therefore, our findings may help to better understand the mechanisms responsible for worse outcome of SAH patients with vascular dysfunction and stress the importance of research efforts directed towards novel therapeutic options to counteract the chronic lack of endothelial NO production in patients at risk for SAH.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X20973787 - Supplemental material for Role of endothelial nitric oxide synthase for early brain injury after subarachnoid hemorrhage in mice
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X20973787 for Role of endothelial nitric oxide synthase for early brain injury after subarachnoid hemorrhage in mice by Irina J Lenz, Nikolaus Plesnila and Nicole A Terpolilli in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was supported by the “Fakultätsförderprogramm für Forschung und Lehre” (FöFoLe; project #31/2016) to IJL/NP/NAT by the Medical Faculty of the University of Munich and the Friedrich-Baur-Foundation (NAT, project #63/13) and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy – ID 390857198).
Acknowledgements
The authors would like to thank Andreas Lenz, Institute for Communications Engineering, Technical University Munich, for writing the codes for automated assessment of hemorrhage area/perfused blood volume and Janina Biller for technical assistance/animal genotyping.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
IJL performed all experiments, analyzed, and interpreted the data, performed statistical analysis. NAT planned and ideated experiments, analyzed and interpreted the data, performed statistical analysis. NP planned and ideated experiments, interpreted the data. The manuscript was written by IJL, NAT, and NP.
Supplementary material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.