
Abstract
Caffeine, the most widely consumed psychoactive drug and a weak adenosine receptor antagonist, can be neuroprotective or neurotoxic depending on the experimental model or neurologic disorder. However, its contribution to pathophysiology and outcome in traumatic brain injury (TBI) in humans is undefined. We assessed serial cerebrospinal fluid (CSF) concentrations of caffeine and its metabolites (theobromine, paraxanthine, and theophylline) by high-pressure liquid chromatography/ultraviolet in 97 ventricular CSF samples from an established bank, from 30 adults with severe TBI. We prospectively selected a threshold caffeine level of ≥1 μmol/L (194 ng/mL) as clinically significant. Demographics, Glasgow Coma Scale (GCS) score, admission blood alcohol level, and 6-month dichotomized Glasgow Outcome Scale (GOS) score were assessed. Mean time from injury to initial CSF sampling was 10.77 ± 3.13 h. On initial sampling, caffeine was detected in 24 of 30 patients, and the threshold was achieved in 9 patients. Favorable GOS was seen more often in patients with CSF caffeine concentration ≥ versus < the threshold (55.6 versus 11.8%, P = 0.028). Gender, age, admission CGS score, admission blood alcohol level, and admission systolic arterial blood pressure did not differ between patients with CSF caffeine concentration ≥ versus < the threshold. Increases in CSF concentrations of the caffeine metabolites theobromine and paraxanthine were also associated with favorable outcome (P = 0.018 and 0.056, respectively). Caffeine and its metabolites are commonly detected in CSF in patients with severe TBI and in an exploratory assessment are associated with favorable outcome. We speculate that caffeine may be neuroprotective by long-term upregulation of adenosine A1 receptors or acute inhibition of A2a receptors.
Introduction
Outcome after severe traumatic brain injury (TBI) is influenced by many mechanisms involved in the evolution of secondary damage and repair. Superimposed on these intrinsic and extrinsic mechanisms, effects of commonly consumed drugs, such as ethanol, illicit drugs, and sex steroids may also play a role. Caffeine, the most widely consumed psychoactive drug (Jacobson et al, 1996), may contribute beneficial or detrimental effects after TBI. A large gourmet coffee may contain as much as 550 mg of caffeine (Nutrition Action Newsletter, Center for Science in the Public Interest, 2007).
Caffeine is a weak and nonselective adenosine receptor antagonist. Studies in adenosine receptor knockout (ko) mice suggest that many of the acute effects of caffeine may be mediated by the adenosine A2a receptor (A2AR) (Huang et al, 2005). However, effects of caffeine can also be mediated at the adenosine A1 receptor (A1AR), and results depend on whether acute or long-term exposure is involved (Johansson et al, 1993; Georgiev et al, 1993; Al Moutaery et al, 2003). Adenosine A2b receptor and A3AR may also play a role (Conde et al, 2006; Pugliese et al, 2006).
Adenosine levels are markedly increased both in brain interstitial fluid and cerebrospinal fluid (CSF) after severe TBI in humans (Bell et al, 2001; Clark et al, 1997; Robertson et al, 2001). Adenosine has complex effects in experimental models of central nervous system (CNS) trauma—both neuroprotection and neurotoxicity have been reported (Cassada et al, 2002; Varma et al, 2002). Pignataro et al (2007) reported that transgenic overexpression of adenosine kinase—the primary adenosine-metabolizing enzyme—rendered the mouse brain more susceptible to ischemic cell death. Similarly, A1AR ko mice develop lethal acute status epilepticus after experimental TBI (Kochanek et al, 2006). Also supporting a neuroprotectant role for adenosine, administration of an A2AR agonist conferred beneficial effects on outcome or cerebral blood flow after experimental spinal cord injury or TBI (Cassada et al, 2002; Reece et al, 2004; Kochanek et al, 2005), and A1AR upregulation and activation can attenuate neuroinflammation and demyelination in experimental multiple sclerosis (Tsutsui et al, 2004), and reduce β-amyloid deposition in experimental Alzheimer's disease (Arendash et al, 2006). However, A2AR ko mice show reduced infarct size versus wild type in stroke (Chen et al, 1999; Yu et al, 2004), suggesting complex effects of adenosine in CNS injury.
In experimental TBI, caffeine administration has produced beneficial or detrimental effects, depending on the dose, model, and timing. Al Moutaery et al (2003) reported that acute administration of extremely large doses of caffeine immediately before injury worsens outcome after experimental TBI in rats, although the doses used (100 to 150 mg/kg) would equate to a human acutely consuming massive amounts of caffeine. In contrast, Dash et al (2004) reported beneficial effects of the combination of caffeine and ethanol after experimental TBI, mirroring the work of Strong et al (2000) in stroke. An important interaction between caffeine and ethanol has been suggested by these studies. Long-term caffeine administration reduces neuronal injury in rats after ischemic insults (Sutherland et al, 1991). Upregulation of A1AR number or function, by long-term intermittent exposure, may mediate this effect (Johansson et al, 1993; Georgiev et al, 1993), along with other potential mechanisms.
In clinical studies, caffeine consumption is powerfully associated with reduced risk for development of Parkinson's disease. Coffee or tea intake at levels of three cups per day for 10 years was associated with a 22 to 28% risk reduction of Parkinson's disease (Tan et al, 2003). Ross et al (2000) reported a two to threefold increase in risk of Parkinson's disease for noncoffee drinkers, adjusted for age and tobacco use. High striatal levels of A2AR may play a role in effecting this relationship. These and other epidemiologic studies have assessed caffeine consumption through dietary recall. However, to our knowledge, neither the direct measurement of caffeine in human samples nor an assessment of the association between caffeine consumption and outcome in TBI has, been investigated. There have also been no studies of CSF levels of caffeine metabolites, such as theobromine or paraxanthine after TBI in humans.
We sought to test the hypotheses that clinically relevant levels of caffeine would be detectible in the CSF of patients after a severe TBI and that its presence would be associated with favorable outcome. To this end, we used our established repository of CSF samples from adults with severe TBI (Bayir et al, 2004) and established methods to quantify levels of caffeine and its metabolites (Frye et al, 1998).
Materials and methods
IRB approval from the University of Pittsburgh Medical Center was obtained for use of CSF samples and associated demographic and outcome data that was collected from adult patients with written informed consent from next of kin. All patients were treated with a standard protocol based on the Guidelines for the Management of Severe TBI in adults (1996). Patients were selected for inclusion in our study from our CSF repository based on the requirement that sufficient CSF was available from the acute (a minimum of two samples in the initial 24 h) period after severe TBI (GCS score ≤ 8). Cerebrospinal fluid samples were used to study caffeine and the caffeine metabolites theobromine, paraxanthine, and theophylline. We studied 97 CSF samples from 30 adult patients (age = 17 to 76 years), an average of 3.2 samples per patient. Samples were stored at −80°C before batch analysis. Baseline demographic data that were collected included age, gender, admission GCS score, admission blood alcohol concentration, and admission systolic arterial blood pressure.
We assayed CSF samples (0.1 mL) for caffeine and its metabolites by high-pressure liquid chromatography (Hewlett Packard HPLC system model 1050, Palo Alto, CA, USA) with detection of ultraviolet absorbance at 274 nm using the method described previously by Frye et al (1998). The internal standard was β-hydroxyethyl theophylline, and the retention times of the analytes of interest were: theobromine 3.0 mins, paraxanthine 4.5 mins, theophylline 5.2 mins, internal standard 5.7 mins, and caffeine 7.8 mins. The sensitivity of the assay permitted detection of caffeine and its metabolites if the levels were at least 0.18 μmole/L in the undiluted CSF sample.
We prospectively chose a threshold caffeine level of ≥ 194 ng/mL (1 μmol/L) as clinically significant; a level approximating the lowest level that can discriminate CNS effects in normal human volunteers based on the work of Evans and Griffiths (1999).
We used a dichotomized 6-month Glasgow Outcome Scale (GOS) to quantify outcome with unfavorable outcome defined as GOS 1, 2, or 3, and favorable outcome defined as GOS 4 or 5. Glasgow Outcome Scale was assessed by clinicians masked to CSF caffeine concentration.
Statistical methods included descriptive statistics, Fisher's exact test, univariate, and multivariate logistic regression—controlling for admission GCS score, and admission blood alcohol concentration. Demographic and individual group comparisons were made by the Mann—Whitney or Student's t-test, where appropriate. Spearman's rank correlation was also used, where appropriate.
Results
Mean patient age was 32.33 ± 2.71 years (mean ± s.e.m.); there were 22 men and 8 women. The median admission GCS score was 7. Six-month GOS score was available on 26 of the 30 patients. The four patients in whom GOS data were not available were in the low caffeine group. The mean time from injury to initial CSF sampling was 10.77 ± 0.57h. In the initial sample, caffeine was detected in 24 of the 30 patients, and caffeine or any of its metabolites were detected in 27 of the 30 patients. Overall, caffeine was detected in 72 of the 97 samples. The clinically relevant caffeine threshold level of 1 μmol/L (194 ng/mL) CSF was achieved or exceeded in 9 of the 30 patients. The average peak CSF caffeine concentration was 220.98 ± 79.04 ng/mL. Caffeine concentrations as high as 2,005.40 ng/mL were detected, greater than 20 times the threshold level. Peak CSF concentrations of the caffeine metabolites theobromine, paraxanthine, and theophylline were 156.5 ± 32.7, 167.6 ± 39.8, and 18.9 ± 3.3 ng/mL, respectively. The time course of CSF caffeine concentration in patients after severe TBI is shown in Figure 1.
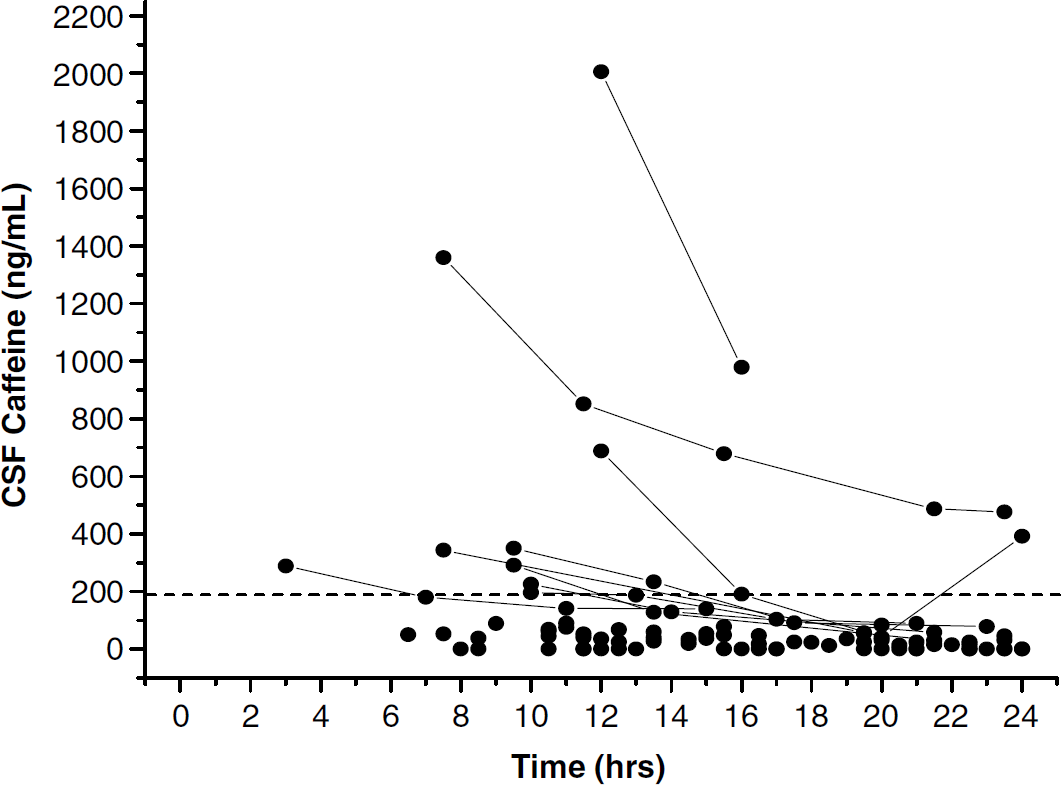
Time course of caffeine concentrations in cerebrospinal fluid (CSF) in patients with severe TBI. The dotted line represents the clinically significant caffeine threshold of 1 μmol/L (194 ng/mL). Solid lines connect values in patients that had an initial caffeine value greater than the predefined threshold.
Age, GCS score, admission blood alcohol concentration, and admission systolic arterial blood pressure did not differ between groups either greater than or less than or equal to the threshold caffeine concentration (Table 1). Admission blood alcohol concentration was assessed in 27 of the 30 patients. Alcohol was detected in 17 of the 27 patients. The mean admission blood alcohol concentration was 90.00 ± 18.85 mg/dL, and 11 patients had an admission blood alcohol concentration greater than 100 mg/dL. Gender did not also differ between groups; notably, 81% of the patients with CSF caffeine levels greater than or equal to the threshold were men, whereas 56% of the patients with levels less than the threshold were male (P = 0.15).
Demographic data in patients with CSF concentration less than or greater than or equal to the caffeine thresholda
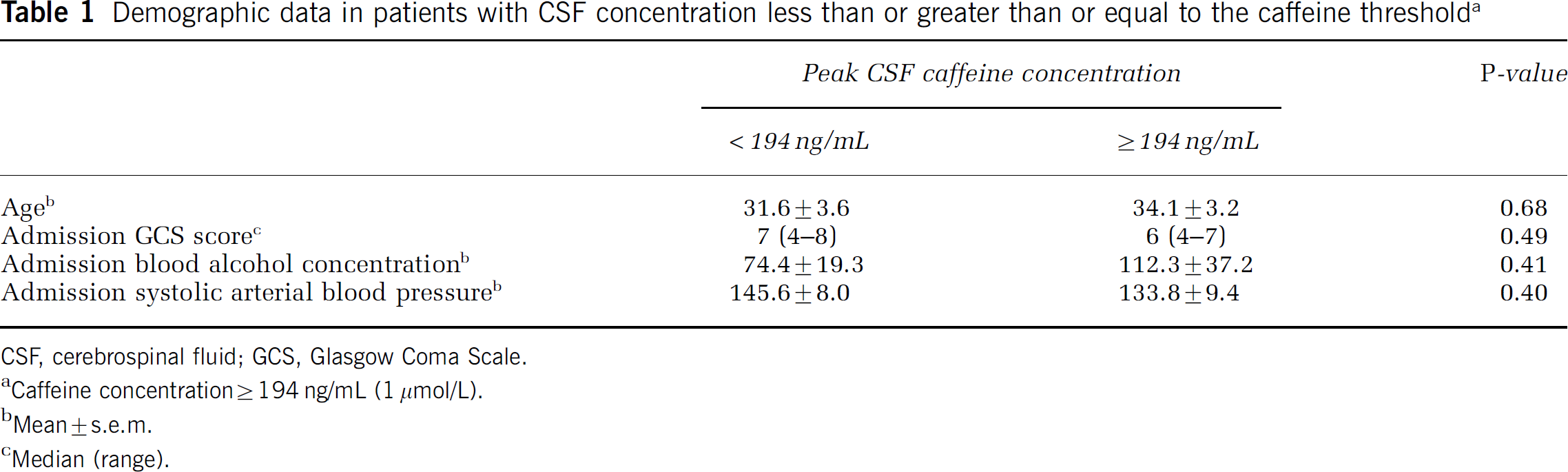
CSF, cerebrospinal fluid; GCS, Glasgow Coma Scale.
Caffeine concentrations ≥ 194 ng/mL (1 μmol/L).
Mean ± s.e.m.
Median (range).
Figure 2 shows the relationship between peak CSF caffeine concentration and 6-month GOS score. Favorable GOS score was seen more often in patients with CSF caffeine concentration greater than or equal to versus less than the threshold caffeine concentration of 1 μmol/L (55.6 versus 11.8%, P = 0.028, Fisher's exact test). Similarly, a multivariate analysis, adjusting for GCS score and admission blood alcohol level suggested an association between the presence of a CSF caffeine concentration greater or equal to the threshold and favorable 6-month GOS score (odds ratio (OR) = 141.204, P = 0.054; Table 2). In that analysis, admission GCS score also exhibited a trend toward association with outcome (OR = 8.885, P =0.074), whereas admission blood alcohol level did not (OR = 0.998, P = 0.78). Blood alcohol level was included in the multivariate analysis because of the important interaction suggested in previous studies in experimental models of stroke and TBI (see Introduction). Of the four patients in whom GOS score was missing, one patient had an admission blood alcohol level >100 mg/dL, two had a blood alcohol level ≥ 100 mg/dL, and one patient did not have an admission blood alcohol level assessed. Of note, the median GOS score was 3 in both the blood alcohol level groups and 3/10 (30.0%) had a favorable outcome with blood alcohol level >100 mg/dL while 4/14 (28.6%) had a favorable outcome with blood alcohol level ≤ 100 mg/dL. Two patients with poor outcome did not have blood alcohol level assessed.
Association of 6-month GOS with CSF caffeine concentration controlling for admission GCS score and admission blood alcohol concentration using a caffeine threshold a
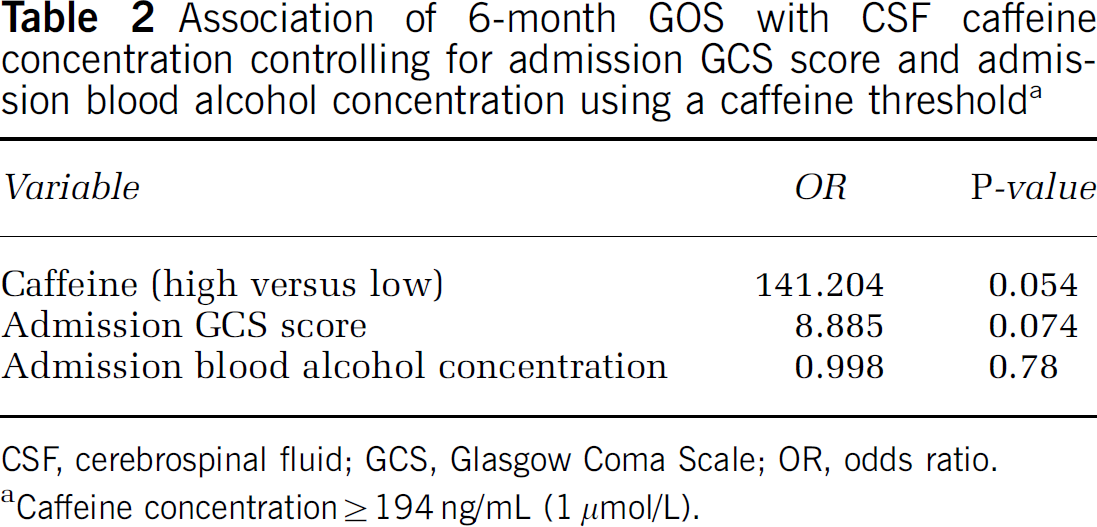
CSF, cerebrospinal fluid; GCS, Glasgow Coma Scale; OR, odds ratio.
aCaffeine concentration ≥ 194 ng/mL (1 μmol/L).
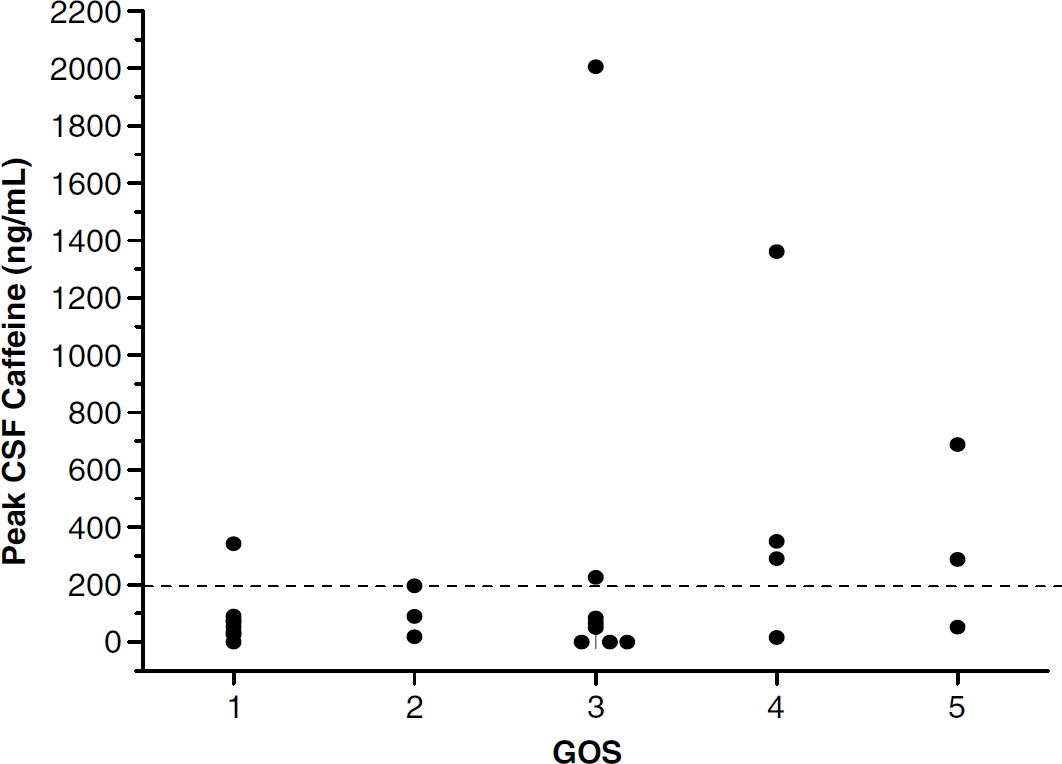
Peak cerebrospinal fluid (CSF) caffeine concentration plotted versus outcome (6-month Glasgow Outcome Scale score (GOS)) in patients with severe TBI. The dotted line represents the clinically significant caffeine threshold of 1 μmol/L (194 ng/mL). Favorable GOS was seen more often in patients with CSF caffeine concentration greater or equal to versus less than the threshold (55.6 versus 11.8%, P = 0.028).
In the absence of a known physiologically relevant threshold value for CSF theobromine, paraxanthine, and theophylline concentrations, each was analyzed as a continuous variable. There was no difference in peak CSF theobromine concentration in women versus men (159.5 ± 33.3 versus 155.4 ± 43.4ng/mL, P = 0.96). Similarly, there was no association between peak CSF theobromine concentration and either age or admission GCS score (r = −0.026, P = 0.89 and r = 0.021, P = 0.91, respectively). However, peak CSF theobromine concentration was significantly increased in patients with favorable versus unfavorable outcome, specifically, 308.0 ± 97.04 in the seven patients with favorable outcome versus 115.4 ± 30.4 in the 19 patients with unfavorable outcome (P = 0.018).
The initial CSF paraxanthine concentration represented the peak value in each case. There was no difference in peak CSF paraxanthine concentration in women versus men (216.0 ± 247.2 versus 150 ± 209.5 ng/mL, P = 0.47). Similarly, there was no association between peak CSF paraxanthine concentration and either age or admission GCS score (r = 0.11, P = 0.58 and r = 0.02, P = 0.91, respectively). Peak CSF paraxanthine concentration was 320.44 ± 115.49 in the seven patients with favorable outcome versus 126.52 ± 41.39 in the 19 patients with unfavorable outcome (P = 0.056). Univariate analysis also revealed a trend toward a significant association between peak CSF paraxanthine concentration and favorable outcome (OR = 1.41 for a 100U increase in paraxanthine concentration, P = 0.086).
Peak CSF concentrations of the caffeine metabolite theophylline were relatively low and did not differ significantly between the seven patients with favorable outcome versus the 19 with unfavorable outcome (29.9 ± 9.4 versus 16.4 ± 3.7, respectively, P = 0.12).
Discussion
This is, to our knowledge, the first study assessing either CSF caffeine concentration or the relationship between CSF caffeine concentration and outcome in severe human TBI. Although significant relationships between caffeine consumption and favorable outcome have been reported for both Parkinson's and Alzheimer's disease (Ross et al, 2000; Ascherio et al, 2001; Tan et al, 2003; Lindsay et al, 2002), in those reports, the investigators relied on dietary history to assess caffeine consumption. By having CSF samples available early after injury in our patients and directly quantifying the concentration of caffeine and its metabolites, our study in TBI is thus unique and provides important direct evidence of a relationship between increases in caffeine concentration and favorable outcome; this time in another CNS disorder, namely TBI.
After oral intake and absorption, caffeine rapidly crosses the blood—brain barrier in normal volunteers. At 60 mins after ingestion of 300 mg of caffeine, CSF concentrations were 2.9 ± 1.1 μg/mL (∼50% of plasma concentration) and were highly correlated with plasma caffeine concentration (Soto et al, 1994). We selected a clinically relevant CSF caffeine concentration of 1 μmol/L based on the work of Evans and Griffiths (1999), who reported effects of caffeine doses as low as 25 mg in patients who were chronically consuming caffeine.
Caffeine was detected in the initial sample in 24 of the 30 patients highlighting its common presence in the CSF of adult patients after a severe TBI. Caffeine was present in a higher percentage of patients than alcohol—a drug that has received much study in TBI. Peak CSF caffeine concentration in our study also underestimates the CSF caffeine levels at the time of injury, because the mean time between injury and initial CSF sampling was >10 h. Blood alcohol concentrations, however, were routinely obtained in the emergency room. Estimates of CSF caffeine concentration at the time of injury could be extrapolated from Figure 1; our reported values represent a minimum level in the critical early postinjury period.
We observed a significant association between the presence of a CSF caffeine concentration ≥1 μmol/L (194 ng/mL) and favorable 6-month outcome. In addition, multivariate analysis that included two other key parameters, admission GCS score and admission blood alcohol concentration suggested a powerful favorable effect of caffeine (OR= 141.2). Differences in gender, age, or baseline severity of injury are also unlikely to explain the differences in outcome between the groups with and without clinically relevant CSF caffeine concentration. Similarly, differences in admission systemic arterial blood pressure do not explain the observed effect of caffeine on neurologic outcome.
Caffeine metabolites were also increased in CSF in most patients in this study. Theobromine is a metabolite of caffeine and its levels were relatively high, although it can also be derived from other dietary sources such as chocolate. Theobromine has similar (∼0.5 times) affinity as caffeine for adenosine receptors (Shi and Daly, 1999), supporting the parallel association between peak CSF theobromine concentration and favorable outcome that we noted. Peak CSF theobromine concentration was approximately threefold higher in patients with favorable versus unfavorable outcome. Paraxanthine was also detected at levels somewhat lower than those seen for caffeine. It also has similar affinity as caffeine for adenosine receptors (∼2-fold greater than caffeine). Peak CSF paraxanthine levels were also more than 2-fold greater in patients with favorable versus unfavorable outcome; a difference that was nearly significant (P = 0.056). These results also mirror our findings with caffeine. Theobromine and paraxanthine deserve additional study in experimental and clinical TBI, and other CNS disorders. Finally, although theophylline has higher affinity than adenosine for adenosine receptors (Shi and Daly, 1999), it is a minor caffeine metabolite and CSF levels were low.
How might caffeine, a nonselective weak adenosine receptor antagonist be beneficial after severe TBI? This finding might be construed as paradoxical given the fact that adenosine is considered to be an endogenous neuroprotectant (Ribeiro, 2005). It has been suggested that long-term exposure to caffeine mediates its beneficial effects by upregulating adenosine receptor number or function, particularly A1AR. Numerous studies in mice or rats have shown that long-term caffeine administration increases the number of A1AR (Boulenger et al, 1983; Fredholm, 1982). Also, long-term caffeine exposure increases sensitivity of A1AR possibly via enhanced coupling of adenylate cyclase to Gi protein (Ramkumar et al, 1988). This could magnify the benefit anticipated from the massive adenosine surge that routinely follows TBI (Clark et al, 1997), and is also seen in response to secondary insults in humans (Bell et al, 2001). The putative beneficial actions of adenosine at A1AR are suggested to include anti-excitotoxic and anti-inflammatory effects (Ribeiro, 2005; Tsutsui et al, 2004). Acute deleterious consequences of the adenosine receptor antagonist effects of caffeine may not manifest because of the extremely high levels of adenosine observed after TBI (Clark et al, 1997; Bell et al, 2001) and the relatively weak antagonist properties of caffeine and its metabolites at adenosine receptors (Jacobson et al, 1996).
An alternative possibility, however, is that acute effects of caffeine at A2AR mediate this beneficial effect. Chen et al (1999) reported that A2AR ko mice are protected in experimental stroke. Yu et al (2004), using bone marrow chimeras of A2AR wt and ko mice, further reported that this neuroprotection is mediated by the inhibition of A2AR-mediated proinflammatory effects of leukocytes in brain or microcirculation. Other mechanisms of protection by caffeine may be operating (Azam et al, 2003; Dassesse et al, 1999). Our study is not able to discriminate between the effect of long-term or acute caffeine exposure in conferring favorable outcome.
Caffeine and ethanol produce synergistic benefit in experimental TBI and ischemia (Strong et al, 2000; Dash et al, 2004). However, despite a favorable effect of caffeine, and a considerable number of patients with detectable admission blood alcohol concentrations, we did not observe a benefit from alcohol in our study. We recognize that admission blood alcohol concentration was numerically higher in the patients with high caffeine levels, although the difference was not statistically significant. Our exploratory multivariate analysis suggested an independent effect of caffeine, although further study with a larger sample is needed.
Despite assessing 97 CSF samples, we recognize that our study was limited to 30 patients. This limited our ability to assess more than three parameters on multivariate analysis. Indeed, our multivariate analysis is exploratory rather than definitive, and a substantially larger study is essential to examine adequately the role of the many other potential confounders. Nevertheless, it was consistent with the significant association between caffeine and outcome seen using Fisher's exact test. Lack of a difference in admission systolic blood pressure between groups argues against important extracerebral effects and only one patient had an admission systolic blood pressure < 90 mm Hg. We also recognize that the caffeine threshold was exceeded in only nine patients in the study, suggesting the need to further examine our findings in a larger cohort. Although the retrospective design has limitations, the availability of CSF samples, expertise of our pharmacology group with the caffeine assay, and the long-standing use of a standardized clinical treatment protocol in our center support the value of this provocative exploratory study.
What are the potential implications of these findings? First, the presence of caffeine and its metabolites is extremely common in human TBI and that relevant concentrations were seen in nearly one-third of our patients even when sampled as late as 10 h after injury. Second, if the association between caffeine and favorable outcome is confirmed in a larger study, it would suggest implications on prognostication and might suggest new therapeutic possibilities. For example, A2AR antagonists are promising therapies for Parkinson's disease (Schapira et al, 2006) and might merit additional preclinical testing in TBI if the benefit of caffeine is mediated by acute effects. Alternatively, strategies to enhance A1AR or A2AR activation or resultant signal transduction pathways might be pursued if it is long-term effects of caffeine exposure that mediate this protection (Phillis and Goshgarian, 2001). There are two other issues that should be considered in the light of our findings. First, in clinical care, patients extensively exposed to caffeine routinely have it abruptly stopped—and the implications of that after TBI have not been studied even in an animal model. Second, experimental models of TBI do not consider the fact that many patients with severe TBI have appreciable caffeine levels, which could influence clinical translation of therapies (Doppenberg et al, 2004).
We conclude that caffeine and its major metabolites are commonly increased in CSF in adult patients with severe TBI. In an exploratory analysis, the presence of caffeine levels ≥1 μmol/L (194 ng/mL) in CSF is associated with favorable 6-month outcome. Increased levels of caffeine metabolites in CSF were also associated with favorable outcome. Our findings mirror the association between caffeine consumption and neuroprotection in other CNS disorders such as Parkinson's disease. Future studies are needed in a larger sample to confirm this stimulative finding.