
Abstract
Adenosine, acting at A1 receptors, exhibits anticonvulsant effects in experimental epilepsy—and inhibits progression to status epilepticus (SE). Seizures after traumatic brain injury (TBI) may contribute to pathophysiology. Thus, we hypothesized that endogenous adenosine, acting via A1 receptors, mediates antiepileptic benefit after experimental TBI. We subjected A1-receptor knockout (ko) mice, heterozygotes, and wild-type (wt) littermates (n = 115) to controlled cortical impact (CCI). We used four outcome protocols in male mice: (1) observation for seizures, SE, and mortality in the initial 2 h, (2) assessment of seizure score (electroencephalogram (EEG)) in the initial 2 h, (3) assessment of mortality at 24 h across injury levels, and (4) serial assessment of arterial blood pressure, heart rate, blood gases, and hematocrit. Lastly, to assess the influence of gender on this observation, we observed female mice for seizures, SE, and mortality in the initial 2 h. Seizure activity was noted in 83% of male ko mice in the initial 2 h, but was seen in no heterozygotes and only 33% of wt (P < 0.05). Seizures in wt were brief (1 to 2 secs). In contrast, SE involving lethal sustained (>1 h) tonic clonic activity was uniquely seen in ko mice after CCI (50% incidence in males), (P < 0.05). Seizure score was twofold higher in ko mice after CCI versus either heterozygote or wt (P < 0.05). An injury-intensity dose–response for 24 h mortality was seen in ko mice (P < 0.05). Physiologic parameters were similar between genotypes. Seizures were seen in 100% of female ko mice after CCI versus 14% of heterozygotes and 25% wt (P < 0.05) and SE was restricted to the ko mice (83% incidence). Our data suggest a critical endogenous anticonvulsant action of adenosine at A1 receptors early after experimental TBI.
Introduction
Study of early posttraumatic seizures has been limited, despite the fact that excitotoxicity is believed to play a key role early after experimental traumatic brain injury (TBI) (Faden et al, 1989; McIntosh, 1993). Posttraumatic seizures in the acute phase after experimental TBI was reported by Dixon et al (1987) in the midline fluid percussion injury (FPI) model. However, only a 13% incidence of posttraumatic seizures was observed when rats received respiratory support to prevent apnea/hypoxemia, and it was concluded that convulsions were not a prominent feature of midline FPI. Nilsson et al (1994) also reported seizure activity, assessed by electroencephalogram (EEG), early (in the initial 2 mins) after parietal compression TBI in rats, which was correlated temporally with increases in brain interstitial levels of aspartate, taurine, glutamate, and glycine. Behavioral seizure activity was not assessed. Most investigation has focused on delayed posttraumatic seizures after TBI. Feeney et al (1981) suggested the development of delayed epilepsy as a result of cortical damage after cortical weight-drop injury. Golarai et al (2001) observed electrographic seizure activity at 3 and 15 weeks after experimental TBI produced by weight-drop to the parietal cortex in rats. A key role for seizure discharge for dentate gyrus (a region selectively vulnerable to cell death after TBI) (Clark et al, 2001; Lowenstein et al, 1992) was also shown. D'Ambrosio et al (2004) reported a 92% incidence of epilepsy at 10 weeks after FPI in rats, with an important role for the cortex as the focus of origin for the epileptiform activity. Coulter et al (1996) studied kindling of seizure activity in hippocampal–entorhinal cortex slices at 1 week after FPI and reported that a prior incident of TBI in rats produces an enhanced predilection of the limbic system to trigger epileptic seizure discharges in the dentate gyrus. In addition, Zanier et al (2003) showed increased CA3 sensitivity to kainic acid-induced seizure activity after lateral FP. Despite these findings, early posttraumatic seizures or status epilepticus (SE) have not been routinely reported in the most commonly used contemporary models of TBI in rodents, including controlled cortical impact (CCI), lateral FPI, or impact acceleration (McIntosh et al, 1989; Dixon et al, 1991; Marmarou et al, 1994).
Brain interstitial levels of adenosine increase dramatically early after experimental TBI, including studies in both the CCI model and lateral FPI (Headrick et al, 1994; Bell et al, 1998). The A1 adenosine receptor (A1AR) has the highest affinity for adenosine and is suggested to mediate antiexcitotoxic effects, along with a number of other potentially beneficial actions. Notable among the effects of adenosine at the A1AR are potent antiepileptic effects (Knutsen and Murray, 1997). It has been suggested that adenosine is a critical endogenous metabolite that causes seizure arrest in a number of paradigms (Franklin et al, 1989; Knutsen and Murray, 1997; Avsar and Empson, 2004). Presynaptic inhibition of excitatory amino-acid release from hippocampus via adenosine effects at A1AR (Emptage et al, 2001) and postsynaptic hyperpolarization of neurons via A1AR-mediated G-protein-coupled inwardly rectifying potassium channel currents (Takigawa and Alzheimer, 2002; Avsar and Empson, 2004). Consistent with this notion, administration of A1AR agonists (Gouder et al, 2003; Franklin et al, 1989) or inhibition of adenosine breakdown by administration of inhibitors of adenosine deaminase (Southam et al, 2002) exhibits protection against the development of seizures in a variety of models. Conversely, reduction in endogenous adenosine levels by the induction of adenosine kinase in gliotic regions exacerbates kainic acid-mediated seizures (Gouder et al, 2004). A loss of A1 receptors is seen in brain tissue taken from patients suffering from temporal lobe epilepsy (Glass et al, 1996), supporting a clinical role for the antiepileptic actions of A1AR activation. Thus, an endogenous antiexcitotoxic and anticonvulsant role for adenosine could be pivotal in experimental and clinical TBI, reducing early posttraumatic excitotoxicity and preventing early posttraumatic seizures.
Adenosine A1AR activation has been implicated as an endogenous neuroprotectant in a variety of CNS insults (Fredholm et al, 2005); however, studies with AR agonists or antagonists have yielded conflicting results, likely related to effects of these agents at multiple ARs (Jiang et al, 1997; von Lubitz, 1999; Chen et al, 1999; Phillis and Goshgarian, 2001; Higashi et al, 2002; Olsson et al, 2004; Fredholm et al, 2005). In experimental TBI in rats and mice, both nonselective and selective A1AR agonists have been shown to exhibit beneficial effects on functional and histopathological outcome (Headrick et al, 1994; Varma et al, 2002). However, because of the hemodynamic side effects of systemically administered adenosine-related therapies, beneficial effects in TBI have only been reported with local administration of these agents injected either intracerebroventricullarly or directly into brain parenchyma. In addition, variable distribution and concentration of adenosine agonists has been recently reported after parenchymal injection in rat brain, making this approach less conclusive than desired (Kochanek et al, 2005). Recent development of the adenosine A1AR knockout (ko) mouse (Sun et al, 2001; Fredholm et al, 2005) provided an excellent opportunity for our laboratory to examine the role of this receptor in the acute phase after experimental TBI in a well-characterized CCI model in mice (Smith et al, 1995; Whalen et al, 2000; Sinz et al, 1999).
We hypothesized that endogenous adenosine, acting via A1AR, mediates endogenous antiexcitotoxic and antiepileptic effects after CCI. To test this hypothesis, we subjected A1AR ko mice, heterozygotes, and wild-type (wt) littermates to CCI and studied the incidence of behavioral and electrographic seizure activity, and mortality. We also examined the role of both injury severity and gender on the incidence of seizures and mortality after CCI.
Materials and methods
All experiments were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh School of Medicine.
Generation and Characterization of the A1 Adenosine Receptor Knockout Mouse
A1 receptor ko mice were generated as described by Sun et al (2001) and breeding pairs were obtained from that laboratory. Heterozygote male and female mice bred at our facility generated homozygous wt, ko, and heterozygotes (F4-7). Genotypes were confirmed by polymerase chain reaction (PCR) on tail deoxyribonucleic acid (DNA). Briefly, DNA was isolated from mouse tail by a high salt procedure. Polymerase chain reaction was performed at cycling temperatures of 94°C, 3 mins denaturing, 30 cycles of 94°C, 58°C, 72°C each 1 min annealing, and 72°C, 8 mins extension. Primer pairs for genotyping were as described previously (Sun et al, 2001). Products were separated on a 1% agarose gel with 0.5% ethidium bromide and visualized under ultraviolet (UV) light.
Reverse Transcriptase-Polymerase Chain Reaction
RNA was isolated from mouse brain tissue using a TRIzol reagent (Invitrogen, Eugene, OR, USA) and transcribed using a Superscript First-Strand Synthesis Kit (Invitrogen). Polymerase chain reaction was performed via the conditions described above. Products were separated on a 1% agarose gel with 0.5% ethidium bromide and visualized under UV light. Primer pairs for RT-PCR for A1AR were forward 5'-CAGAGCTCCATCCTGGCTCT-3’ and reverse 5'-CGCTGAGTCACCACTGTCTTG-3'.
A1 Adenosine Receptor Binding
Mice were killed by decapitation and their brains immediately frozen in liquid nitrogen. Twenty micrometer coronal sections were cut, mounted on slides, and were preincubated with Tris-HCl containing 2 U/mL of adenosine deaminase at room temperature for 30 mins to remove endogenase adenosine. Next, slides were incubated with 6 nmol/L [3H]DPCPX for 90 mins at 37°C. Slides were washed 2 × 5 mins in ice-cold TE buffer, followed by three dips in ice-cold distilled water and dried under a stream of air. Sections were exposed for 8 weeks to Hyperfilm (Amersham, NJ, USA). Autoradiograms were analyzed with a MCID System (Imaging Research, St Catherines, Canada).
CCI Model
Mice (12 to 13 weeks of age) were subjected to our CCI model at various injury levels. The mouse CCI protocol was performed as described previously (Smith et al, 1995) with minor modifications (Sinz et al, 1999; Whalen et al, 2000). Mice were anesthetized with 2% isoflurane in N2O/O2 (2:1) by nose cone and placed in a stereotaxic frame. Brain temperature was monitored by a probe inserted through a burr hole in the left frontal cortex and body temperature was monitored by a rectal probe. With brain temperature maintained at 37.0 ± 0.5°C, a 5 mm craniotomy was made over the left parietotemporal cortex with a dental drill and the bone flap was removed. CCI was performed as previously described (Sinz et al, 1999) with a 3-mm flat-tip impounder at a velocity of 6.0 ± 0.2 m/sec and a depth of 1.2 mm. The bone flap was replaced and sealed with dental cement and the scalp was sutured closed. Anesthesia was discontinued, and mice were placed in an oxygen hood for 30 mins, then returned to their cages. The study groups included five outcome protocols.
Two Hours Observation Protocol
Male mice (n = 6/genotype) were subjected to the standard mouse CCI model (velocity 6 m/sec, deformation 1.2 mm). After injury, mice were continuously observed by a technician for incidence of seizures, status epilepticus, and mortality rate—during the initial 2 h interval after injury. Clinical seizures were confirmed based on the work of Golarai et al (2001). Status epilepticus was defined as continuous tonic–clonic seizure activity involving all four extremities lasting greater than 5 mins (Bassin et al, 2002). All mice were either observed until death from SE or were reanesthetized with 4% isoflurane in oxygen and killed by transcardial perfusion with 50 mL of ice-cold saline followed by 50 mL of 4% paraformaldehyde at 2 h after injury.
Two Hours Electroencephalogram Protocol
Male mice (n = 6/genotype) were subjected to the standard CCI protocol described above. Scalp needle electrodes (1 electrode per hemisphere) were inserted. Mice of each genotype were maintained on isoflurane anesthesia (1.5% titrated downward to 1% if necessary to maintain adequate respiratory effort) by nosecone. Brain temperature was continuously monitored and maintained at 37.0 ± 0.5°C. Electroencephalogram activity was recorded 15 mins before CCI to establish a baseline. Electrodes were then removed and mice were subjected to CCI (velocity 6 m/sec, deformation 1.2 mm). Scalp electrodes were then reinserted and EEG continuously recorded for 120 min after trauma. At the end of the monitoring period, mice were killed by decapitation. A neurologist with special expertise in epileptology (PC), masked to treatment, scored the EEGs in 60 mins epochs based on the following scale: 0 = no seizures; 1 = rare spike; 2 ≤ 1 every 2 s; 3 ≤ 1 every second; and 4 ≥ 1 every second. The peak seizure score in either epoch was determined for each mouse.
Twenty-Four Hours Outcome Protocol
Male mice (total n = 44 across the three genotypes) were subjected to CCI using one of two injury levels (either velocity of 4 m/sec, deformation 0.8 mm, or velocity 4 m/sec, deformation 1.2 mm). Both of these injury levels are lower than the standard mouse CCI model that is routinely used in our laboratory and that was used for the 2 h studies (see above). Beginning immediately after injury, mice were continuously observed by a technician for 2 h for incidence of seizures and/or SE, as described above. Subsequently, all surviving mice were returned to their cages and mortality rate was assessed over a 24 h period. Surviving mice were reanesthetized with 4% isoflurane in oxygen and euthanized at 24 h after injury.
Two Hours Physiology Protocol
Male mice (n = 4/genotype) were subjected to the standard mouse CCI model as described above (6 m/sec, 1.2 mm deformation), with the following modifications. Before CCI, a catheter was placed in the femoral artery of each mouse while under anesthesia and arterial pH and blood gases were monitored 15 mins before CCI and at 15 and 60 mins after CCI. EKG was also continuously monitored. Hematocrit (Hct) was measured at 60 mins after CCI. Mean arterial pressure (MAP) and heart rate were monitored throughout the study. Mice were killed by decapitation after the data were collected.
Two Hours Observation Protocol in Female Mice
The initial study design included only male mice, related to the high preponderance of clinical TBI in males. However, once the unique finding of SE was noted in male mice, female littermates were sequentially used in a 2 h observation protocol. Female mice (total n = 17 across genotypes) were subjected to the standard mouse CCI model (velocity 6 m/sec, deformation 1.2 mm). After injury, mice were continuously observed by a technician for 2 h for incidence of seizures and/or SE (as described above), and mortality rate. The remainder of the methods was identical to those described for the 2 h observation protocol in male mice.
Histopathologic Examination
Formalin-fixed brains from male mice in the 2 h observation protocols (described above) were divided into seven coronal slices, and these were processed for embedding in paraffin. Two 5 μm sections were prepared from each paraffin block and stained with hematoxylin and eosin (H&E) and with Fluro-Jade B, a fluorescent stain that optimizes detection of neuron degeneration (Schmued and Hopkins, 2000). Because of the relatively early time point of killing (after CCI), neuropathologic assessment was limited to qualitative rather than quantitative assessment. Substantial mortality in the initial 24 h after CCI in A1AR ko mice precluded histopathological examination at more delayed time points.
Statistical Analysis
Incidence of seizures, SE, and mortality was compared between groups using χ2 test. Seizure scores and physiologic parameters were compared by one-way analysis of variance (ANOVA) and the Dunnett test for multiple comparisons. All values are either percent incidence or mean ± standard deviation (s.d.).
Results
Generation and Characterization of the A1 Adenosine Receptor Knockout Mouse
Genomic tail DNA from wt, heterozygote, and ko mice was used to identify the genotype in each mouse. Wild-type alleles yielded a 444 bp product, while the mutant allele yielded 457 bp product (Figure 1A) (Sun et al, 2001). To confirm the genotype of the A1AR ko mouse, in selected mice (n = 3), RT-PCR of mouse brain messenger ribonucleic acid (mRNA) was performed and yielded the anticipated 92 bp product for A1AR in wt and heterozygote mice (Figure 1B). A1 adenosine receptor ko mice lacked this product. In addition, A1AR binding, as assessed in selected uninjured mice (n = 3) using the tritium-labeled highly selective A1AR antagonist DPCPX (Johansson et al, 1997), confirmed the anticipated relationship with the genotype (Figure 1C).
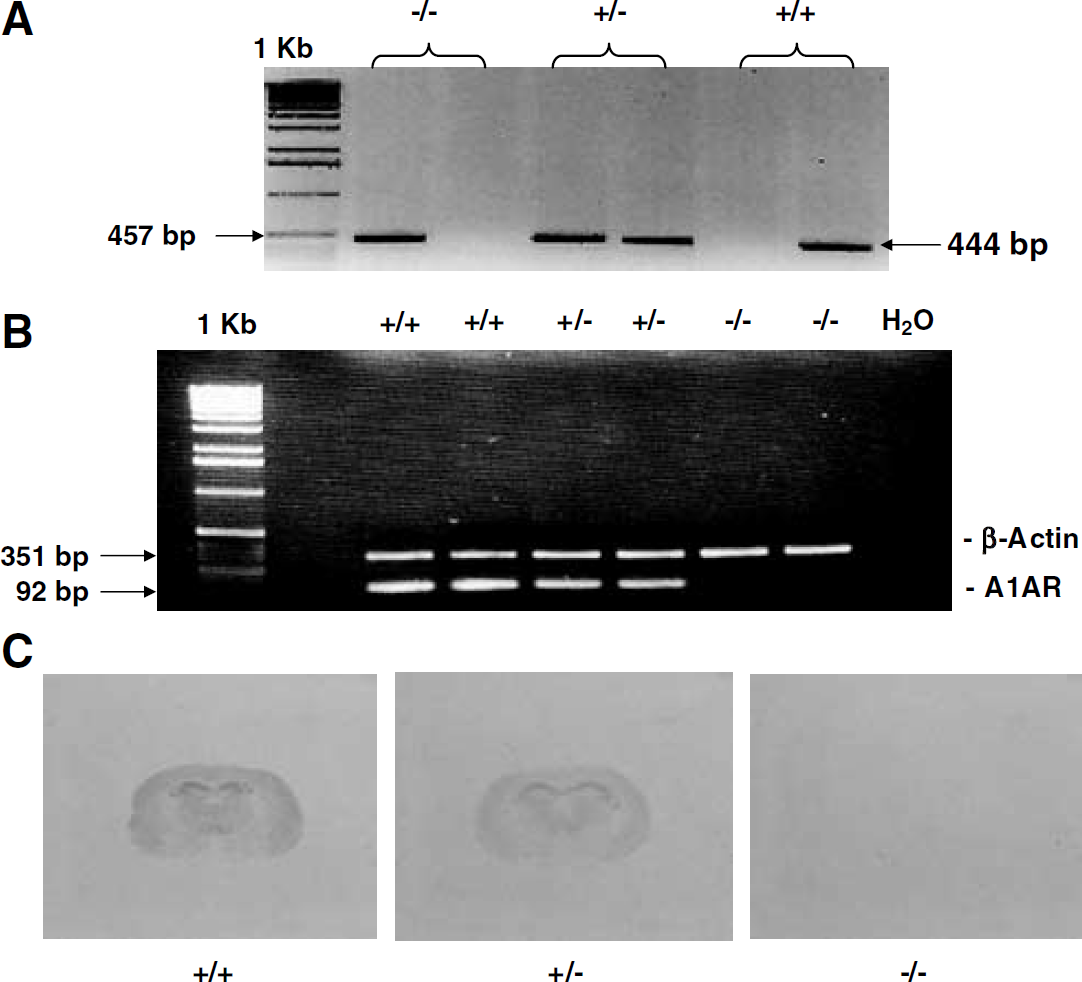
(
Body weights were 28.1 ± 4.0, 29.8 ± 3.6, and 28.6 ± 3.6 in male wt, heterozygote, and A1AR ko littermates, respectively, P = 0.19, including all mice across all protocols. Body weights were 22.8 ± 1.3, 21.6 ± 2.6, and 23.5 ± 1.9 g in female wt, heterozygote, and A1AR ko littermates, respectively, P = 0.30 for all female mice in this report.
Two Hours Observation Protocol
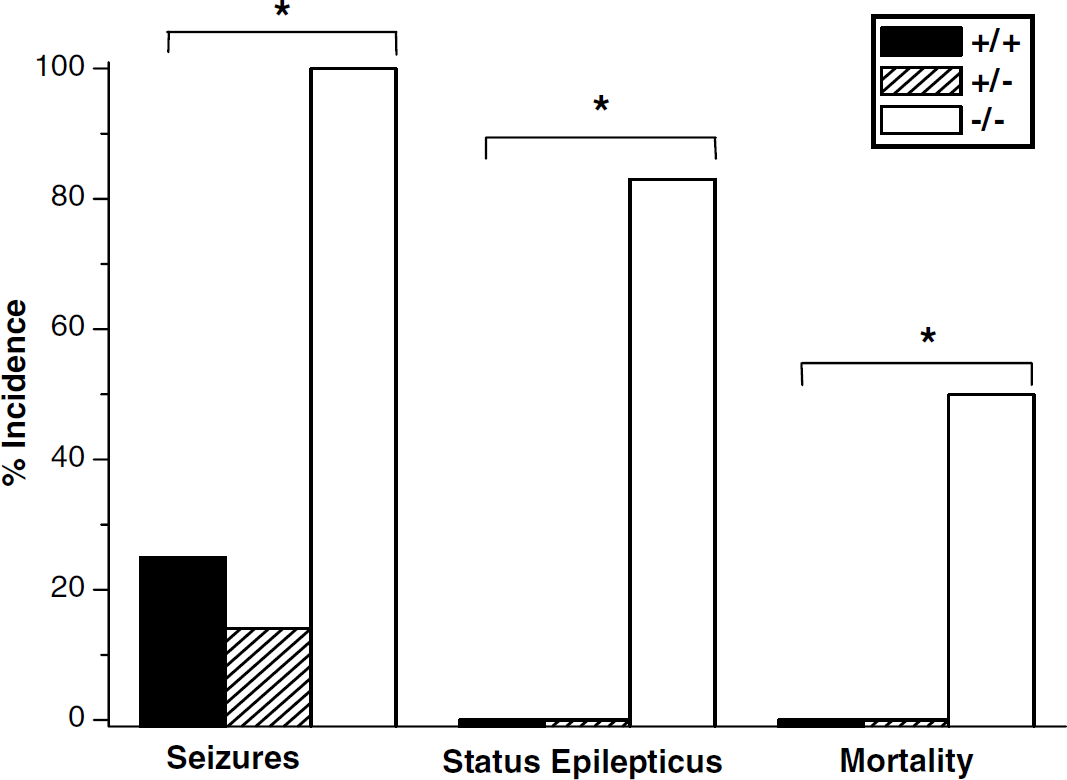
Seizure activity was observed in 83% of male A1AR ko mice after our standard CCI, as studied in the 2 h observation protocol, but was not seen in heterozygotes and seen in only 33% wt littermates (Figure 2, P < 0.05 by χ2 test). Seizure activity did not generally begin until approximately 1 h after recovery from anesthesia. The seizures that were observed in 2/6 wt mice were very brief (lasting 1 to 2 secs), were only detectable on careful observation, were comprised of localized twitching, and resolved spontaneously. In contrast, SE was dramatic when it occurred and produced sustained generalized tonic–clonic seizure activity. It was uniquely seen in A1AR ko mice after CCI. Status epilepticus occurred in 50% of male A1AR ko mice after severe CCI and was lethal, lasting at least 60 min, in all of the male mice in which it occurred (Figure 2, P < 0.05 by χ2 test) and progressively led to an unresponsive, moribund condition with severely compromised respiratory effort and death. Status epilepticus accounted for all of the observed mortality. Mortality is rarely seen in wt C57/BL6 mice in this model at any of the injury levels studied.
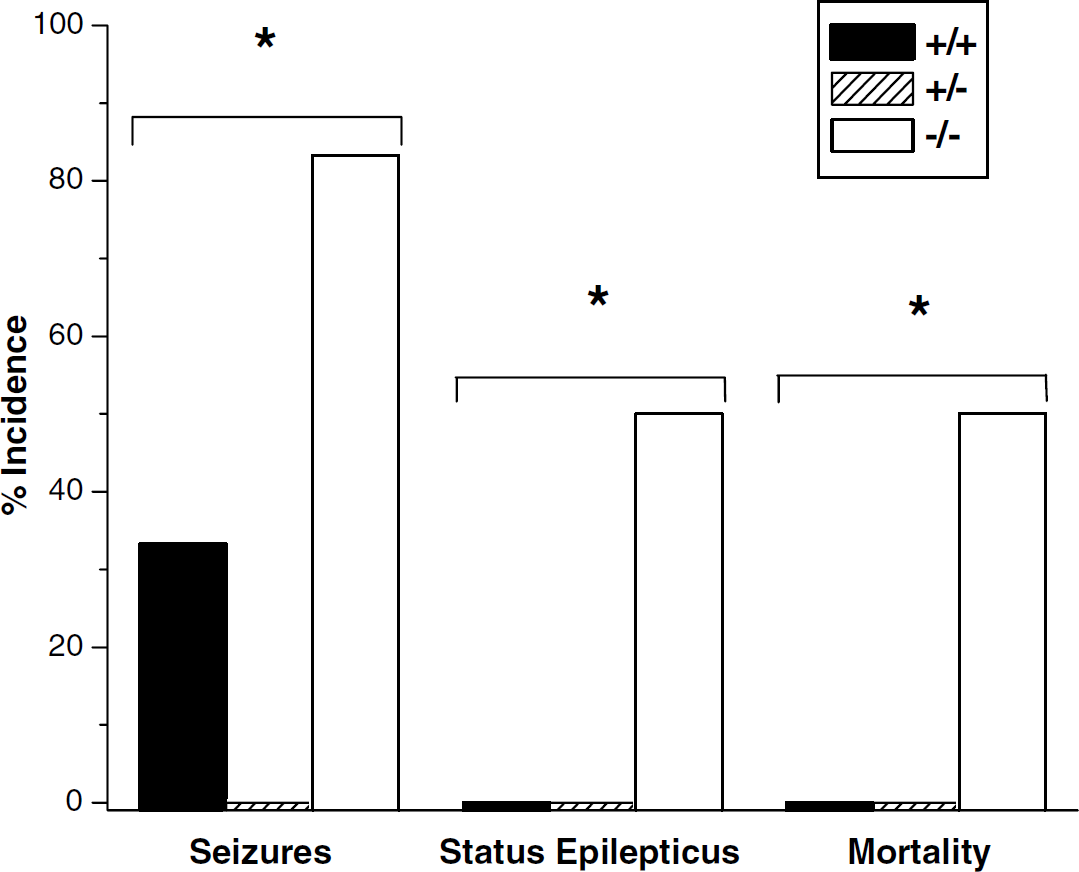
Incidence (percent) of seizures and status epilepticus (SE), and mortality rate after controlled cortical impact in male adenosine A1 receptor +/+, /-, and -/- mice (n = 6/group). A1 receptor knockout mice showed an increased incidence of seizures (*P < 0.05 across genotypes by χ2 test) and SE (P < 0.05 across genotypes by χ2 test), and mortality rate (*P < 0.05 across genotypes by χ2 test). Status epilepticus accompanied 2 h mortality in all cases, and was observed exclusively in A1 receptor knockout (ko) mice.
Two Hours Electroencephalogram Protocol
Peak seizure score was over twofold higher in homozygote A1AR ko mice after CCI than either heterozygote or wt littermates (Figure 3, P < 0.05 for wt versus ko by ANOVA and the Dunnett test).
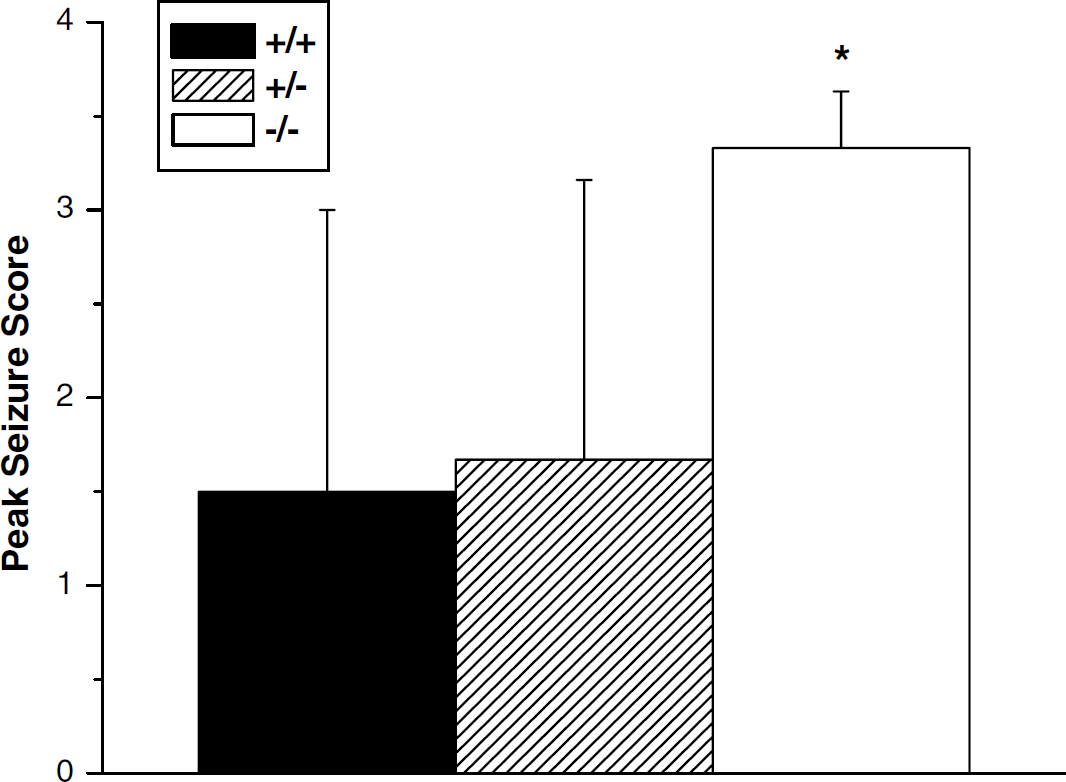
Male adenosine A1 receptor knockout mice showed an increase in peak seizure score versus either wild-type or heterozygote littermates as assessed during the initial 2 h after injury (*P < 0.05 by one-way analysis of variance and the Dunnett test for multiple comparisons).
Twenty-Four Hours Outcome Protocol
The effect of injury severity on mortality rate after CCI in A1AR +/+, +/-, and -/- mice (total n = 44 across the three genotypes and two injury levels) is shown in Figure 4. The overall χ2 for mortality rate across injury levels and genotypes was P < 0.05. There were no deaths in any of the 22 wt or heterozygote littermates that were injured. An injury dose–response for mortality rate was observed in A1AR ko mice, with 25% (4/16) and 83% (5/6) 24 h mortality at 4 m/sec, 0.8 mm and 4 m/sec, 1.2 mm injury levels, respectively (†P < 0.05 by the Fisher exact test).
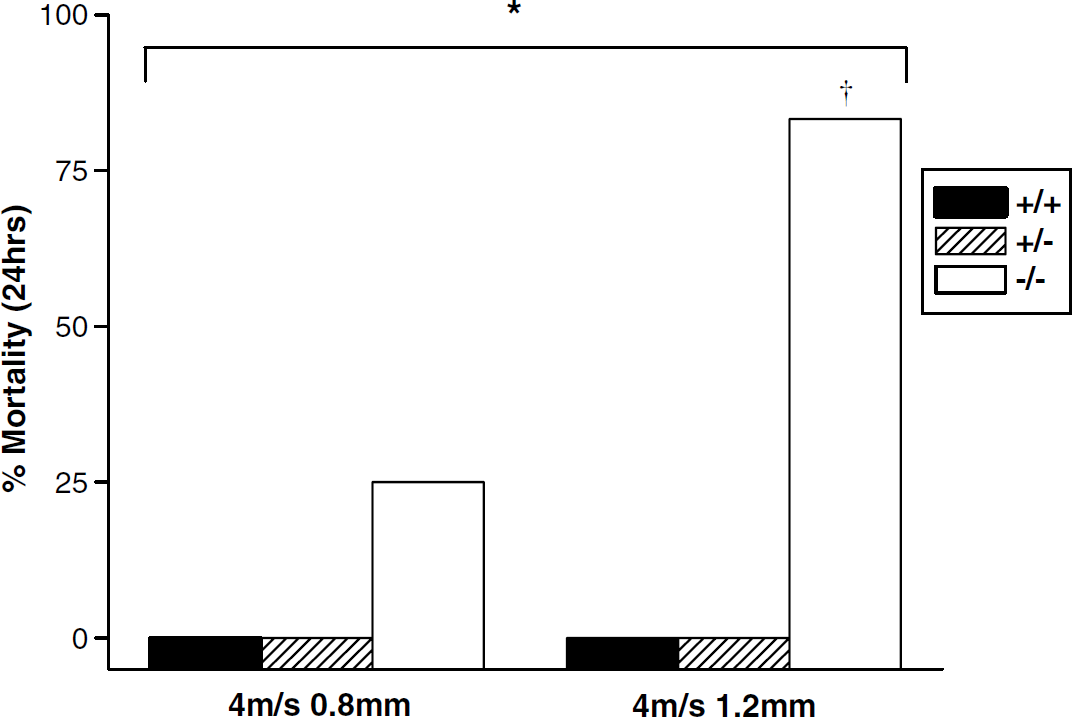
Effect of injury severity on mortality rate after controlled cortical impact in male A1 receptor +/+, +/-, and -/- mice (total n = 44 across the three genotypes and two injury levels). The overall χ2 for mortality rate across injury levels and genotypes was *P < 0.05. A1 receptor knockout mice showed an increase in mortality rate at 1.2 mm depth versus 0.8 mm depth (†P < 0.05 by the Fisher exact test).
Physiology Protocol
Body weight was lower in wt versus either heterozygote or ko (both P < 0.05 versus wt). No baseline differences were seen between genotypes in heart rate, arterial pH, PaCO2, PaO2, or MAP (Table 1). These same physiologic parameters did not differ between genotypes at either 15 or 60 mins after CCI, with the exception of a modestly lower pH in wt versus either heterozygote or ko (both P < 0.05 versus wt) at 15 mins after injury and a lower Hct at 60 mins after injury in wt versus either heterozygote or ko (both P < 0.05 versus wt)—although all mice had Hets in the physiologically normal range.
Physiological data across genotypes in male mice
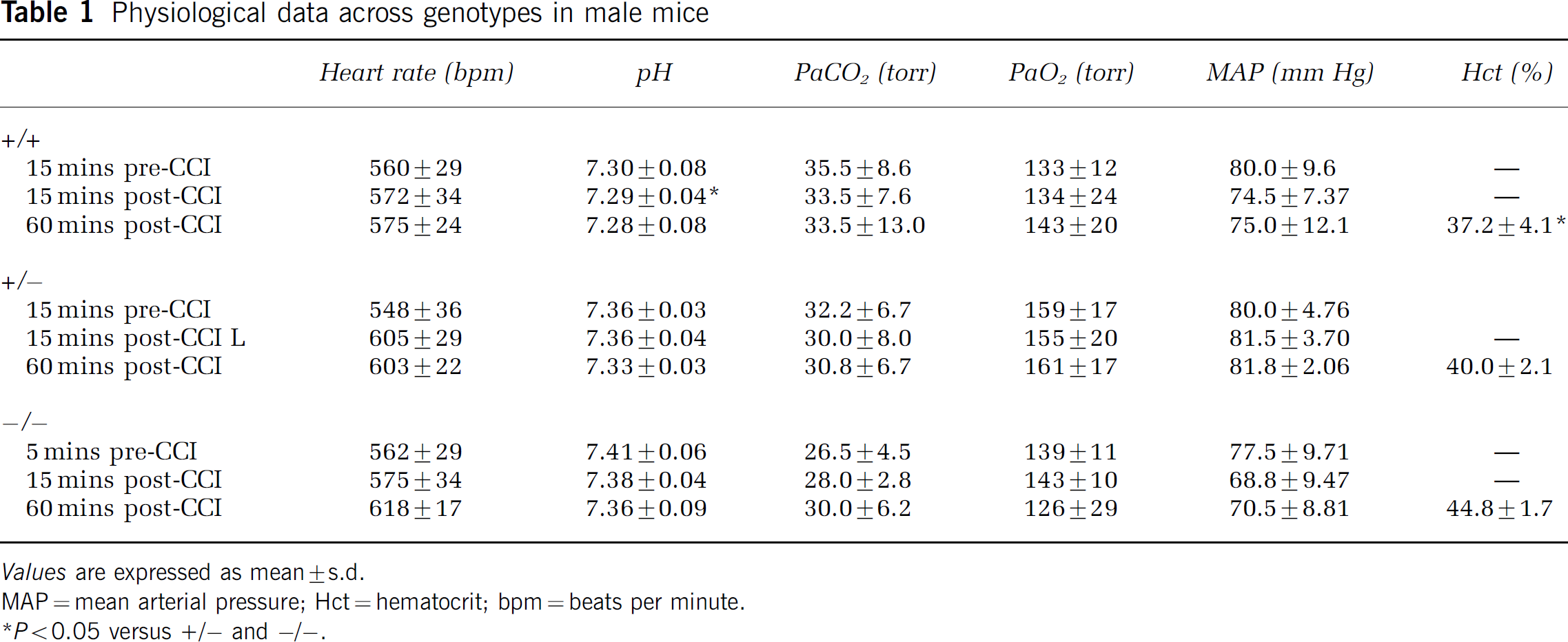
Values are expressed as mean ± s.d.
MAP = mean arterial pressure; Hct = hematocrit; bpm = beats per minute.
P < 0.05 versus +/- and -/-.
Two Hours Observation Protocol in Female Mice
Seizure activity was observed in 100% of female A1AR ko mice after our standard CCI, versus 14% of heterozygotes and 25% wt littermates (Figure 5, P < 0.05 by χ2 test). Similar to that observed in males, seizures were not generally seen until ~1 h after injury. Status epilepticus was similarly dramatic as seen in males and produced sustained generalized tonic–clonic seizure activity. As in males, it was uniquely seen in female A1AR ko mice versus either heterozygotes or wt littermates after CCI. Status epilepticus occurred in 83% of female mice after severe CCI and was lethal in 50% (Figure 5, P < 0.05 by χ2 test). Two female mice had SE that was not lethal at 2 h after injury; however, both of these mice were moribund at 2 h after CCI. As in male mice, the seizures that were observed in heterozygote and wt female mice were very brief (lasting 1 to 2 secs), were only detectable on careful observation, were comprised of localized twitching, and resolved spontaneously. Status epilepticus accounted for all of the observed mortality.
Incidence (percent) of seizures, status epilepticus (SE), and mortality in female adenosine A1 receptor +/+, +/-, and -/- mice (n = 17 across genotypes). As seen in males after controlled cortical impact, A1 receptor knockout mice show an increased incidence in seizures (*P < 0.05 across genotypes by χ2 test) and SE (*P < 0.05 across genotypes by χ2 test), and mortality rate (*P < 0.05 across genotypes by χ2 test).
Neuropathology Examination
At 2 h after CCI, evidence of neuron degeneration in either the H&E- or Fluoro-Jade B-stained sections was limited to the side of the contusion, and this degeneration was most prominent within the dorsal hippocampus immediately underlying the contusions. Here, darkly stained to eosinophilic neurons (with H&E stain) and Fluoro-Jade B-postitive cells were found within most of the pyramidal neuron sectors, as well as within the granule neuron layer of the dentate gyrus (Figures 6A to F). The patterns of injury were similar both within the contusion and hippocampus as assessed in male mice across genotypes at 2 h after CCI. The contusions involved the left frontal, parietal, and visual cortex in all mice. However, at this early time point, Fluoro-Jade B staining was only minimal to not present within the contusions per se.
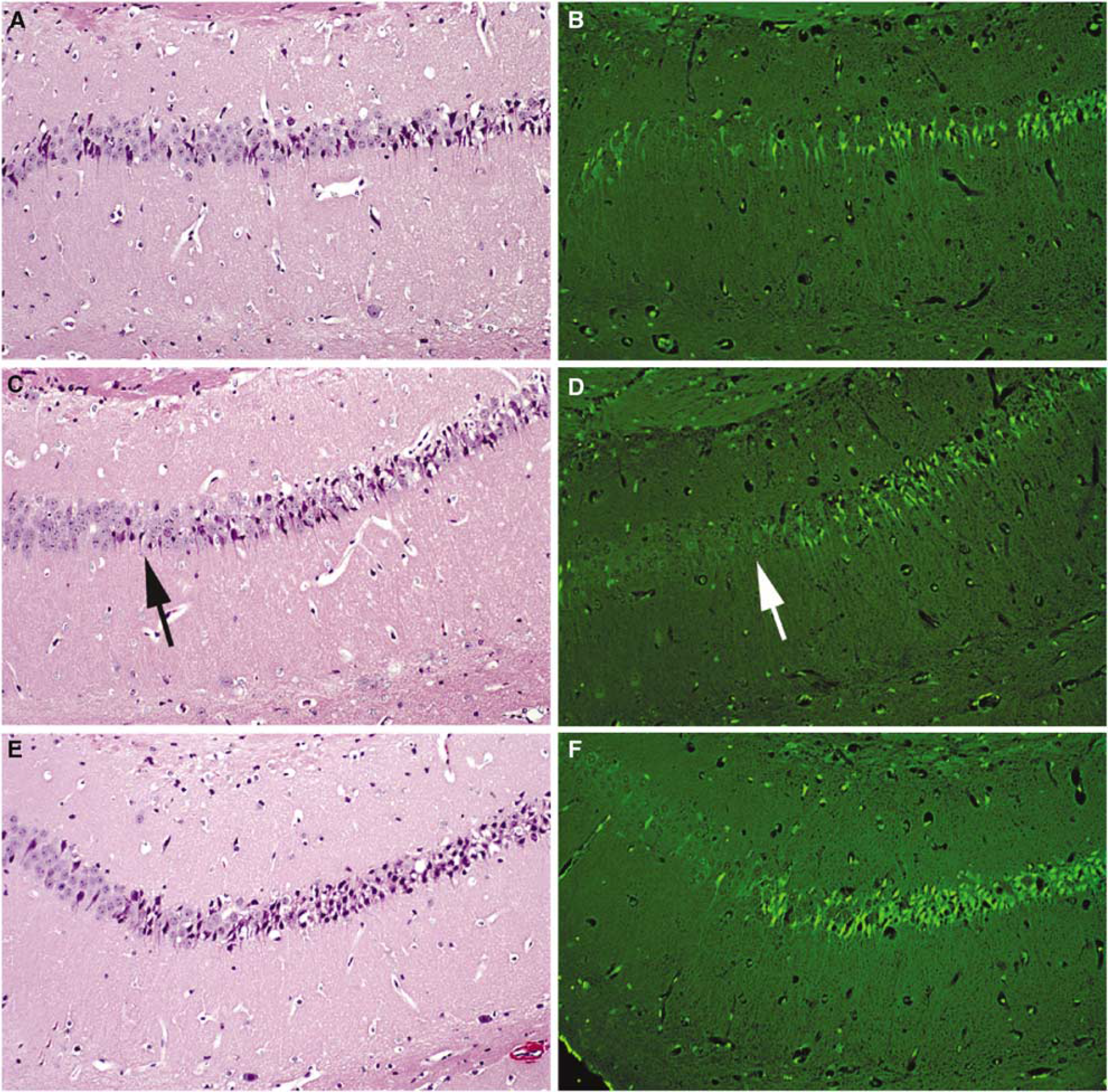
(
Discussion
A1 Adenosine receptor Activation as an Endogenous Anticonvulsant in TBI
Our data support a powerful antiepileptic effect of adenosine via A1AR early after experimental TBI. A rich literature over the past 20 years strongly supports an important role for adenosine—acting at A1AR as an endogenous anticonvulsant across experimental models of epilepsy (Dragunow et al, 1985; Murray et al, 1985; Barraco et al, 1986; von Lubitz et al, 1994; Pourgholami et al, 1997a, b ; Harrison et al, 2003; Gouder et al, 2004). Although both adenosine acting at the A1AR and γ-aminobutyric acid (GABA) share key roles in maintaining inhibitory tone, it has been suggested that adenosine is the endogenous agent responsible for seizure arrest in a number of paradigms (Knutsen and Murray, 1997; Avsar and Empson, 2004). Young and Dragunow (1994) similarly suggested that adenosine plays a critical role in both terminating brief seizures in brain and in blocking the progression to SE, reporting that adenosine antagonists were highly effective in producing SE (versus vehicle) in an electrical stimulation model in rats. A1 adenosine receptor activation is coupled to activation of Gi proteins with reduction in intracellular cyclic adenosine monophosphate (cAMP) and resultant presynaptic inhibition of excitatory amino-acid release and postsynaptic hyperpolarization of neurons. Consistent with this mechanistic paradigm, they also reported that administration of pertussis toxin, which inactivates Gi proteins, leads to SE. A1 adenosine receptor also has important neuromodulatory effects via modulation of phospholipase C of excitatory metabotropic receptors, which could have implications on the observed potentiation of excitation and seizures in the A1AR ko (Fredholm et al, 2001). Highlighting the purported domination of adenosine at the A1AR in producing seizure arrest, administration of GABA-A, GABA-B, or adenosine A2a receptor antagonists did not prevent the development of SE after recurrent electrical stimulation. Consistent with these studies in experimental models of SE, the critical importance of adenosine actions on A1AR in preventing the development of fulminate seizure activity early after TBI was shown in our study by the unique observation of lethal SE in A1AR ko mice. This is strengthened by the fact that prior reports show marked increases in brain interstitial levels of adenosine early after experimental TBI (Bell et al, 1998, Headrick et al, 1994)—increases that closely parallel the temporal increases in excitatory amino acids such as glutamate (Palmer et al, 1993) after experimental TBI. Reports of SE after experimental TBI in rodents with an intact adenosine A1AR axis are lacking across the wealth of experimental models—further supporting an important endogenous anticonvulsant role for A1AR in TBI (Smith et al, 1995; Marmarou et al, 1994; McIntosh et al, 1989; Whalen et al, 2000). Finally, that A1AR ko mice fail to exhibit spontaneous epileptic activity in the absence of TBI is consistent with the findings of Chesi and Stone (1997) who suggested that a substantial number of A1AR are inactive under physiologic conditions because of the absence of a basal purinergic tone. The A1AR antagonist DPCPX indeed does not induce seizures when administered in vivo.
In contrast to the dramatic development of SE in A1AR ko mice after CCI, heterozygote mice were completely protected against SE—which was not seen in any of the 23 heterozygotes (across injury levels and gender) that were injured in this report. A1 adenosine receptor number has been shown to be directly proportional to genotype (Fredholm et al, 2005). This suggests the need for a high degree of inhibition of the A1AR in the development of SE after experimental TBI. Consistent with this finding, we previously reported that administration of DPCPX directly into the injured hippocampus immediately after CCI does not lead to SE in our model (Varma et al, 2002). Although a key role for dentate and CA3 hippocampal regions in seizure kindling has been shown in experimental TBI (Golarai et al, 2001), we cannot, however, exclude cortical or other foci for the origin of the epileptic activity in our model.
The critical effect of A1AR in preventing the development of posttraumatic seizures and SE appears to be independent of gender—since a 50% mortality rate from SE, in the initial 2 h after injury, was seen in both male and female A1AR ko mice after the 6 m/sec 1.2 mm deformation insult. Similar rates for both SE (50% in male and 83% in female ko mice) and seizures (83% in male and 100% in female ko mice) were also observed. This finding contrasts the emerging trend of important gender-related differences for a variety of important mechanisms in the response to ischemic and TBI in a variety of experimental models (McCullough et al, 2005; Roof and Hall, 2000; Hoffman et al, 2003). It suggests that the anticonvulsant actions of adenosine at the A1AR are an important endogenous mechanism independent of gender differences.
A substantial direct effect of refractory SE on acute (2 h) mortality was seen in A1AR ko mice. Consistent with our prior studies in hundreds of commercially available C57BL/6 mice in this model at the standard injury level (6 m/sec, 1.2 mm deformation), in wt and heterozygote littermates, acute (2 h) or 24 h mortality is rare (Whalen et al, 2000; Sinz et al, 1999; Varma et al, 2002; Bay***ir et al, 2005). However, in the 24 h outcome protocol, mice were observed for only 2 h before being returned to their cages and we did not prove that seizures or SE during the subacute period lead to the increase in mortality rate from 50% to 83% in male A1AR ko mice after CCI. Use of 24 h video or EEG monitoring would be required. Mice of all genotypes in this study that were returned to their cages were ambulatory and appeared otherwise to have recovered from the injury.
A1 Adenosine Receptor and Neuroprotection After TBI
We did not see differences in patterns or degrees of neuronal degeneration after CCI between genotypes in our study; however, the histologic assessment was made very early (2 h) after injury and is a preliminary and descriptive approach in this regard. The focus of this report was on the role of the A1AR in the development of seizures and SE after experimental TBI. The high incidence of acute mortality from SE in the A1AR ko precluded our ability to assess neuropathologic alterations at a more appropriate—delayed—time after injury such as at 24 h, or 7 or 21 days.
Seizures have been shown to accelerate anoxia-induced neuronal death in rat hippocampus (Dzhala et al, 2000), but the brief duration of observation in our study may have limited the ability to detect either this mechanism of injury exacerbation or the loss of A1AR-mediated neuroprotection. However, recently, Olsson et al (2004) reported that A1AR deficiency failed to increase neuronal death in the hippocampus, cortex, or striatum after global cerebral ischemia in adult A1AR ko versus wt mice. In contrast, intraperitoneal administration of the A1AR antagonist 8-cyclopentyl theophylline before the ischemic insult exacerbated the neuronal damage in their model. It was suggested that compensatory mechanisms may have developed in the ko that afforded an alternative mechanism of neuroprotection. However, in our CCI model, intrahippocampal administration of the A1AR antagonist DPCPX after injury had no deleterious effect versus vehicle on hippocampal neuronal survival and administration of the A1AR agonist 2-chloro-N-cyclopentaladenosine afforded only modest protective effects on hippocampal neuronal survival assessed at 21 days after injury (Varma et al, 2002). A definitive study of long-term histopathology and functional outcome in the A1AR ko mouse (versus heterozygote and wt) is ongoing in our laboratory, but has been complicated by the obvious need for much lower injury levels that are below the threshold for the development of SE. Alternatively, long-term outcome could be studied in ko and wt mice treated with anticonvulsants to facilitate survival.
Extracerebral Physiology
There were no clinically significant cardiovascular differences between genotypes and blood gasses did not differ between groups with the exception of a modest difference in arterial pH at 15 mins after injury. However, pH values for all groups were within acceptable physiologic ranges for each parameter for mice (Sheng et al, 2000). The only surprising finding was that Hct, assessed at 60 mins after CCI, was significantly lower in the wt versus either the heterozygote of ko. However, again values for all groups were within ranges consistent with normal physiology and the number of mice in these subgroups was small. Although speculative, we cannot rule out the possibility that the higher Hct in A1AR ko mice is related to higher erythropoietin levels in the ko versus wt. Ohigashi et al (1993) have reported that A1 receptor activation is associated with a reduction of erythropoietin levels in a human hepatocellular carcinoma cell culture model. Related to this finding, we cannot rule of the possibility that differences in erythropoietin levels between genotypes could have contributed to either the development of posttraumatic seizures or neuroprotection in our model.
Potential Clinical Relevance
Excitotoxicity is believed to be important to the development of seizures early after clinical TBI (DeLorenzo et al, 2005). Marked increases in adenosine are observed in brain interstitial fluid and cerebrospinal fluid (CSF) after severe TBI in humans—a finding that may be important to the lack of reports of fatal SE early after clinical injury (Clark et al, 1997; Bell et al, 2001; Robertson et al, 2001). Recent preliminary studies have shown an association between the presence of the weak A1AR antagonist caffeine in CSF early after severe TBI in humans and favorable long-term outcome (Sachse et al, 2004). Chronic caffeine consumption is known to upregulate both A1AR number and their coupling to Gi proteins in brain—particularly hippocampus (Johansson et al, 1993, 1997; Georgiev et al, 1993). Factors that increase A1AR number or function could have important effects on posttraumatic excitotoxicity, seizures, and long-term outcome after TBI.
Conclusions
We report the unique development of acute and lethal SE after experimental TBI in A1AR ko mice—versus either wt or heterozygote littermates. The development of seizures, SE, and mortality in A1AR ko mice was dependent on the severity of injury but did not show a gender difference. Our findings suggest a critical endogenous anticonvulsant action of adenosine at A1AR in brain early after TBI.
Footnotes
Acknowledgements
We thank Carolyn Ferguson for expert technical assistance. We also thank Rosalyn Garman and Rita Gabel for the histologic preparations, and Marci Provins and Fran Mistrick for assistance with preparation of the manuscript.