
Abstract
The temperature threshold for protection by brief postischemic cooling was evaluated in a model of transient focal ischemia in the Spontaneously Hypertensive Rat, using an array of epidural probes to monitor regional brain temperatures. Rats were subjected to 90 mins tandem occlusion of the right middle cerebral artery (MCA) and common carotid artery. Systemic cooling to 32°C was initiated 5 mins before recirculation, with simultaneous brain cooling to temperatures ranging from 28°C to 32°C within the MCA territory by means of a temperature-controlled saline drip. Rewarming was initiated at 2 h recirculation and was complete within 30 mins. Tissue damage and edema volume showed clear temperature-dependent reductions when evaluated at 3 days survival, with no protection evident in the group at 32°C but progressive effects on both parameters after deeper cooling. A particularly striking effect was the essentially complete elimination of edema progression between 1 and 3 days. Temperature at distal sites within the MCA territory better predicted reductions in lesion volume, indicating that protection required effective cooling of the penumbral regions destined to be spared. These results show that even brief cooling can be highly protective when initiated at the time of recirculation after focal ischemia, but indicate a substantially lower temperature threshold for hypothermic protection than has been reported for other strains, occlusion methods, and cooling durations.
Introduction
Moderate intraischemic hypothermia is strikingly protective in all transient focal ischemia models, but results obtained with postischemic cooling are expectedly more modest (Ginsberg et al, 1992; Lyden et al, 2006; Miyazawa et al, 2003; Morikawa et al, 1992). Some data can be interpreted to suggest that cooling initiated before reperfusion and extending into the early recirculation period may be particularly effective (Xue et al, 1992). The temporal window for hypothermic protection shows significant strain and model dependence (Ren et al, 2004). Relatively few studies have examined temperature effects in the Spontaneously Hypertensive Rat (SHR), which is characterized by a limited capacity for collateral perfusion in brain (Coyle 1986). As in other strains, moderate intraischemic hypothermia is clearly protective during transient middle cerebral artery (MCA) occlusion in SHR (Aronowski et al, 1994; Barone et al, 1997; Ridenour et al, 1992). Cooling in the absence of reperfusion has been generally found not to be protective (Ren et al, 2004; Ridenour et al, 1992), in contrast to its beneficial effects in Wistar rats that maintain better collateral perfusion during occlusion (Baker et al, 1992; Kader et al, 1992; Ren et al, 2004). The one exception employed profound 24°C cooling of SHR during the initial hour of permanent occlusion of the MCA alone (Onesti et al, 1991), which would have allowed appreciable residual perfusion even in this strain. A more robust, well-characterized model of ischemia in the SHR involves MCA occlusion together with ipsilateral common carotid artery (CCA) occlusion to further limit collateral flow (Brint et al, 1988; Jacewicz et al, 1992; Kaplan et al, 1991). Using this model it was found that transient (2 h) postischemic hypothermia to 32°C failed to reduce infarct volume when initiated at the time of reperfusion after 1.5 h MCA occlusion, although further brain cooling to 30°C could provide significant protection (Ren et al, 2004). This suggests a notably lower temperature threshold for postischemic hypothermic protection than has been observed in studies involving other strains, different occlusion methods, and/or longer cooling durations (Colbourne et al, 2000; Huh et al, 2000; Kollmar et al, 2002; Zhang et al, 1993a), including those that specifically targeted cooling during initial reperfusion (Ding et al, 2004), in which temperature reductions to 32°C to 34°C were typically effective. This study examined in detail the relationship between lesion size and regional temperature change at sites within and outside the MCA territory in a model of brief postischemic cooling in the SHR.
Materials and methods
Animal Preparation
All animal procedures were approved by the Animal Care and Use Committee, University of Tennessee. Male SHR (220 to 300 g, Harlan Lab Animals Inc., Indianapolis, IN) were fasted overnight with free access to water. Anesthesia was induced with 5% halothane in air and maintained after endotracheal intubation using 1% halothane in 70% N2, 30% O2 under mechanical ventilation. The tail artery was cannulated to monitor blood pressure and sample blood for determinations of pH, PaCO2, PaO2, hematocrit, and glucose concentration. Body temperature was kept at 36.8°C to 37.2°C with a rectal thermistor connected to a heating pad. Fine bare-wire thermocouples (IT-1E, Physitemp Instruments, Inc., Clifton, NJ, USA) were inserted via 1 mm burr holes for subsequent measurement of epidural temperature, with all drilling performed under a saline drip to avoid thermal injury to the brain. Probe positions are illustrated in the inset of Figure 1. Three probes were placed in the right parietal cortex spanning the territory of the MCA to be occluded, at the following positions relative to bregma: 5 mm lateral, 1 mm posterior (T1); 4 mm lateral, 5 mm posterior (T2); and 3 mm lateral, 7 mm posterior (T3). An additional probe was positioned opposite T1 in contralateral cortex (T4). Lead wires were routed through a manifold of polyethylene tubing segments glued together to form an orderly bundle for transit through the skin. Burr holes were covered with bone wax and the scalp incision was closed. Each probe was connected to a thermocouple amplifier board (TC-12-B, Physitemp Instruments Inc., Clifton, NJ), from which temperature was digitized and recorded at 2 secs intervals via an interface board (instruNet, GW Instruments, Somerville, MA).
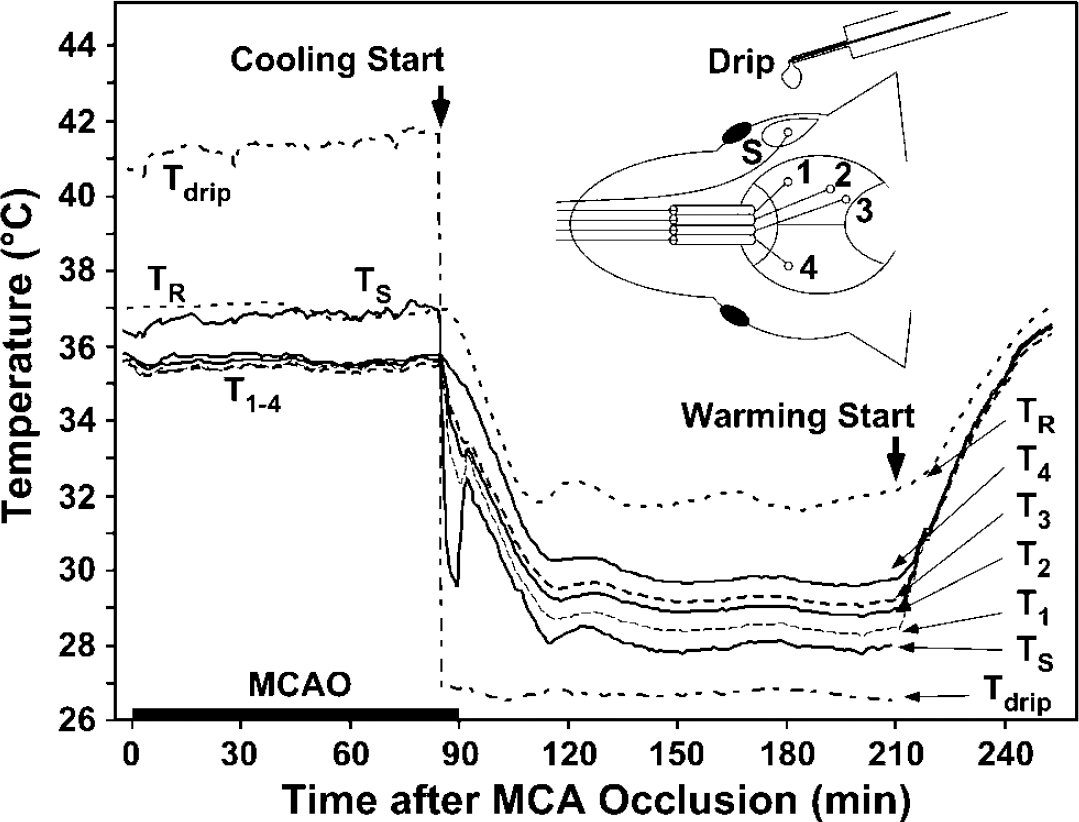
Regional epidural temperature recording during postischemic hypothermia. Animals were prepared as described in the text, with prior placement of temperature probes at positions indicated in the inset diagram. The time course of temperature change at each location is illustrated for a representative animal subjected to normothermic MCA occlusion followed by hypothermic recirculation at a rectal temperature (TR) of 32°C with superimposed brain cooling to 28°C. Initiation of systemic cooling, accompanied by an abrupt transition in superfusate temperature, resulted in a gradual decrease in TR with more rapid brain cooling in proportion to proximity of the recording site to the exposed, superfused surface. Release of occlusion resulted in transient warming followed by equilibrium hypothermia at all sites within 30 mins. Systemic rewarming resulted in simultaneous return to normothermic temperatures at all locations.
Fifty-three rats were successfully instrumented and carried through the study. In a preliminary experiment, three additional animals were fitted with a pair of temperature probes at position T3, one epidural, and one approximately 2 mm deep in adjacent cortex, to compare the patterns of temperature change monitored at these positions. A follow-up study involved another 14 animals in which temperatures were manipulated without regional epidural measurements to examine outcome at a longer postischemic interval.
Surgical preparation for reversible focal ischemia was performed essentially as described previously (Brint et al, 1988; Kaplan et al, 1991). The right CCA was isolated and fitted with a Teflon/Silastic occluding device, which was then tightened to produce occlusion. The right temporalis muscle was partially excised and a 2 mm burr hole was drilled 2 to 3 mm rostral to the point of fusion of the zygoma with the temporal bone to expose the MCA. An additional thermocouple was positioned approximately 2 mm dorsal to this burr hole to record surface temperature near the occlusion site, and drip temperature was adjusted to maintain 36.5°C ± 0.5°C in the pool of saline overlying the exposed artery. The dura was opened and the MCA was snared with a stainless-steel hook (80 μm diameter) at the point of the rhinal fissure using a micromanipulator. Focal ischemia was produced by raising the artery 0.5 to 1 mm above the cortical surface, allowing positive verification of flow disruption in the occluded MCA. Normothermic focal ischemia was maintained for 1.5 h with continuous temperature recording from the rectal probe as well as all brain sites, after which recirculation was achieved by lowering the MCA, removing the hook, and releasing the loop around the CCA.
Temperature Manipulations
In hypothermia studies, cooling was initiated 5 mins before recirculation by applying a wet ice pack to the body surface and switching the saline drip superfusing the MCA. Rectal temperature (TR) was lowered to 32.0°C ± 0.5°C in all hypothermic groups, with saline drip temperatures chosen to target surface temperatures (TS) of 28°C, 30°C, or 32°C, respectively (n = 7 per group evaluated at 24 h, n = 6 to 7 per group at 72 h). Normo thermic controls (n = 6 at both 24 and 72 h) were maintained at TR = 37°C, TS = 36.5°C. Animals were then subjected to 2 h of normothermic or hypothermic recirculation under continued anesthesia. At the end of this interval the saline drip was stopped, the incision at the occlusion site was closed, and cooled animals were rewarmed to 37°C using a heating pad and lamp. At the end of the recording period the skull surface was briefly reexposed to remove temperature probes, the scalp incision was closed, anesthesia was discontinued, and the animal was weaned from the respirator. Body temperature was monitored and maintained through 4 h reperfusion, by which time animals had regained mobility and temperature control. Rats were returned to their cages for survival intervals of 24 or 72 h.
Tissue Sampling and Analysis
Animals were briefly halothane anesthetized and decapitated, and brains were quickly removed and frozen in hexane precooled to −40°C. Coronal sections (20 μm) were cut on a cryostat at −25°C and collected at 1 mm intervals throughout the extent of the MCA territory. For routine histology slides were dried, fixed in 95% ethanol for 10 mins, and stained with hematoxylin/eosin.
For immunocytochemistry, freshly cut wet thaw-mounted sections were fixed in methanol at −20°C for 5 mins, briefly air-dried at room temperature, and stored desiccated at −80°C. Microtubule-associated protein 2 (MAP2) was detected by standard immunocytochemical procedures, essentially as described previously (Vass et al, 1988). Slides were washed in 0.9% NaCl buffered with 20 mmol/L sodium phosphate (pH 7.4; phosphate-buffered saline (PBS)), and blocked 2 h in normal goat serum supplemented with 4% bovine serum albumin. Sections were incubated for 2 h in mouse monoclonal anti-MAP2 antibody (M4403, Sigma Chemical Co., St Louis, MO) diluted 1:500 in PBS containing 10% goat serum and 0.4% bovine serum albumin. Sections were washed in PBS and endogenous peroxidase activity was blocked with 0.3% H2O2, 10% methanol in PBS, followed by further washes in PBS. Sections were then incubated 1 h with peroxidase-conjugated anti-mouse IgG (115-035-146, Jackson Immuno Research Laboratories, West Grove, PA) diluted 1:200 as above. After several washes in PBS, peroxidase activity was detected by incubation in 0.05% diaminobenzidine, 0.015% H2O2 in PBS. Slides were dehydrated through graded alcohols and xylene and coverslipped in Permount.
Volumetric assessments of ischemic injury were performed essentially as described previously (Jacewicz et al, 1990). Calibrated digital images of hematoxylin/eosin- and MAP2-stained sections were captured using the program NIH Image, regions of pallor were demarcated, and the lesion areas (in mm2) determined at 1 mm intervals from each brain were summed to obtain total lesion volume (mm3). In addition, total cortex volumes were determined for ipsilateral and contralateral hemispheres of hematoxylin/eosin-stained sections, from which edema volume as well as infarct volume corrected for edema were determined. Statistical comparisons of lesion size, as well as all physiologic parameters, employed analysis of variance and Scheffé's F-test, with P < 0.05 considered significant. Comparison of normothermic and hypothermic groups in the separate follow-up study involved an unpaired t-test.
Results
Hypothermia and Temperature Recording
Temperature probe placements and tracings from a representative animal are illustrated in Figure 1. Stable normothermic rectal temperature was well maintained during ischemia. Drip temperature varied slightly, but that of the exposed brain surface typically drifted < 0.5°C. Temperatures at the remote epidural recording sites were unchanged before experimental temperature manipulations and were consistently 1.5°C lower than TR. The switch in superfusate resulted in an essentially instantaneous decrease in temperature at the site of the exposed MCA, with progressively more gradual cooling at more distal recording sites. Because of the differential rate of brain and systemic cooling, recirculation was associated with a transient rewarming of the brain, detectable at the exposed surface, and proximal recording sites at which the initial cooling had been most rapid. Conversely, the brain continued to cool for approximately 10 mins after stable TR had been achieved. Equilibrium temperatures were reached at all sites within approximately 30 mins recirculation, with a stable gradient between TS and TR appropriate to thermocouple position. Homogeneous rewarming occurred when the saline drip was discontinued, the surgical site was closed, and systemic heating was initiated. Epidural probe placement was chosen to avoid potential complications in both experiment and interpretation that could arise from the acute trauma accompanying insertion of multiple probes into cortex. In preliminary studies it was established that epidural temperatures were well correlated with those measured in nearby cortex during the course of all manipulations involved in these studies, but were expectedly cooler by 0.5°C ± 0.2°C.
The impact of superfusate temperature on the time course of temperature change at each site is documented in Figure 2. TR was stable in the normothermic group, and identical systemic cooling and warming profiles were obtained in the three hypothermic groups. A temperature of approximately 40°C at the tip of the saline drip was required to maintain a temperature of 36.5°C at the exposed brain surface, and transitions to mean drip temperatures of 26.7°C, 29.6°C, and 34.6°C resulted in mean surface temperatures of 28.8°C, 30.0°C, and 32.4°C, respectively, in the cooling studies. A plot of the difference between TS and TR emphasizes the relative rapidity of local brain cooling, as well as the stability of equilibrium temperature decrements. Recordings at ipsilateral sites 1 to 3 showed similar temperature difference profiles, exhibiting a progressive attenuation of the differential cooling by the superfusate with distance from the site of surgical exposure, although even the contralateral cortex (site 4) was 1°C cooler in the nominally 28°C group versus the 32°C group.
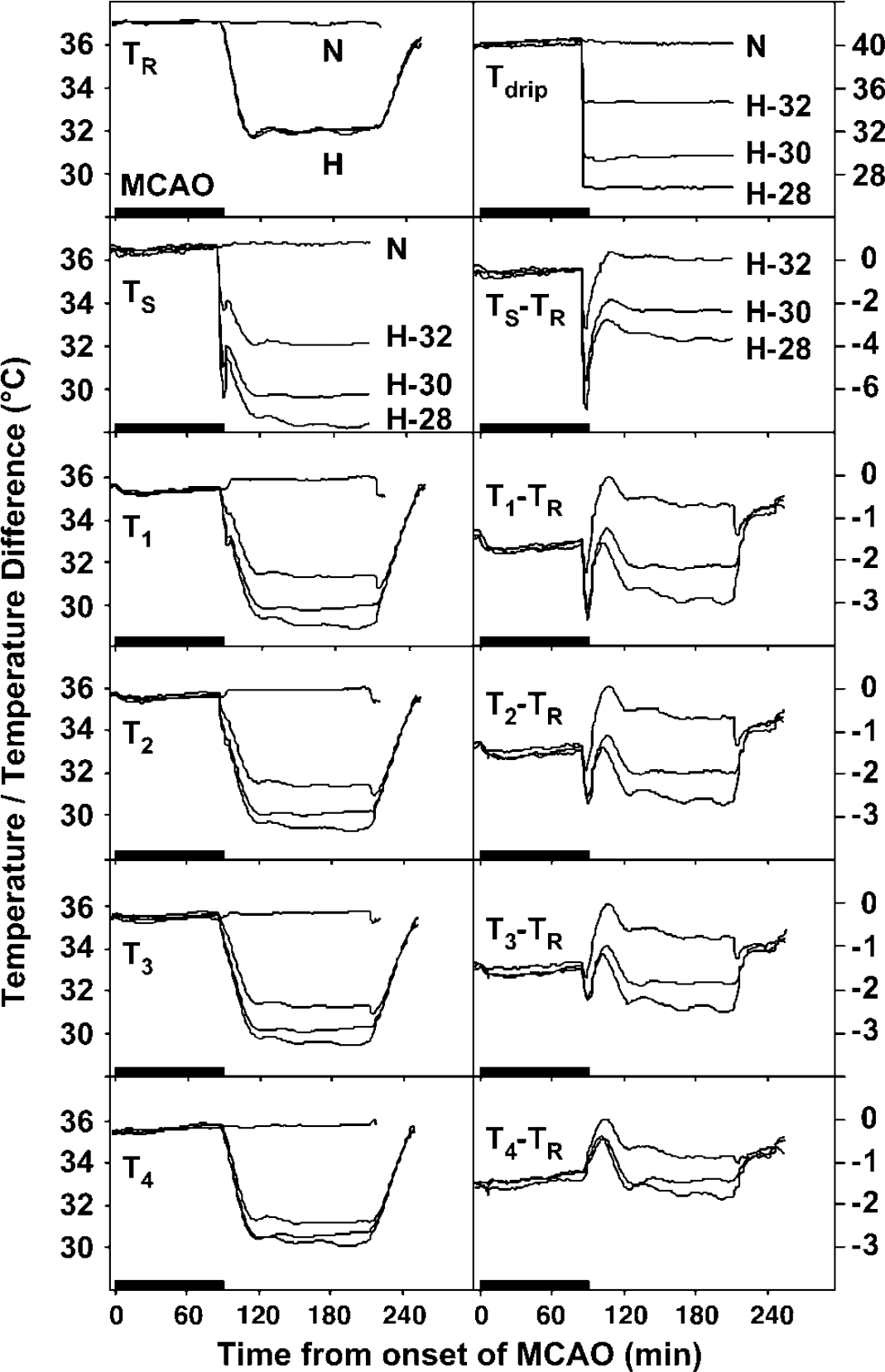
Regional temperature profiles during normothermic and hypothermic recirculation. Average temperatures recorded at each site are shown for normothermic animals (N) as well as for hypothermic animals that experienced systemic cooling to 32°C, with simultaneous transition to target brain surface temperatures of 28°C, 30°C, or 32°C (H-28, H-30, and H-32, respectively). Regional temperature difference profiles of the three hypothermic groups are also compared in relation to TR, illustrating the variation in effective cooling superimposed at different brain sites as a result of lowering superfusate temperature.
Physiological Parameters During Cooling and Rewarming
Physiological variables were evaluated in anesthetized, ventilated animals at the end of surgical preparation (just before occlusion), during ischemia (10 mins before recirculation), at 2 h recirculation (just before rewarming of the hypothermic groups), and finally at 4 h recirculation (by which time animals had fully recovered from anesthesia). There were no significant differences among the three hypothermic groups for any parameter at any time point (Table 1), and all four groups showed comparable physiologic stability during spontaneous respiration at 4 h recirculation. The only consistent difference from normothermic animals observed in the three cooled groups was a decrease in apparent pH (measured at the instrument temperature of 37°C) at the end of the cooling period. This was accompanied by slight increases in measured PCO2 that reached significance for the 30°C and 32°C groups. Final measurements in awake animals showed higher blood pressure, as expected in the absence of anesthesia, and elevated glucose reflecting the stress of brief restraint required for blood sampling. The hematocrit progressively increased during the course of the procedure but this change was identical in all groups.
Physiological variables during MCAO protocol in normothermic and hypothermic recirculation groups
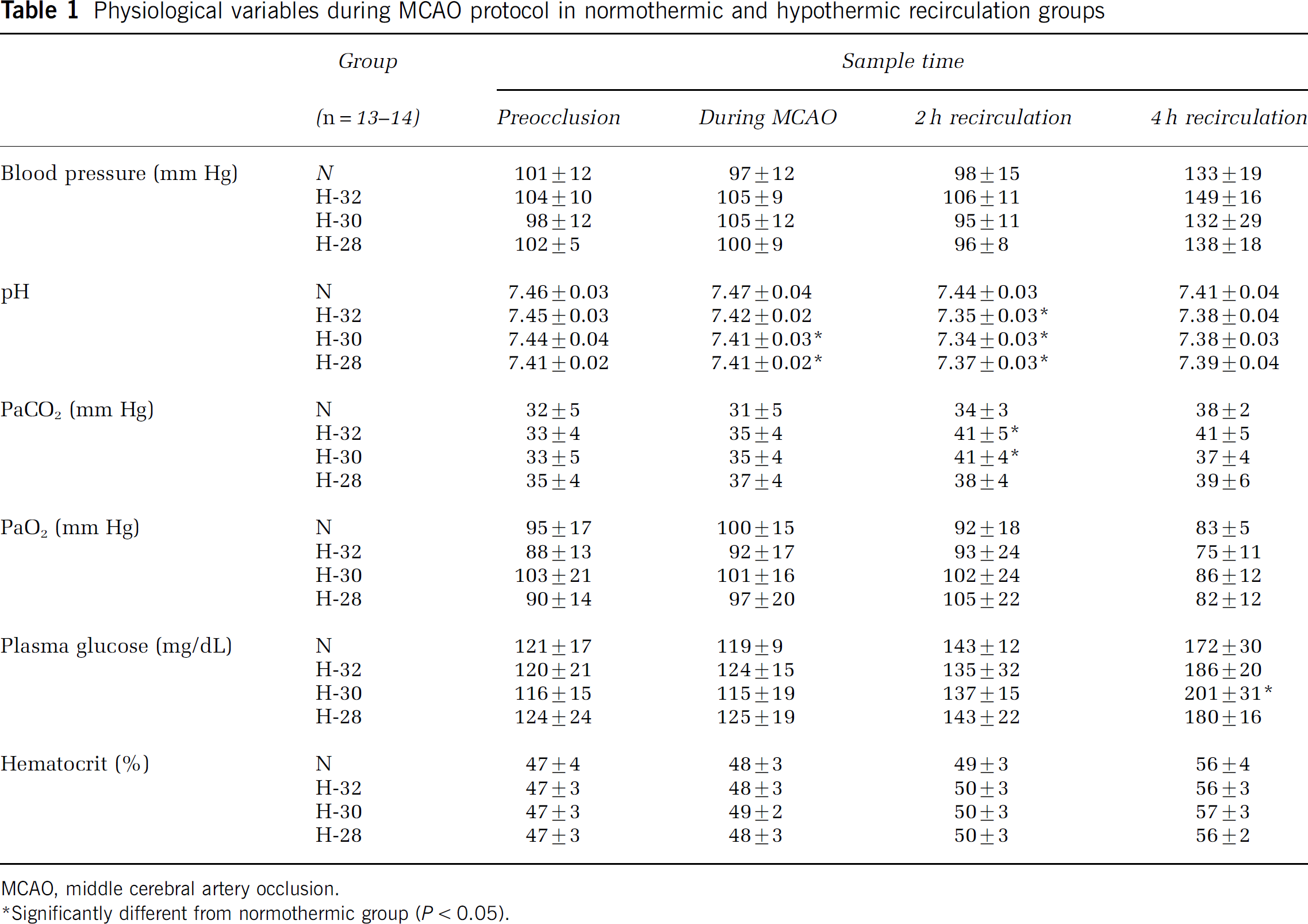
MCAO, middle cerebral artery occlusion.
Significantly different from normothermic group (P < 0.05).
Temperature Dependence of Hypothermic Protection
The impact of hypothermia on infarct evolution is illustrated by photomicrographs of hematoxylin/eosin- and MAP2-stained sections from representative animals in Figure 3A, and comparisons of lesion volumes measured using these two end points are shown in Figure 3B and Table 2. At 24 h recirculation, islands of preserved MAP2 signal remained within regions of pallor included in the lesion defined by histology (Figure 3A). These were more prominent in the 28°C and 30°C hypothermic groups, resulting in a detectable variation in lesion volume determined using these two methods (Figure 3B). In contrast, MAP2 and histologic lesion volumes were well correlated at 72 h, although the latter remained consistently larger. Quantitative values for lesion parameters in the several experimental groups are provided in Table 2. Systemic hypothermia of 32°C without additional brain cooling had no statistically significant effect on any of the measures of lesion volume at either 24 or 72 h. Aggressive brain cooling with 28°C superfusate resulted in striking protection at 72 h, as indicated by decreases in total lesion volume determined using both MAP2- and hematoxylin/eosin-stained sections, as well as edema and infarct volume corrected for brain swelling. Significant effects were also observed with the 30°C superfusate, although the reduction in edema did not reach statistical significance. Effects were not statistically significant at the 24 h time point, but there were clear trends toward reduced lesion volumes in both 28°C and 30°C groups. A subsequent follow-up study by a separate investigator, applying 28°C superfusate cooling but without regional temperature measurements, confirmed a persistent reduction in corrected infarct volume at 1 week survival from 117721 to 80 ± 21 mm3 in normothermic and hypothermic groups, respectively (n = 7 animals per group, P < 0.01).
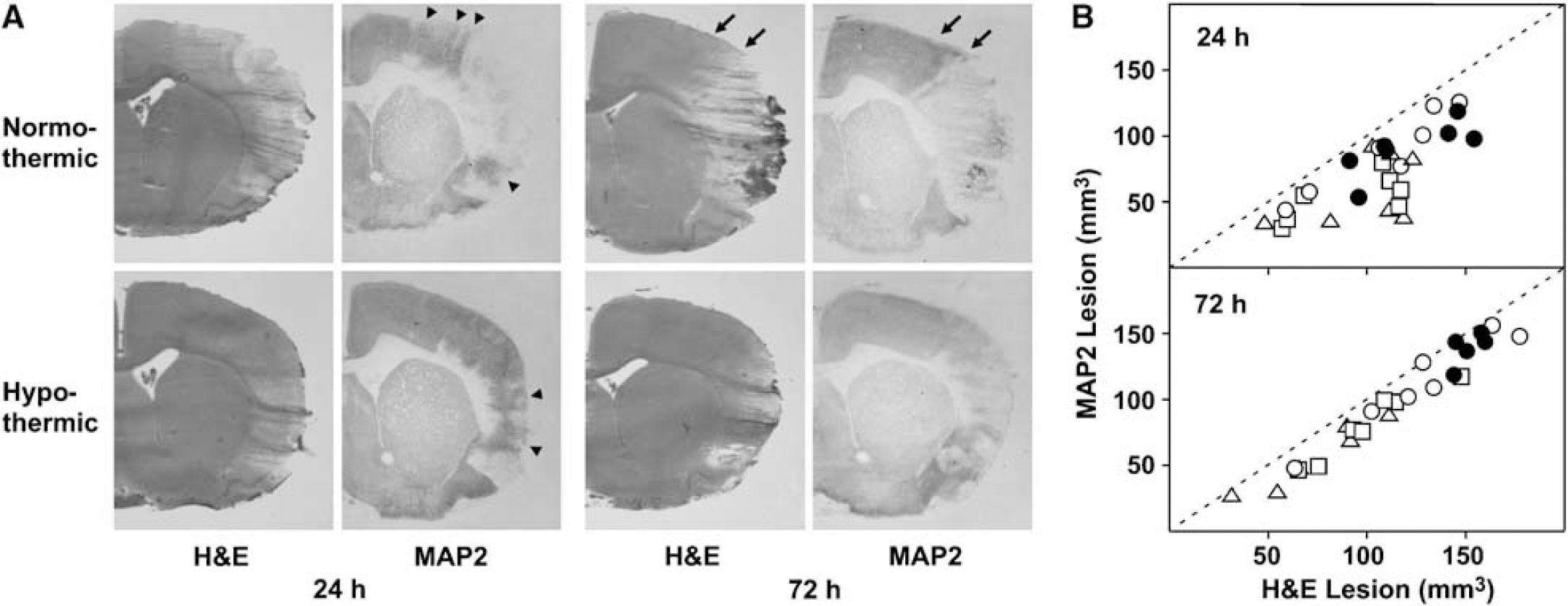
Effect of postischemic cooling on infarction evaluated by hematoxylin/eosin (H&E) and MAP2 staining. (
Effect of selective postischemic brain cooling on infarct evolution after 90 min MCAO
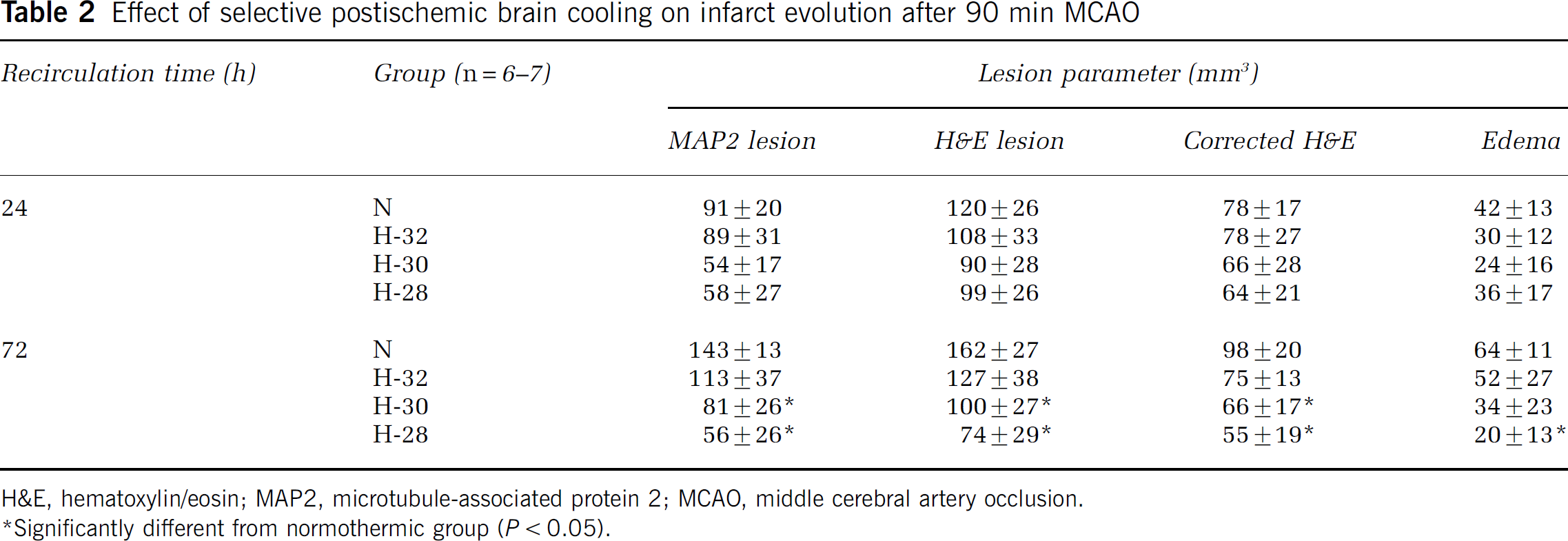
H&E, hematoxylin/eosin; MAP2, microtubule-associated protein 2; MCAO, middle cerebral artery occlusion.
Significantly different from normothermic group (P < 0.05).
There was no evidence of increased bleeding in association with the cooling protocol used in this study. Rather, hemorrhage incidence and severity were well correlated with the severity of tissue damage across groups. Small bleeds were evident at 1 day in 50% of the normothermic and 32°C animals, decreasing to only 7% (1/14) in the pooled 28°C and 30°C groups that showed protection. Hemorrhage frequency remained lower at 3 days in the more deeply cooled animals (70% and 46% in unprotected and protected pools, respectively), and those that did occur were markedly smaller. This indicates that brief, modest systemic cooling was not associated with overt coagulopathy, and there was no apparent adverse impact of deeper local cooling.
The regional temperature data provided an opportunity to examine the relationship between the distribution of brain temperature change and subsequent infarct size. A comprehensive examination of the MAP2 data at 72 h is presented in Figure 4. The decrease in lesion volume was well correlated with decreasing temperature at all recording sites evaluated at 60 mins recirculation, and the slope of this relationship increased progressively with distance from the site of surgical exposure, amounting to a reduction by 36 mm3/°C at site 3. Although there is no reason to expect a linear relationship between temperature and lesion parameters, this provides a simple approach to visualizing the data. Essentially equivalent correlations were obtained at this recording site at time points of 20 mins or longer. On the basis of this analysis, the relationships between temperature and each of the quantitative lesion parameters examined in this study were evaluated using mean site 3 temperature values recorded during 30 to 60 mins recirculation (Figure 5). Weak trends were seen at 24 h, with neuron loss monitored by MAP2 staining showing the strongest effect, although edema was unaffected. In contrast, clear temperature-dependent protection was evident at 72 h. Rats with epidural temperatures of 32°C showed no protection, but the coolest animals exhibited greater than 80% decreases in lesion volume determined by either MAP2 or hematoxylin/eosin. A significant component of this protection arose from decreased brain swelling, because infarct volume corrected for edema showed a smaller proportionate decrease (approximately 50%), but edema was reduced more than threefold by deep cooling. All lesion parameters indicated substantial injury expansion in normothermic animals between 24 and 72 h (Table 2, Figure 5). Conversely, the most deeply cooled group showed no significant increase in either MAP2 lesion or edema-corrected infarct volume during this interval. Therefore, a major impact of early cooling was to attenuate delayed injury progression.
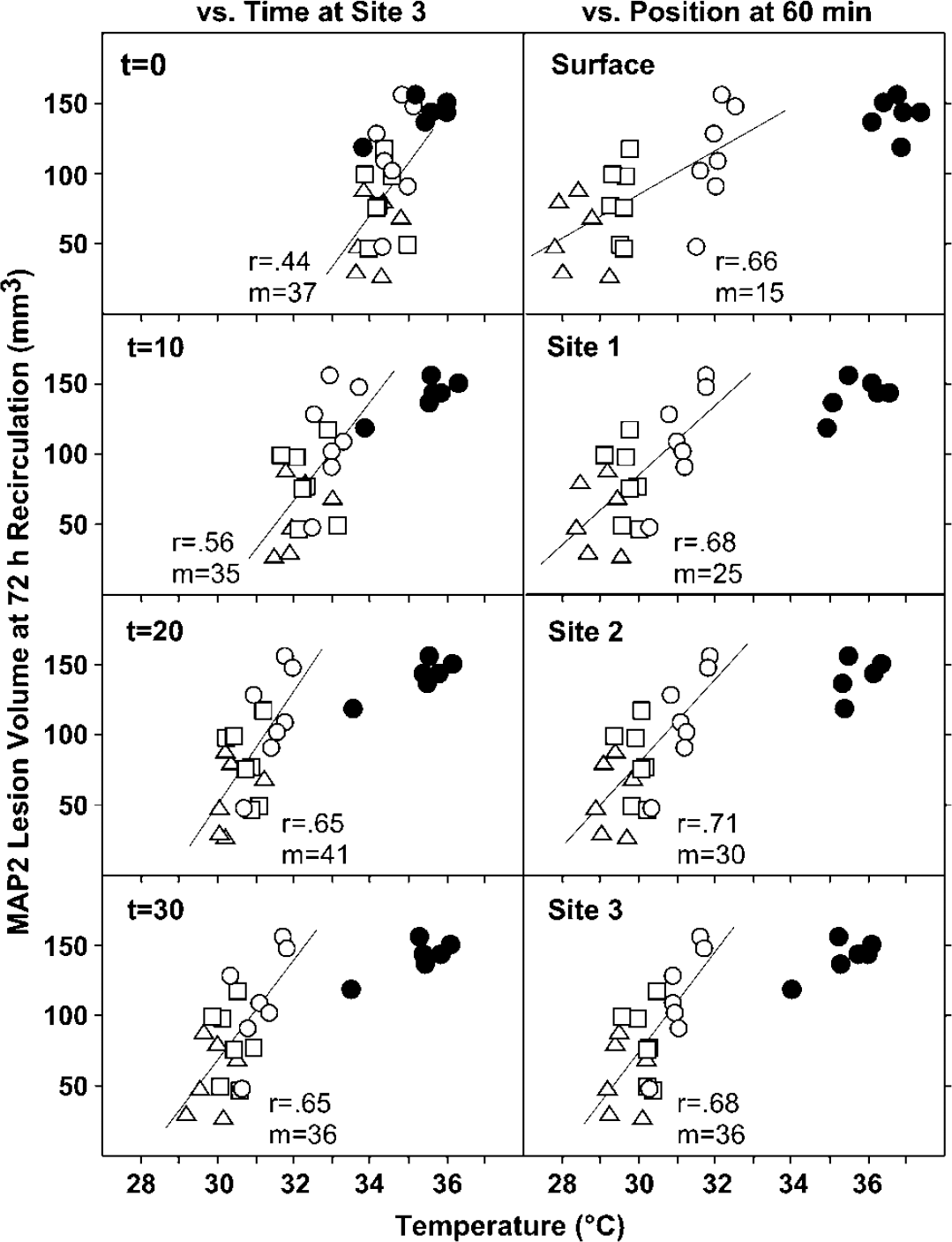
Temperature dependence of hypothermic protection as a function of recirculation time and recording site. MAP2 lesion volumes at 72 h are plotted versus temperatures of individual animals recorded in the distal MCA territory (site 3) at varied recirculation times, and at each brain site at 60 mins recirculation. Stable correlation coefficients (r) were obtained at site 3 at recirculation times of 20 mins or longer. The reduction in lesion volume was well correlated with the extent of cooling at all recording sites, with increasing slope (m) at more distal locations.
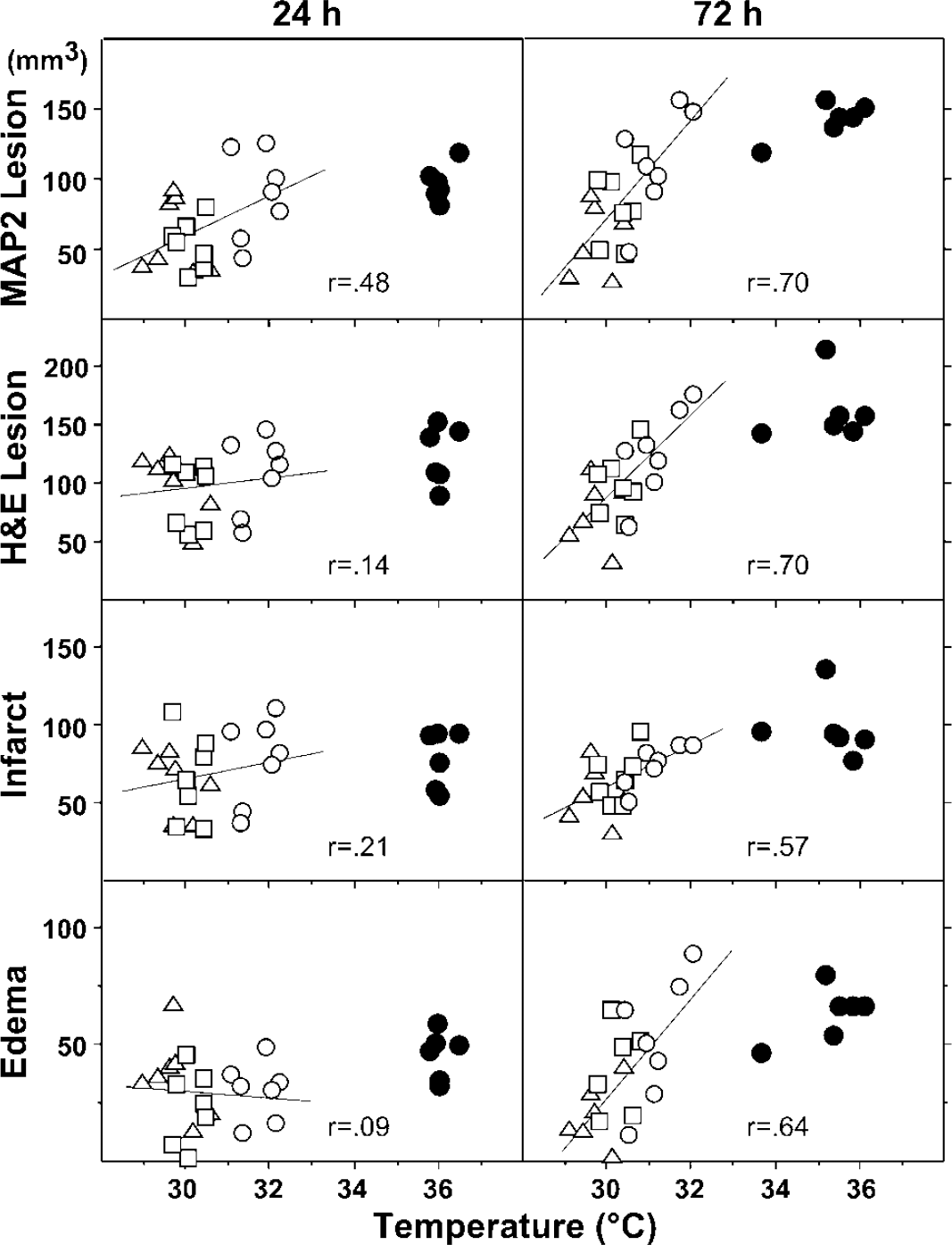
Temperature dependence of hypothermic protection for all lesion parameters. Volumetric estimates of MAP2 lesion, hematoxylin/eosin (H&E) lesion, corrected infarct, and edema were determined as described in the text and plotted as a function of mean equilibrium temperature (during 30 to 60 mins recirculation) in the distal MCA territory (site 3) for individual animals. Strong temperature-dependent protection was evident for all parameters at 72 h recirculation.
Discussion
The present results confirm that transient 2 h cooling initiated at the time of reperfusion can be protective in the SHR, and provide a detailed characterization of the brain temperature dependence of this protection. Infarct volume at 3 days recirculation was strikingly reduced by post-ischemic hypothermia (Figure 3, Table 2), and showed a clear relationship to the magnitude of temperature reduction (Figures 4 and 5). Temperature became more predictive of lesion volume at more distal thermocouple sites (Figure 4) suggesting that effective cooling of the entire MCA territory, specifically including penumbral regions, was required to achieve maximal protection. Temperature continued to decrease through 20 to 30 mins recirculation (Figures 1 and 2), after which there was a close relationship between infarct volume and epidural temperature in the range of 29°C to 32°C (Figure 5). A particularly striking aspect of this protection was the marked reduction in edema swelling at 3 days in brains that had achieved the lowest temperatures.
Methodologic and Analytical Considerations
The distribution and magnitude of brain cooling achieved in this study reflects a composite mechanism involving several sites of heat exchange. Most efficient cooling occurred at the point of surgical exposure, where blood flowed through a vessel directly bathed in the superfusing saline. In addition, there would certainly have been heat transfer through the exposed skull that remained interposed between the superfusate pool and a somewhat larger region of brain surface, also contributing to the gradient in local cooling. More widespread disturbance of the scalp to place recording thermistors may have permitted some diffuse saline migration and facilitated general head cooling, perhaps contributing to the slight differences in contralateral temperature among groups (Figure 2).
Several observations from this study are of general relevance to the choice of methods and time points for evaluating infarct progression after focal ischemia. MAP2 loss is a consistent correlate of neuron death after ischemia, and MAP2 immunocyto-chemistry has been documented to be a reliable means of assessing infarct volume and distribution after permanent or transient focal ischemia (Dawson and Hallenbeck 1996). However, differences in lesion size determined by histology and MAP2 staining have been noted (Zhao et al, 2001). In the present study, there was an excellent correlation between infarct volumes assessed by MAP2 loss and hematoxylin/eosin pallor at 3 days recirculation (Figure 3B). A consistent margin of preserved MAP2 signal within the lesion defined by hematoxylin/eosin (Figure 3A), perhaps associated with periinfarct edema, resulted in a small systematic difference in the absolute lesion volumes measured with these two methods. In contrast, several MAP2 lesions were notably smaller in cooled animals at 24 h recirculation (Figure 3B), resulting from the presence of clearly demarcated islands of preserved MAP2 staining within the MCA territory that could not be as reliably detected in the histologic sections (Figure 3A). Such discrepancies were no longer evident at 3 days, indicating progression to a homogeneous lesion.
The component of systemic cooling produced in the present studies was moderate and brief. There has been considerable debate about the handling of respiratory control during hypothermia in clinical settings that can involve large temperature decreases, decreases in PCO2 because of increased gas solubility and increases in pH. Most experimental studies have employed the simple ‘α-stat’ approach, maintaining physiologic variables as measured at a routine analyzer temperature of 37°C. This was the most practical approach to the present studies in view of the thermal gradients generated in tissues during local cooling, because attempts to control temperature-corrected pH and PCO2 (‘pH-stat’) in a moderately cooled periphery would in any case fall short of doing so at a more deeply cooled location in brain. In the present studies ventilation was not changed once stable conditions had been achieved before occlusion. The only consistent variation noted was slight apparent acidosis progressing during the hypothermic interval, associated with a small increase in PCO2 that became statistically significant in two experimental groups (Table 1). A subtle pH decrease associated with acute halothane exposure in naïve normothermic rats has recently been noted (Zhao and Nowak, 2006), speculated to reflect weak effects at wild-type ryanodyne receptors, mutant forms of which mediate hypermetabolism and malignant hyperthermia in response to this anesthetic. Such effects could be amplified by increases in dissolved anesthetic levels under conditions of systemic hypothermia. It is important to note that none of the changes observed here were far outside the physiologic range, and none correlated with the magnitude of ischemic injury.
Temperature Thresholds for Protection and the Contributions of Brain and Body Cooling
There was a strong temperature dependence of the decrease in lesion volume observed at 72 h (Figures 4 and 5), regardless of the experimental end point evaluated. No protection was evident unless epidural temperature decreased below 32°C within the MCA territory. The greatest benefit was seen at 29°C to 30°C, the lowest tissue temperature reached in these animals, and temperature deeper in the protected cortex would have been only slightly higher. Variations in cooling time course within the MCA territory (Figures 1 and 2), and uncertainty regarding the precise temporal window for protection, preclude identification of an absolute temperature threshold for the effect. The impact of cooling rate was not explicitly evaluated in this study, which rather produced the most rapid controlled temperature decrease possible in the model. The protected, more deeply cooled animals reached an MCA territory temperature of 32°C only 10 mins sooner than did the minimally affected group in which this temperature was targeted (Figure 2), which would not be expected to introduce an appreciable difference in effective insult duration after 90 mins of normothermic occlusion. Therefore, deeper brain cooling in close temporal association with reperfusion appears to be the key element of this intervention. This replicates the essential findings of an initial study using this approach (Ren et al, 2004), which built on earlier observations that cooling extending into the recirculation period was more protective than intraischemic cooling alone (Xue et al, 1992). Instantaneous regional temperature at the moment of recirculation was not strongly predictive of outcome (Figure 4), which correlated best with equilibrium temperature maintained after 20 mins recirculation (Figures 4 and 5). This suggests a window of some tens of minutes after reperfusion during which evolving pathology may remain accessible to transient temperature manipulation.
The surgical model used in these studies requires stringent temperature control of the exposed brain surface to avoid artifact, which at the same time provides an opportunity for convenient intentional modulation (Barone et al, 1997; Ren et al, 2004). Heat transfer to the superfusing saline was efficient, allowing rapid equilibration and precise temperature control near the site of exposure, but a temperature gradient within the MCA territory was detectable (Figures 1 and 2). It was clear from the brain temperature increases observed upon reperfusion that blood from the warmer periphery constituted a significant heat source, and that systemic hypothermia was required to achieve the magnitude of brain cooling obtained in these experiments. This is consistent with available data in neurosurgical patients (Mellergård 1992). One report indicated that selective head cooling in normal rats can be associated with dramatically increased cerebral blood flow during a modest transition in brain temperature from 37°C to 35°C (Kuluz et al, 1993).
Such a perfusion effect would in turn be expected to blunt the efficacy of brain cooling. Whether overt cerebral blood flow increases occur in postischemic brain during cooling, sustained delivery of warm blood may provide some explanation for the lack of further protection observed at temporalis muscle temperatures of 27°C versus 32°C observed in a study that used liquid nitrogen vapor cooling after intraluminal suture occlusion in Sprague-Dawley rats, which preserved a surgically intact head and maintained systemic temperature at 35°C (Huh et al, 2000).
Optimal brain cooling methods for experimental and clinical studies remain to be established. A method for selectively cooling one hemisphere in rats using an implanted extracranial coil has very recently been described and validated using brain temperature telemetry (Clark and Colbourne, 2007), which should be particularly applicable to stroke models in which the MCA itself would not be surgically exposed. One study demonstrating protective effects of postischemic cooling used intravascular saline infusion to reduce brain temperature (Ding et al, 2004), although systemic cooling had not been included and modest temperature reductions of short duration were obtained. The feasibility of applying a comparable approach to cooling human brain using available instrumentation has also been suggested (Slotboom et al, 2004). In the rodent experiments saline administration preceded reperfusion, and theoretical calculations of cooling efficacy in human brain also assumed the absence of additional blood flow. If maintaining tissue hypothermia during initial restoration of perfusion should prove necessary for protection, the present results indicate that adequate cooling of the blood reaching the tissue would require a considerably greater capacity for local intravascular heat exchange than previously appreciated.
Temperature Effects on Delayed Edema Development
A particularly striking aspect of the present study was the difference between normothermic and hypothermic animals with respect to later brain swelling (Figure 5). This pronounced divergence reflected the trend toward progressive swelling in the normothermic group as well as a more robust temperature-dependent decrease in edema than in other measured parameters. Indeed, edema-corrected infarct volume remained essentially constant between 1- and 3-day intervals in the cooled groups. This is generally consistent with early literature documenting the temperature dependence of ischemia effects on brain vasculature and blood-brain barrier function (Dietrich et al, 1990). In contrast to the present results, one earlier study of systemic cooling during 6 h recirculation in SHR reported no significant effect on edema at 2 days survival, even though infarct size and blood-brain barrier leakage were reduced (Huang et al, 1999). However, a downward trend had been seen in the most deeply cooled group of that study (28°C to 29°C), and persistent CCA occlusion may have altered the temporal window for protection in the model, as will be considered below. The effect observed here as a consequence of early, transient cooling would appear to be mechanistically distinct from the impact of delayed cooling on brain swelling observed in clinical studies of hypothermic intervention in stroke (Schwab et al, 1998).
There were trends toward increased lesion size between 24 and 72 h as assessed using all volumetric parameters, particularly in the normothermic group, although these did not reach statistical significance by the stringent test applied (Table 2, Figure 5). The 90 mins insults employed in the present studies are below the temporal threshold for maximal infarction in this model (Kaplan et al, 1991; Ren et al, 2004), and it is not surprising that infarct evolution would occur more slowly after shorter insults. Secondary energy failure continues to progress between 1- and 3-days recirculation after 1 h focal ischemia in the mouse (Hata et al, 2000). Furthermore, brief hyperthermia initiated 24 h after 1 h focal ischemia increases infarct volume in a Sprague-Dawley model (Kim et al, 1996), and this capacity for delayed exacerbation of injury suggests persistent ongoing pathology. A mechanistic involvement of edema per se in the progression of delayed secondary injury in experimental stroke remains to be delineated, but its apparent temperature sensitivity in this study suggests an approach to further investigations.
Model-Dependent Efficacy of Hypothermic Protection
A study of intraischemic hypothermia in the SHR, using a comparable brain cooling approach, observed detectable reduction in infarct volume at a brain temperature of 34°C, progressing to nearly complete protection by 29°C (Barone et al, 1997). The present results indicate that intervention limited to the time of recirculation and the early postischemic period requires cooling at the lower end of this range to be effective. It is possible that a more prolonged interval of postischemic cooling would show protection at a higher temperature threshold. For example, although maximal blood-brain barrier protection was noted at 28°C to 29°C with a 6 h cooling interval initiated before reperfusion in the SHR (Huang et al, 1999), substantial histologic protection was seen with cooling to 32°C to 33°C. Another investigation in this strain also showed significant protection with prolonged 32°C cooling initiated after a delay of 1 h after 90 mins occlusion (Colbourne et al, 2000). However, in both of the latter studies permanent CCA occlusion was maintained after release of the MCA, leading to a condition of sustained hypoperfusion during which prolonged, moderate cooling might be expected to be more beneficial.
Studies in other strains have also been interpreted to show the efficacy of postischemic hypothermic protection that involved more modest temperature reduction or greater delays in cooling. In a particularly informative example, moderate hypothermia (32°C to 33°C) was initiated before reperfusion after prolonged (3 h) occlusion of the MCA and bilateral carotid arteries in Sprague-Dawley rats (Yanamoto et al, 1996). Significant reductions in infarct volume were obtained when rectal temperatures were maintained at 33°C to 35°C for 24 h by housing in a cold room. Rewarming at 1 h recirculation eliminated protection, but also permitted the development of persistent hyperthermia comparable with that of the ‘normothermic’ group. Conversely, rats in which persistent cooling was delayed until 30 mins recirculation exhibited sustained rectal temperatures of 36°C to 37°C, but only slightly larger infarcts than those in which hypothermia was initiated before reperfusion. Therefore, the benefit of cooling in this study derived primarily from the avoidance of postischemic hyperthermia, and similar conclusions can be drawn from subsequent studies involving severe ischemic insults and prolonged postischemic cooling (Yanamoto et al, 1999, 2001). An earlier report indicating benefits of mild postischemic cooling in a comparable model had not included long-term temperature measurements (Markarian et al, 1996).
Changes in temperature regulation do not occur using the occlusion method of this study (Zhao and Nowak, 2006). In contrast, hyperthermia as a consequence of hypothalamic ischemia is a frequently recognized complication of intraluminal filament occlusions (Ma et al, 2006). This is the experimental approach used in most studies reporting benefits of modest (30°C to 34°C) postischemic cooling (Ding et al, 2004; Kollmar et al, 2002; Maier et al, 2001; Zhang et al, 1993a, 1993b). In one such study, 33°C hypothermia was initiated before reperfusion and sustained at 35°C by housing at 5°C, but the control group reached hyperthermic brain temperatures in excess of 39°C during the 24-h interval evaluated (Kawai et al, 2000). However, in no other report was temperature monitored during an extended recirculation interval after recovery from anesthesia. Furthermore, as typically implemented such models also have the potential to produce unrecognized vascular trauma and a resultant prothrombotic state, which would extend the effective duration of cerebral blood flow reduction and thereby increase sensitivity to prolonged, moderate cooling. In addition, filament models typically use strains that exhibit sufficient collateral perfusion to tolerate prolonged obstruction of the internal carotid artery, and brains of such animals can be protected by transient hypothermia even in the absence of overt reperfusion (Baker et al, 1992; Kader et al, 1992; Morikawa et al, 1992; Ren et al, 2004; Yanamoto et al, 2001), provided that cooling is initiated soon enough after occlusion (Baker et al, 1992). It should also be noted that occasional studies of moderate postischemic cooling after filament occlusions have been negative (Huang et al, 1998), including the finding of delayed lesion evolution without significant long-term protection (Inamasu et al, 2000). Finally, although brain temperatures at the upper limit of the protective range of the current experiments were reached during 30°C systemic cooling in some early filament studies (Zhang et al, 1993a, 1993b), the reported efficacy of delayed transient cooling at 1 h recirculation is not consistent with experience in the model used here (Ren et al, 2004).
Conclusions
These results support the interpretation that cooling of sufficient magnitude at the time of reperfusion can be protective after transient focal ischemia, even in a model as robust as the SHR. The temperature threshold for protection is lower than reported for other strains and conditions, particularly in models employing intraluminal filament occlusions. In translating hypothermic protection studies to clinical stroke it is important to consider that cooling requirements may differ substantially with variations in the time course and magnitude of cerebral blood flow reduction within an evolving ischemic lesion, and with model-dependent differences in temperature response to the insult. The notable effect of acute, deep cooling on subsequent edema development suggests specific attenuation of vascular injury initiated at the time of reperfusion, which may be particularly relevant as a therapeutic target.