
Abstract
During reperfusion after ischemia, deleterious biochemical processes can be triggered that may antagonize the beneficial effects of reperfusion. Research into the understanding and treatment of reperfusion injury (RI) is an important objective in the new era of reperfusion therapy for stroke. To investigate RI, permanent and reversible unilateral middle cerebral artery/common carotid artery (MCA/CCA) occlusion (monitored by laser Doppler) of variable duration in Long-Evans (LE) and spontaneously hypertensive (SH) rats and unilateral MCA and bilateral CCA occlusion in selected LE rats was induced. In LE rats, infarct volume after 24 hours of permanent unilateral MCA/CCA occlusion was 31.1 ± 34.6 mm3 and was only 28% of the infarct volume after 120 to 300 minutes of reversible occlusion plus 24 hours of reperfusion, indicating that 72% of the damage of ischemia/reperfusion is produced by RI. When reversible ischemia was prolonged to 480 and 1080 minutes, infarct volume was 39.6 mm3 and 16.6 mm3, respectively, being indistinguishable from the damage produced by permanent ischemia and significantly smaller than damage after 120 to 300 minutes of ischemia. Reperfusion injury was not seen in SH rats or with bilateral CCA occlusion in LE rats, in which perfusion is reduced more profoundly. Reperfusion injury was ameliorated by the protein synthesis inhibitor cycloheximide or spin-trap agent N-tert-butyl-alpha-phenylnitrone pretreatment.
The most obvious strategy to counteract ongoing ischemia caused by vascular occlusion is to recanalize the obstructed vessel. Spontaneous recanalization caused by endogenous thrombolysis is a complex process that depends on the size, composition, and anatomic location of the thrombus, and frequently takes hours to days to occur. Numerous preclinical and clinical studies using thrombolytic agents, especially tissue plasminogen activator, demonstrated pharmacologic recanalization and restoration of cerebral circulation (reperfusion) within minutes of the onset of treatment. Tissue plasminogen activator recently was shown to be effective and approved for urgent treatment of ischemic stroke in humans (NINDS, 1995). Because recanalization of occluded vessels in the clinic is becoming a daily practice, careful inspection of the possible negative influence of reperfusion (apart from the risk related to hemorrhage) must be taken into consideration.
During reperfusion after ischemia, while restoration of oxygen and glucose supply reinstates the oxidative phosphorylation that helps to normalize energy demanding physiologic processes (Pulsinelli and Duffy, 1983; Kinouchi et al., 1990), a parallel cascade of deleterious biochemical processes can be triggered that may paradoxically antagonize the beneficial effect of reperfusion. This phenomenon has been demonstrated in various tissues, especially in the heart, and has been termed reperfusion injury (RI) (Jennings et al., 1960; Karmazyn 1991). The understanding and treatment of RI is an important objective in the new era of reperfusion therapy for stroke.
Yang and Betz (1994) recently demonstrated that 3 hours of focal ischemia followed by 3 hours of reperfusion in the rat produced more damage than 6 hours of continuous ischemia without reperfusion. However, no well-characterized model of pure early RI analogous to what might occur after thrombolysis has yet been described.
The cause of early RI may be multifactorial. The major possibilities for causing cerebral RI include a secondary wave of excitatory amino acid release (Matsumoto et al., 1996, Phillis et al., 1994), Ca2+ influx producing further elevation of cytosolic free Ca2+ concentration (above an already high ischemic level) (Uematsu et al., 1988), free radical formation (Floyd, 1990; Traystman et al., 1991), disaggregation of polyribosomes and depression of protein synthesis (Morimoto and Yanagihara, 1981; Orunesu et al., 1980; Krause and Tiffany, 1993; Hossmann, 1991), blood–brain barrier injury (Yang and Betz, 1994), elevation of leukotrienes and prostaglandins (Gaudet et al., 1980; Kiwak et al., 1985; Tegtmeier et al., 1990), and expression of endothelial adhesion molecules (Okada et al. 1995; Zhang et al., 1995).
In the present study, we describe a model of unilateral middle cerebral artery/common carotid artery (MCA/CCA) occlusion in Long-Evans (LE) rats, wherein ischemic damage can be augmented dramatically by reperfusion initiated during the early hours of ischemia. Finally, we describe preliminary probes into the mechanism of this RI.
MATERIALS AND METHODS
Production of Ischemia
Focal ischemia of variable duration in male 332- to 382-g LE rats and 269- to 294-g spontaneously hypertensive (SH) rats was induced by left MCA and left CCA occlusion as described previously (Aronowski et al., 1994). In addition to unilateral left MCA/CCA occlusion, in selected LE rats the right CCA was occluded (three-vessel occlusion [LE-3 hereafter]).
Briefly, all animals were fasted overnight with free access to water and then anesthetized with an intraperitoneal injection of chloral hydrate in 1 mL of saline. A single 0.45 g/kg intraperitoneal bolus provided anesthesia lasting at least 120 minutes. Animals subjected to ischemia ranging from 180 to 240 minutes were administered an additional 0.1 to 0.15 mg/kg of chloral hydrate approximately 120 minutes after the first injection. For occlusion lasting longer than 240 minutes, rats were allowed to recover from anesthesia before being reanesthetized for the restoration of cerebral blood flow. The femoral artery was cannulated for blood pressure measurement. Temperature from the right temporalis muscle was monitored and maintained at 37.0 ± 0.4°C during ischemia and first hour of reperfusion using a feed-forward temperature controller (YSI Model 72, Yellow Springs Instrument Company Inc, Yellow Springs, OH, U.S.A.) that used a heating lamp and warming blanket. The CCA was isolated through a midline incision and tagged with a suture. An incision was made through the left temporalis muscle perpendicular to a line drawn between the external auditory canal and the lateral canthus of the left eye. Under direct visualization with the surgical microscope, six burr holes were made with a hand-held drill: a 1- × 3-mm rectangular burr hole, situated 2 to 3 mm rostral to the fusion of the zygomatic arch with the squamosal bone to expose the left MCA rostral to the rhinal fissure and five 1-mm burr holes for cerebral perfusion (CP) measurement (Fig. 1). The beveled edge of a 23-gauge hypodermic needle was used to pierce and open the dura along the entire length of the rectangular burr hole. A 0.005-inch diameter stainless steel wire (Small Parts Inc., Miami, FL, U.S.A.) was placed underneath the left MCA rostral to the rhinal fissure, proximal to the major bifurcation of the MCA, and distal to the lenticulostriate arteries. Then the artery was lifted, and the wire was rotated clockwise. Then the left CCA was occluded using atraumatic Heifetz aneurysm clips for reversible ischemia and ligation with a 5-0 silk suture for permanent ischemia. Reperfusion was established by first removing the aneurysm clips from the CCA, and then rotating the wire counterclockwise and removing it from beneath the MCA. Interruption of flow through the MCA was inspected under the microscope and verified by CP measurement using a laser Doppler flowmeter (LDF) (model BPM2, Vasamedics Inc., St. Paul, MN, U.S.A.). Only those rats that displayed reduction of CP after ischemia to less than 15% of the preischemic value or those with a CP reading of 10 or less on the LDF at location 2 (see following section) were included in the analyses.
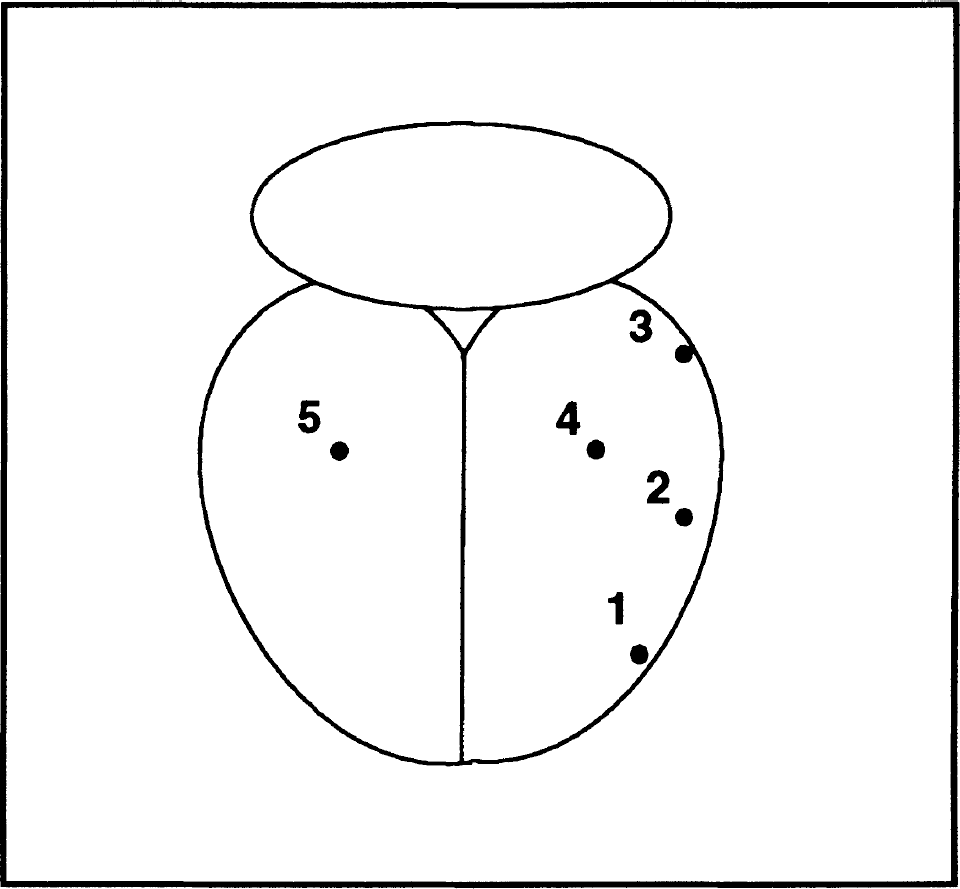
Schematic illustration of spatial distribution of locations of cerebral perfusion (CP) measurement on the surface of cerebral cortex.
Cerebral Perfusion Measurement
Cerebral perfusion at four locations of cortex ipsilateral to the occluded MCA (left; locations 1, 2, 3, and 4) and one location on the contralateral side (location 5) was measured using LDF (Fig. 1).
One-millimeter diameter burr holes were drilled through the skull with special care to avoid damage to the dura to provide access for the 0.8-mm diameter LDF probe. The coordinates for the location of burr holes were as follows: location 1 (anterior central core) was 2 mm anterior to the bregma and together with locations 2 (posterior central core) and 3 (distal core) was placed 1 mm under the left squamosal and temporal bone suture. The distance between locations 1 and 2 and locations 2 and 3 was 4.2 mm and 4.8 mm, respectively. The loci for the remaining burr holes 4 (penumbra) and 5 (contralateral cortex) were 3.8 mm posterior and 4 mm lateral (left and right, respectively) from the bregma. The main trunk of the MCA was approximately halfway between locations 1 and 2, with the occlusion site approximately 2 to 2.5 mm below the locations.
Cerebral perfusion was measured with the LDF probe, in a vertical position that, under dissecting microscope guidance, was placed touching but not compressing intact dura, moistened with 0.9% saline underlying each burr hole. Twenty (2 seconds apart) readings of the CP readout from the digital display of the LDF were collected and averaged to provide a value of CP for each location before occlusion (baseline) and 15 minutes after induction of ischemia (occlusion).
Infarct Volume Analysis
Twenty-four hours after MCA/CCA occlusion, or as indicated otherwise, animals under deep chloral hydrate anesthesia were cardiac-perfused with 60 mL of 0.9% sodium chloride delivered under 110 mmHg pressure. The dissected brains were cut into 2-mm thick coronal sections using a Jacobowitz brain slicer and stained in 2% 2,3,5-triphenyltetrazolium chloride (TTC) in phosphate-buffered saline for 30 minutes at room temperature. Stained sections were fixed in phosphate-buffered 10% formalin before the morphometric analysis. Morphometric determination of the infarct size was achieved using a computed based image analyzer operated by “Brain” software (Drexel University, Philadelphia, PA, U.S.A.), as described previously (Aronowski et al., 1994). Infarcts produced by our protocol were restricted to cortical tissue. The infarct volume (mm3) was calculated from the difference between the volume of contralateral cortex and the volume of the TTC-stained portion (nonischemic) of ipsilateral cortex of each rat. This indirect measure of infarct volume, based on the assumption that the volume of the ipsi- and contralateral cortices are the same before ischemia, corrects the total infarct volume for the edema component (Swanson et al., 1990).
Other Techniques
To ensure that infarct size measurement based on TTC staining actually represented morphologic cell death, selected sections were cryosectioned into 40-μm sections and stained with hematoxylin and eosin (H&E). The area of abnormal H&E staining was compared with that measured by TTC to detect any apparent difference between TTC and H&E staining after permanent and 12 hours of unilateral MCA/CCA occlusion.
All animals in which hemorrhagic transformation developed, identified by the presence of uniformly intensive dark pink/brown color of the infarcted portion of the cortex (before TTC staining) under visual inspection of 2-mm thick coronal sections, were excluded from the analysis (approximately 2–3% of animals). We have observed previously that this discoloration correlated positively with engorgement of cerebral vessels with adjacent microhemorrhages (Aronowski and Grotta, unpublished data, 1996). Such hemorrhagic transformation always was associated with overexaggerated edema and infarct volume.
Pharmacologic agents were administered to groups of rats to determine their effect on ischemic damage compared with untreated controls.
N-tert-Butyl-alpha-phenylnitrone (PBN) (Aldrich), a spin-trapping agent, was administered at 300 mg/kg intraperitoneally 15 minutes before MCA occlusion or 120 minutes after occlusion (30 minutes before reperfusion).
Cycloheximide (1 mg/kg), a protein synthesis inhibitor, was administered intraperitoneally 60 minutes before ischemia. Because cycloheximide results in profound hypothermia that has been implicated in its neuroprotective effects after ischemia (Kiessling et al., 1991), close attention was directed to ensure that our thermoregulation system provided normothermic conditions.
RESULTS
Cerebral Perfusion After Arterial Occlusion
Cerebral perfusion at the surface of the cortex after ischemia produced by unilateral MCA/CCA occlusion in LE rats (LE-2) and SH rats and after unilateral MCA plus bilateral CCA occlusions in LE (LE-3) rats was studied using LDF in the central core (locations 1 and 2), distal core (location 3), penumbra (location 4), and contralateral hemisphere (location 5) as illustrated in Figure 1. Percent of the baseline (preischemic) CP during ischemia for all five locations are indicated in Table 1.
Intraischemic cerebral perfusion (% of control)

LE-2, Long Evans 2; LE-3, Long Evans 3; SHR, spontaneously hypertensive rats.
Unilateral MCA/CCA occlusion in LE rats (LE-2) resulted in significantly less (P < 0.05; determined for each location by analysis of variance followed by Newman-Keuls test) of a decrease of CP in the central core (locations 1 and 2) and penumbra (location 4) than in SH or LE-3 rats. However, there was no difference in reduction of CP between the LE-3 and SH groups (locations 1–4).
Augmentation of Ischemic Damage by Reperfusion
A total of 71 LE rats were used in the experiment. Randomly selected animals were subjected to variable durations of reversible unilateral MCA/CCA occlusion ranging from 45 to 1080 minutes before reperfusion. Each animal was killed 1440 minutes after the beginning of ischemia.
Ischemic durations shorter than 90 minutes were well tolerated by all animals, with 90 minutes of ischemia producing only very small infarct volume, 17.1 ± 20.5 mm3 (n = 5; Fig. 2). Prolongation of the ischemic duration to 120 minutes resulted in a dramatic increase of infarct volume to 113.9 ± 39.3 mm3 (n = 10). Any further prolongation of ischemic duration to either 180, 210, or 300 minutes produced infarct volumes that were statistically indistinguishable from that seen after 120 minutes of ischemia, demonstrating that in this model, MCA/CCA occlusion lasting 90 to 120 minutes represents a threshold amount of ischemia above which full expression of damage occurs (Fig. 2).
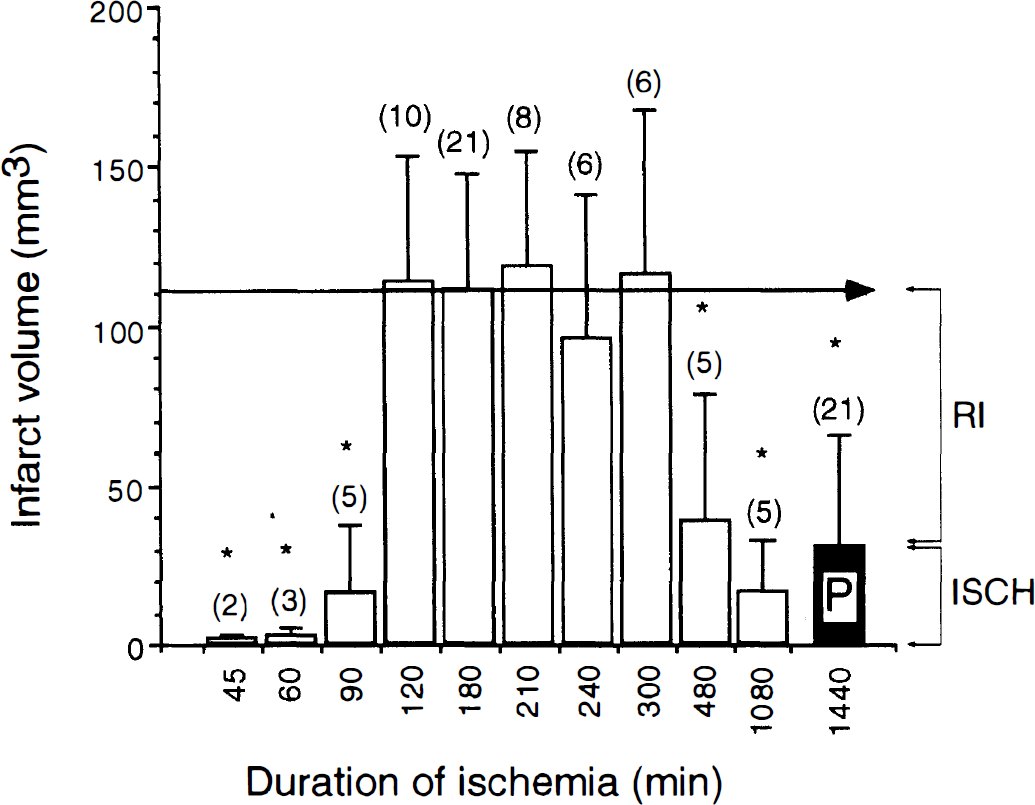
Average infarct volume ± standard deviation (mm3) at 24 hours of maturation after variable (45–1080 minutes) duration of reversible (open bars) or permanent (closed bar; P) left unilateral middle cerebral artery/common carotid artery occlusion in Long-Evans rats. The number of animals in each group is indicated in parentheses above the bars. The magnitude of reperfusion (RI) and ischemic (ISCH) damage is indicated by the brackets to the right of the figure. *P < 0.05 versus average maximal infarct volume produced by reversible ischemia of 120- to 300-minute duration (denoted by horizontal arrow) was determined by analysis of variance followed by Newman–Keuls test.
Because 120-, 180-, 210-, 240-, and 300-minute durations of ischemia resulted in a similar, large infarct (Fig. 2), it was surprising to find that further prolongation of the ischemic duration to 480 and 1080 minutes resulted in dramatically smaller infarct volumes. The infarct volume after 480 minutes (n = 5) and 1080 minutes (n = 5) of reversible ischemia was approximately 36% and 15% of the average maximal infarct volume (110.7 ± 37.4 mm3; average infarct volume of all rats after 120–300 minutes of ischemia; n = 51), respectively (Fig 2). After 1080 minutes of reversible ischemia, rats were analyzed only 360 minutes after reperfusion, a duration of reperfusion that might not be long enough for full maturation of reperfusion damage. To allow for longer infarct maturation time, after 1080 minutes of ischemia, additional randomly selected rats were allowed to survive and were analyzed after 168 and 336 hours of reperfusion. The infarct volumes of these rats were 57.2 ± 17.3 mm3 (n = 3) and 40.1 ± 30.2 mm3 (n = 5), respectively (data not shown), which was smaller than the average maximal infarct volume and indistinguishable from permanent ischemia (see following section), confirming that reperfusion started after prolonged ischemia does not produce RI.
Permanent Unilateral Middle Cerebral Artery/Common Carotid Artery Occlusion in Long-Evans Rats
Because processes triggered during reperfusion were postulated to contribute to the overall amount of reversible ischemic damage, we compared infarct volume after reversible ischemia with that produced by permanent ischemia. As demonstrated in Figure 3, 24 hours of permanent unilateral occlusion of MCA/CCA produced an infarct volume of 31.1 ± 34.6 mm3 (n = 21). This volume is 28% of the average maximal infarct volume (110.7 ± 37.4 mm3) produced by 120- to 300-minute ischemia and 22 to 19 hours of reperfusion. The difference between 110.7 mm3 and 31.1 mm3 (P < 0.001) can be considered the magnitude of RI in this model, which is 79.6 mm3.
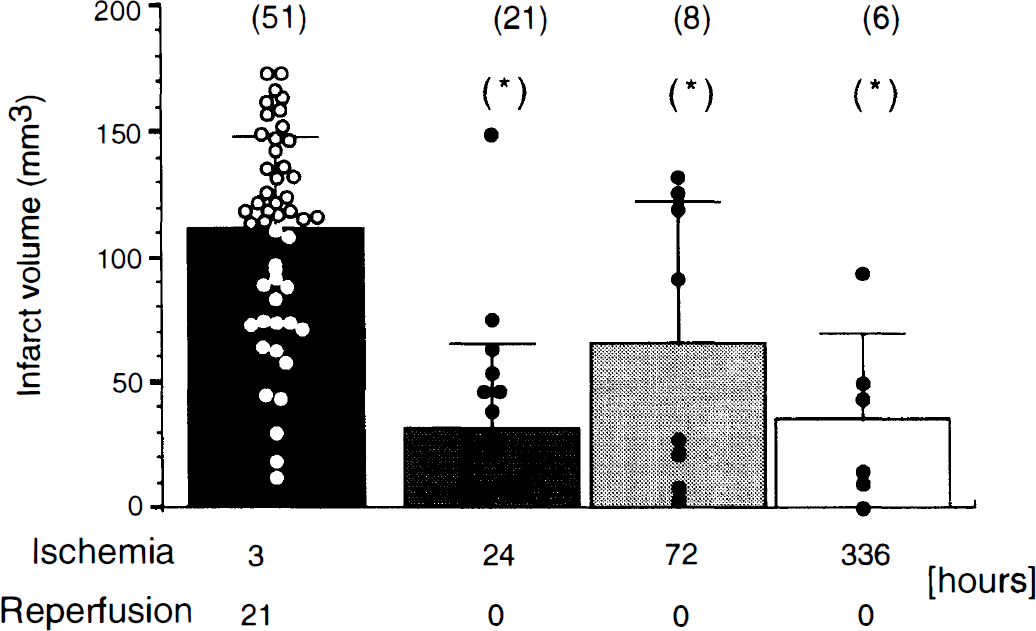
Comparison between infarct volumes after reversible (2–5 hours occlusion followed by 22–19 hours of reperfusion) (closed bar) and permanent 24-hour (dark shaded bar), 72-hour (light shaded bar), and 336-hour (open bar) unilateral middle cerebral artery/common carotid artery occlusion in Long-Evans rats. Cortical infarct volume of individual rats in each group is indicated as a circle. The number of animals is indicated in parentheses above the bars. Level of significance determined by analysis of variance followed by Newman–Keuls test; *P < 0.05 versus reversible ischemia is indicated.
In randomly selected animals, a parallel analysis between TTC and H&E staining was performed to ensure that the lack of TTC staining represented ischemic histologic damage. A third rostral section was stained with H&E and compared with adjacent TTC-stained sections. In all animals tested, TTC staining 24 hours after permanent unilateral MCA/CCA occlusion demarcated the same amount of tissue as measured with H&E (data not shown) in agreement with a previous report (Lin et al., 1993).
It generally is believed that an ischemic infarct may mature for more than 24 hours. Therefore, we analyzed brains of animals after 72 hours of permanent occlusion (Fig. 3). The infarct volume in these animals was 66.1 ± 56.4 mm3 (n = 8) and was significantly smaller (P < 0.005) than infarcts after 120 to 300 minutes of reversible ischemia, but not different from the infarcts after 24 hours of permanent occlusion (P < 0.052). The infarct size in animals after 72 hours of ischemia either was large (more than 90 mm3; 50% of animals) or small (less than 30 mm3; 50% animals), with no animals having intermediate infarct volume. This is in contrast to the infarct volumes in animals after 24 hours of reperfusion, which either had small (<30 mm3; 67% of animals) or intermediate (30–90 mm3; 30% of animals) infarcts, with only one having a large infarction. These data, based on this particular group of animals, suggest that at 72 hours some of the intermediate-sized infarctions seen at 24 hours underwent further maturation to create larger infarctions, and that the small infarctions seen at 24 hours underwent no further maturation and remained small.
Du et al. (1996) recently demonstrated massive ischemic damage 2 weeks (336 hours) after a mild reversible ischemic insult that did not produce signs of damage after 24 hours of reperfusion and only limited damage after 72 hours. Therefore, in selected LE rats, we performed our infarct measurement after 336 hours of permanent unilateral tandem MCA/CCA occlusion (Fig. 3). Unlike the observations of Du et al., infarct volume after 336 hours of permanent occlusion (35.3 ± 34.8 mm3; n = 6) was not different from damage measured after 24 hours (P < 0.8) or 72 hours (P < 0.26) of permanent occlusion, indicating that no processes delayed beyond 72 hours (e.g., vascular occlusion [Garcia et al., 1994] or delayed cell death, apoptosis [Du et al., (1996]) exacerbate acute damage in our ischemia/reperfusion model.
Permanent Bilateral Common Carotid Artery and Unilateral Middle Cerebral Artery Occlusion in Long-Evans Rats
One explanation for the small infarct volume found after permanent unilateral MCA/CCA occlusion in LE rats is a subthreshold reduction of cerebral perfusion in the penumbral (peri-ischemic) region, the result of effective collateral circulation. If the level of hypoperfusion in penumbral regions is insufficient to produce morphologic damage without reperfusion, then further reduction of CP should augment the damage in this territory.
To test this hypothesis, we increased the severity of ischemia by permanently occluding the right CCA in addition to permanent left MCA/CCA occlusion (LE-3) and analyzed cerebral perfusion and infarct volume 24 hours later. Additional occlusion of the right CCA resulted in a significantly greater reduction of cerebral perfusion in LE rats (Table 1). At 24 hours, in agreement with a previous report (Chen et al., 1986), the LE-3 permanent ischemia model resulted in a very high mortality rate (50%). We did not explore the cause of such high mortality because brain swelling with cerebellar tonsillar herniation was noted at autopsy by Chen et al. 24 hours after LE-3. The mean infarct volume of our animals that survived was 159.9 ± 85.1 mm3 (n = 6), approximately four times larger than the infarct volume after permanent unilateral tandem occlusion. It is likely that the average infarct volume after permanent LE-3 ischemia was even larger because rats in which the most severe (largest) strokes developed probably died and were not included in the volumetric analysis.
Therefore, in LE rats, the greater volume of ischemic injury that we found with relatively brief (120–300 minutes) durations of reversible ischemia compared with permanent occlusion (RI) was peculiar to the unilateral MCA/CCA model.
Unilateral Middle Cerebral Artery/Common Carotid Artery Occlusion in Spontaneously Hypertensive Rats
Spontaneously hypertensive rats are highly prone to ischemic damage and exhibit greater CP reduction than LE rats after unilateral MCA/CCA occlusion (Table 1). Like LE-3, SH rats might not demonstrate greater damage with brief ischemia followed by reperfusion as was seen in LE rats with unilateral MCA/CCA occlusion.
In a similar manner to the reperfusion experiment in LE rats, we subjected 48 SH rats to increasing durations of ischemia followed by reperfusion. Animals were killed 24 hours after induction of ischemia for infarct volume examination. Comparing the susceptibility of SH versus LE rats to increasing durations of ischemia (compare Figs. 2 and 4), the duration of ischemia that initiated visible infarct formation was approximately 45 minutes shorter in SH rats than LE rats (45 versus 90 minutes), indicating decreased acute tolerance to ischemia in SH rats. Infarct volume in both strains became maximal after 120 minutes of ischemia. However, unlike LE rats, infarct volume in SH rats after either short (90–300 minutes) or long (480, 720, or 1080 minutes) durations of ischemia were similar (Fig. 4). Again, in contrast to LE rats, after permanent (1440 minutes) occlusion, large infarcts (124.7 ± 21.8 mm3; n = 8) developed in SH rats that were indistinguishable from the average infarct volume of SH rats after 90 to 1080 minutes of reversible ischemia (138.3 ± 35.8 mm3; n = 26; Fig. 4).
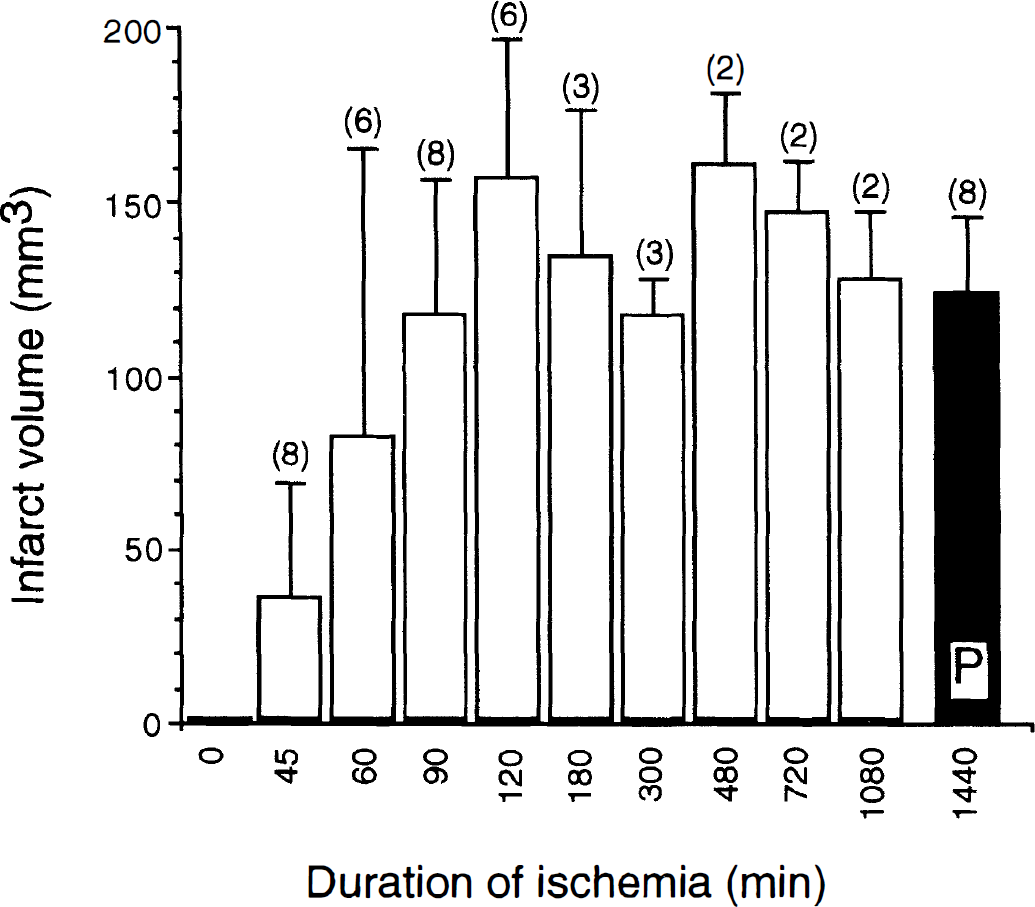
Average infarct volume ± standard deviation (mm3) at 24 hours of maturation after variable (45–1080 minutes) duration of reversible (open bars) or permanent (closed bar; P) left unilateral middle cerebral artery/common carotid artery occlusion in spontaneously hypertensive rats. The number of animals in each group is indicated in parenthesis above the bars.
Protection of Brain from Reperfusion Damage in Long-Evans Rats
To learn more about the nature of the RI we observed after 120 to 300 minutes of reversible unilateral MCA/CCA occlusion in LE rats, we pretreated animals with PBN or cycloheximide and tested the effect on infarct volume 24 hours after 180 minutes of ischemia. N-tert-Butyl-alpha-phenylnitrone almost entirely eliminated RI by reducing infarct volume by 75% from 111.6 ± 36.2 mm3 (n = 21) in controls to 28.0 ± 21.9 mm3 (n = 8) in PBN-treated rats (Fig. 5). However, delay of PBN treatment to 2 hours after occlusion resulted in a loss of the protective effect of the drug (90.6 ± 52.2 mm3; n = 12). Similar to PBN, cycloheximide pretreatment also was very effective in preventing RI, resulting in an 82% reduction of infarct volume to 19.6 ± 17.86 mm3 (n = 7; Fig. 5).
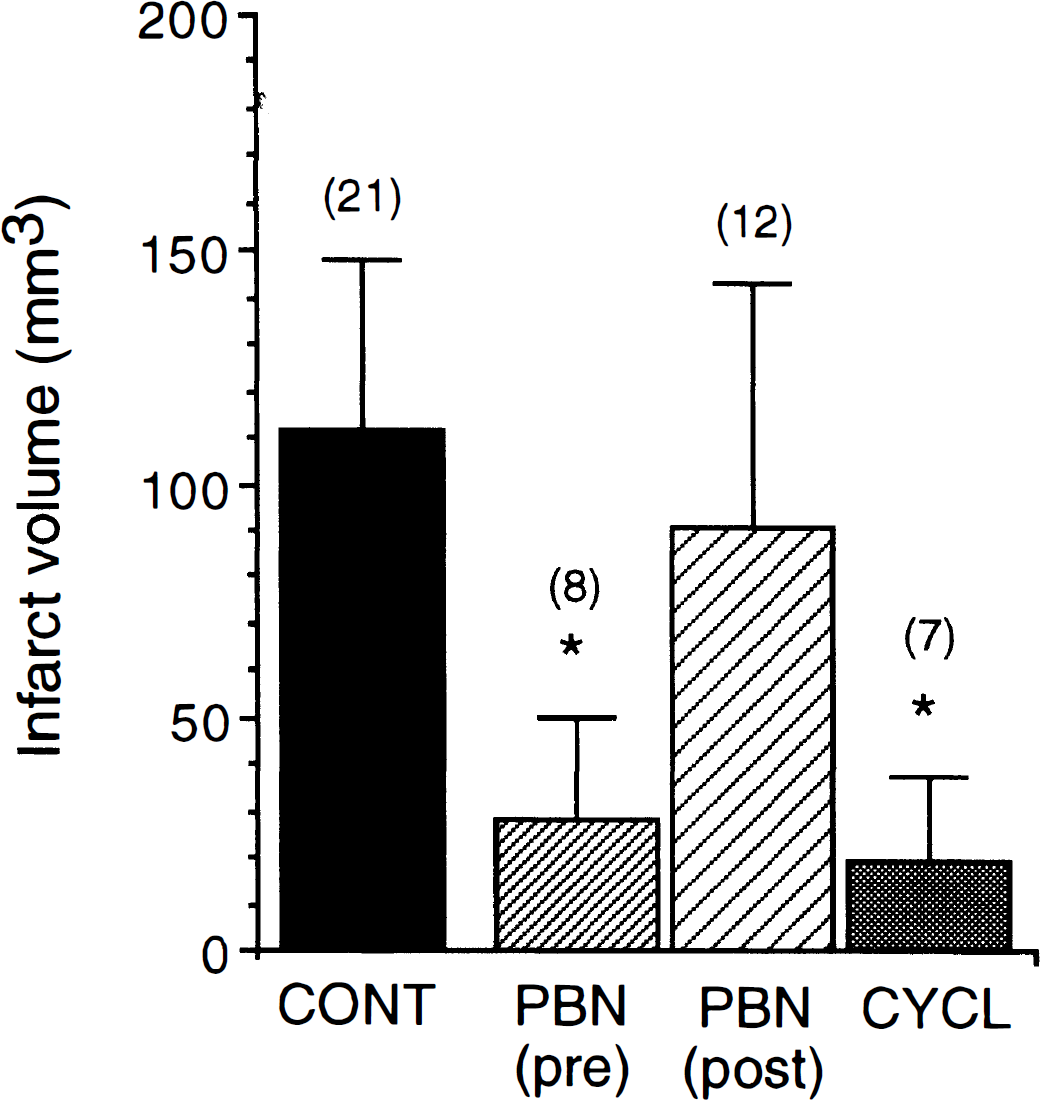
Effect of treatment with 300 mg/kg N-tert-Butyl-alpha-phenylnitrone (PBN) or 1 mg/kg of cycloheximide (CYCL) on infarct volume after 180 minutes of reversible unilateral middle cerebral artery/common carotid artery occlusion in Long-Evans rats. PBN was administered intraperitoneally either 15 minutes before ischemia (pre) or 120 minutes after induction to ischemia (post). Cycloheximide was administered intraperitoneally 30 minutes before ischemia. Animals were killed for 2,3,5-triphenyltetrazolium chloride (TTC) infarct volume analysis 24 hours after induction of ischemia. Level of significance *P < 0.05 versus untreated ischemic control (CONT) determined by analysis of variance followed by Newman–Keuls test is indicated. The number of animals in each group is indicated in parentheses above the bars.
DISCUSSION
Our primary observation is that relatively brief (120–300 minutes) durations of unilateral MCA/CCA ischemia followed by reperfusion in LE rats result in substantially more damage than permanent similar occlusion, and therefore represent a robust model of almost pure RI. The MCA territory irreversibly damaged by permanent ischemia in this model accounts for only 28% of the territory damaged by ischemia/reperfusion, indicating that the level of intraischemic perfusion in the remaining 72% of the MCA territory is sufficient to sustain vital biologic processes and cell survival. However, our finding is clearly strain and model specific. Contrasting our results to other models and strains may help understand some of the constraints needed to produce RI.
The unilateral tandem MCA/CCA occlusion model in LE rats, used in the present study to demonstrate RI, represents a direct adaptation of a method that produces severe ischemia in the SH rat (Brint et al., 1988; Kaplan et al., 1991; Jacewicz, 1992; Buchan et al., 1992; Aronowski et al., 1994). Spontaneously hypertensive rats subjected to reversible ischemia, unlike LE rats (see Fig. 2 versus Fig. 4), responded to reperfusion with no augmentation of damage. The infarct volumes after either 1 to 18 hours of reversible ischemia or 24 hours of permanent ischemia in SH rats were equally large, affected the majority of the MCA territory, and are in agreement with previous reports (Buchan et al., 1992; Kaplan et al., 1991). Spontaneously hypertensive rats in general are more susceptible to ischemic damage than normotensive rats. For instance, the duration of reversible ischemia that produces half maximal infarct volume in LE rats is twice that for SH rats (see Fig. 2 versus Fig. 4) (Aronowski et al., 1994; Aronowski et al., 1996). Furthermore, permanent occlusion of the MCA consistently results in a large infarct volume in SH rats in contrast to no (Coyle and Heistad, 1987) or smaller infarction (Cole, 1996; Grabowski et al., 1988) in normotensive strains. It is thought that this increased vulnerability to damage after MCA occlusion in SH rats is related mainly to a genetic vasculopathy and inefficient collateral circulation (Harper and Bohlen, 1984; Nordborg et al., 1985; Johansson, 1994). In fact, during MCA occlusion, cerebral blood flow/CP is reduced more in SH rats both in the core and penumbral regions. Because ischemia is more severe in SH rats than in LE rats, as demonstrated by morphologic susceptibility and cerebral blood flow reduction, it is logical to anticipate that after any given duration of ischemia (before reperfusion), the extent of irreversible damage in SH rats also will be greater. If we assume that ischemia caused by unilateral MCA/CCA occlusion in SH rats rapidly recruits the entire MCA territory, causing irreversible widespread intraischemic damage, then no further damage in the MCA territory can be seen after reperfusion. This notion essentially is supported by our next observation. Permanent occlusion of the contralateral CCA, in addition to tandem unilateral MCA/CCA occlusion in LE rats, produces more severe blood flow reduction (see Table 1) and results in maximal and more immediate intraischemic damage, similar to the damage in SH rats, and also cannot be further augmented by reperfusion. Again, this observation is in agreement with the finding that in a model analogous to our LE-3, permanent and 3-hour reversible occlusion in normotensive Wistar rats produced equally large infarcts that also were indistinguishable from the infarct volumes after 3 hours of reversible and permanent unilateral MCA/CCA occlusion in SH rats (Buchan et al., 1992). Finally, MCA occlusion produced by an intraluminal filament also does not produce reperfusion damage. In this suture model, there is a profound reduction of cerebral blood flow of the same order of magnitude as occurs during unilateral MCA/CCA occlusion in SH rats (compare Brint et al., 1988 and Memezawa et al., 1992a; Memezawa et al., 1992b). Altogether, these data suggest that an intermediate level of ischemia, although not producing widespread irreversible intraischemic damage, predisposes tissue to RI, as observed in our experiment. More severe intraischemic insults are not associated with reperfusion injury.
To explore possible mechanisms that may contribute to RI in our ischemia/reperfusion model in LE rats, we employed selected pharmacologic interventions aimed at specific cellular processes previously postulated to play a role in ischemia/reperfusion damage, and tested their effect on histologic outcome. We are not postulating that cycloheximide or PBN are specific therapies for reperfusion injury. They may affect intraischemic as well as reperfusion events. The reperfusion phase after cerebral ischemia has been shown to produce high concentrations of free radicals (Floyd, 1990; Traystman et al., 1991). Elevated levels of these highly reactive molecules subsequently have been postulated to participate in ischemic injury through covalent modification of integral constituents of targeted cells, such as proteins, nucleic acids, or lipids (Floyd, 1990). Spin-trapping compounds readily react with a variety of free radicals, forming more stable spin-adduct, the property that was first used to detect free radicals by electron paramagnetic resonance. N-tert-Butyl-alpha-phenylnitrone (PBN), a spin-trapping compound, recently was shown to reduce damage produced by global and focal ischemia, with the latter being reduced even if PBN was administered 5 and 12 hours after MCA occlusion in reversible and permanent models in rats, respectively (Yue et al., 1992; Cao and Phillis, 1994; Zhao et al., 1994). This protective effect of PBN on ischemic outcome was attributed to a scavenging of deleterious free radicals and improvement in recovery of energy state (Folbergrova et al., 1995).
Similar to previous reports, our studies demonstrate that PBN administered before ischemia dramatically reduces RI, resulting in an infarct volume indistinguishable from that produced by permanent ischemia. However, in contrast to PBN pretreatment and reports on PBN neuroprotection after delayed post-treatment (Cao and Phillis, 1994; Zhao et al., 1994), administration of PBN 2 hours after MCA occlusion (1 hour before reperfusion) was ineffective in reducing RI in our ischemia/reperfusion model. The lack of efficacy of PBN posttreatment in our model is surprising, especially because we administered PBN 1 hour before reperfusion, a duration that, based on the known pharmacokinetics of this compound in gerbils, should result in maximal brain concentration during reperfusion (Yue et al., 1992). Because the damaging effect of free radicals should be maximal during reperfusion, it is unclear why PBN post-treatment did not reduce RI in our ischemia/reperfusion model, as compared with reversible intraluminal filament occlusion reported by Zhao et al. (1994). This lack of posttreatment efficacy casts doubt on the central importance of free radicals in causing RI in our model and suggests that PBN pretreatment has a positive effect on RI by some other mechanism.
Because Cao and Phillis (1994) reported an unusually long (12 hours) window of opportunity for protection with PBN using a permanent focal ischemia model with no reperfusion, it is possible that to provide ultimate neuroprotection PBN must interact with a type of cellular processing that occurs during ischemia rather than during reperfusion. We and others described that irreversible loss of Ca2+/calmodulin-dependent protein kinase II (CaM-KII) activity occurring during early ischemia correlated with neuronal damage after global and focal ischemia (Aronowski et al., 1992a; Hanson et al., 1994; Waxham et al., 1996) and that pharmacotherapies such as N-methyl d-aspartate antagonists and hypothermia, which were able to decrease CaM-KII inactivation, were effective in decreasing brain damage (Aronowski et al., 1992b; Churn et al., 1990). Interestingly, PBN has been shown to completely inhibit inactivation of CaM-KII after cerebral ischemia in gerbils (Hiestand et al., 1992), suggesting that prevention of CaM-KII inactivation or improvement of its recovery is one possible way that PBN may protect the brain from ischemia. Other possible mechanisms of PBN neuroprotection exist. N-tert-Butyl-alpha-phenylnitrone may improve late ischemic events by interfering with secondary microvascular occlusion (Folbergrova et al., 1995). It also was demonstrated after endotoxin-induced shock in mice that treatment with PBN before the endotoxin is capable of reducing nitric oxide production through downregulation of expression of inducible nitric oxide synthase hours after treatment with endotoxin (Miyajima and Kotake, 1995). Interestingly, corresponding with the lack of protection of PBN posttreatment on RI in our model, treatment with PBN 2 hours after administration of endotoxin was ineffective in reducing shock-induced pathology, including induction of inducible nitric oxide synthase. One unifying hypothesis is that after endotoxin shock or ischemia, PBN—through an unidentified mechanism—inhibits expression of a deleterious unidentified protein of some kind that participates in RI.
If the neuroprotective effect of PBN was in fact due at least partially to its interference with expression of new noxious proteins in the region affected by RI, then protein synthesis inhibitors also should ameliorate RI. Cycloheximide administration before ischemia, in a dose that previously was shown to inhibit protein synthesis (Pavlik and Teisinger, 1980) and progression of delayed cell death after mild focal ischemia (Du et al. 1996), dramatically reduced RI. This supports the notion that translation of some noxious protein is involved in expression of reperfusion-triggered damage.
One very perplexing finding is that ischemic durations longer than 300 to 480 minutes do not produce RI in our model. We have excluded the possibility that longer durations of reperfusion are needed to allow the lesion to mature by extending histologic analysis to 1 week after ischemia and finding no increase in infarct volume, indicating no secondary necrotic or apoptotic cell death. One hypothesis is that expression during the early phase of ischemia of a putative “noxious/killer protein” (prevented by PBN and cycloheximide) that contributes to RI may be very short-lived and may not be produced or may not remain after longer durations of ischemia. Alternatively, perhaps a second ameliorating (tolerance) factor occurs after more prolonged ischemia. These explanations are conjectural, and much work needs to be done to understand and effectively treat RI.
We have observed and described a robust model of pure cerebral reperfusion injury. Although the mechanism of this phenomenon is unclear, it can be reduced by an inhibitor of protein synthesis. Because RI may be an unwanted consequence of newly approved reperfusion stroke therapies, such as thrombolysis, the model may prove useful for exploring possible therapies that might be advantageously combined with drugs such as tissue plasminogen activator.