
Abstract
To explore the effects of reperfusion on evolution of focal ischemic injury, spontaneously hypertensive male rats were subjected to photothrombotic distal middle cerebral artery occlusion (MCAO) with or without YAG laser-induced reperfusion. The volume of fodrin breakdown zone, water content, and brain tissue levels of sodium (Na+) and potassium (K+) were measured in the ischemic core and penumbra. Reperfusion attenuated fodrin breakdown, and the volume containing fodrin breakdown product at 3 h after reperfusion (5 h after MCAO) (30±7 mm3) was significantly smaller than the 42±3 mm3 of the permanent occlusion group. After 3 to 6 h of ischemia, Na+ increased, and K+ decreased in the ischemic core. Reperfusion after 2 h of MCA occlusion did not mitigate the ischemia-induced changes in brain tissue electrolytes and water content at 3 to 6 h of ischemia. Even in reperfusion after comparatively long periods of occlusion where brain infarction size, assessed 3 days after MCAO, was not significantly reduced by reperfusion, and the precipitating indicators of the ischemic core (Na+, K+, water content) did not improve, temporary improvement or a delay in progression of ischemic injury was discernible in the penumbra. These results indicate the possibility that treatment with reperfusion is permissive to the effects of neuroprotection.
Introduction
Although there is general agreement that thrombolysis of acute stroke patients is effective (The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group, 1995; Furlan et al, 1999; Saver, 2007), the major drawback of thrombolytic therapy is its frequently observed narrow time window of opportunity shown both in rats (2 to 3 h) (Ginsberg, 1995; Yao et al, 2002) and in humans (3 to 6 h) (The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group, 1995; Furlan et al, 1999). Various reperfusion-induced processes are believed to cause reperfusion injury (Pan et al, 2007; Aronowski et al, 1997; also see Discussion by Watson et al, 2002), and have been studied extensively in experimental focal ischemia models. Although some degree of reperfusion could occur in permanent occlusion models due to possible clot lysis or collateral perfusion, careful comparisons of evolving ischemic injury between permanence and reperfusion in the same model are extremely important. Of current interest, neuroprotection may be most effective when reperfusion has occurred by means of thrombolysis. For example, Ginsberg (2007) recommended studying thrombolysed versus nonthrombolysed models of clot-induced ischemia based on the observation in pilot phase clinical data of possible synergism between reperfusion therapy and albumin neuroprotection. To expand the window of opportunity for neuroprotection, we need to investigate what kinds of ischemic injury progress during the immediate period after reperfusion and before final neuronal cell death or infarction. However, one major problem in the analysis of pathologic conditions in experimental reperfusion injury is the frequent failure to set up a suitable control group for the reperfusion group (i.e., a permanent occlusion group in which the animals can survive this insult).
Because the morphologic expression of ischemic injury lags behind the actual biochemical/metabolic processes that determine cell fate, we decided to evaluate the ischemic injury process as it expands from the ischemic core to the penumbra, using fodrin breakdown products as an index (Saido et al, 1993; Yokota et al, 1995). Fodrin (spectrin) is a major structural protein of the neuronal cytoskeleton and a preferred substrate for the calcium-activated cysteine protease calpain. Calpain is one of the key players in the ischemic cascade, and fodrin breakdown products are good indicators of the progression of ischemic damage. Another approach was to use brain tissue sodium (Na
Materials and methods
Care and handling of the animals was approved by the Hizen Psychiatric Center Ethics Committee (no. 19-9).
Surgical Preparation
In spontaneously hypertensive rats (SHR), both blood pressure levels and global ischemia-induced deterioration of brain metabolism (i.e., increased lactate and decreased adenosine triphosphate) progress with age, and reaches a plateau at the age of 5 months (Tamaki et al, 1979). Blood pressure of SHR rises between 1 and 5 months, and blood pressure and also body weight remained constant thereafter (Mori et al, 1995). Survival rate of male SHR declines steeply after 15 months of age. On the basis of these observations, we use SHR at 5 to 7 months for adult models of focal ischemia.
A total of 52 spontaneously hypertensive male rats (SHR/Izm) were purchased from Japan SLC (Shizuoka, Japan) at the age of 3 months, and used at age of 5 to 7 months. Rats were anesthetized with halothane (3% for induction, 1.5% during the surgical preparation with a face mask, 0.75% after intubation, and 0.5% for maintenance) in a mixture of 70% nitrous oxide and 30% oxygen. The right femoral artery and vein were cannulated using PE 50 tubing. The rats were endotracheally intubated with PE 240 tubing. Pancuronium bromide (an initial dose of 0.3 mg followed by 0.1 mg every 30 min) was intravenously injected, and the rats were mechanically ventilated. Mean arterial blood pressure was continuously monitored. Rectal and head temperatures were maintained approximately at 37.5°C and 36.5°C, respectively, by means of a warming lamp.
Rats were mounted on a stereotaxic head holder in the prone position, and a 2 cm incision was made vertically midway between the right orbit and the right external auditory canal. The temporalis muscle was separated and retracted, and a burr hole 3 mm in diameter was made 1 mm rostral to the anterior junction of the zygoma and squamosal bone under an operating microscope (OPMI 111; Carl Zeiss Japan, Tokyo, Japan), revealing the distal segment of the middle cerebral artery (MCA) above the rhinal fissure. A thin bone layer was preserved to prevent injury to the brain and was carefully removed with forceps. The dura was thereby left intact.
Regional cerebral blood flow (CBF) was measured at 1 mm posterior and 4 mm lateral to the bregma with laser—Doppler flowmetry (ALF 21D; Advance Co. Ltd, Tokyo, Japan). Changes in CBF were expressed as a percentage of the average of 2 or 3 baseline values.
Distal MCAO and Reperfusion
The photothrombotic MCAO and YAG laser-induced reperfusion model was described previously (Yao et al, 2003). Briefly, a krypton laser operating at 568 nm (643-Y-A01; Melles Griot Inc., Albuquerque, NM, USA) was used to irradiate the distal MCA at a power of 20 mW for 4 min. The laser beam was focused with a 30-cm-f.l. convex lens (KPX 112; Newport Corporation, Irvine, CA, USA) and positioned onto the distal MCA after reflection from a mirror. The photosensitizing dye rose bengal (15 mg/mL in 0.9% saline; Wako Pure Chemical Industries Ltd, Osaka, Japan) was administered intravenously to a body dose of 20 mg/kg over 90 secs starting simultaneously with 4 min of laser irradiation. A round laser beam with a Gaussian intensity distribution was focused at the Y-shaped juncture of the frontal and parietal branches for 2 min, and then the laser beam was moved to an additional site just proximal to the first irradiated site for 2 min (two-point hit).
After 2 h of MCAO, the beam from a Q-switched, frequency-tripled YAG/Nd laser operating at 355 nm (16 mW, 15 Hz, average intensity 2.3 W/cm2) (Minilite II; Continuum Inc, Santa Clara, CA, USA) was focused with a 30-cm-f.l. cylindrical lens (CSX 300; Newport Corporation) and positioned with a mirror to envelop the occluded distal MCA.
Experiment 1: Immunohistochemistry
At 3 or 5 h after distal MCAO (Figure 1A), the rats were perfused through the aorta with 20 mmol/L Tris/HCl buffer (pH 7.6) containing 5 mmol/L EDTA, 5 mmol/L β-mercaptoethanol, 250 mmol/L sucrose, and 0.1 mmol/L leupeptin followed by 2% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4) under deep halothane anesthesia. The perfused brains were removed and fixed with the same fixative for 1 day. Next, the brains were immersed in 0.01 mol/L phosphate-buffered saline containing 20% sucrose for 1 day, and processed for immunohistochemistry.
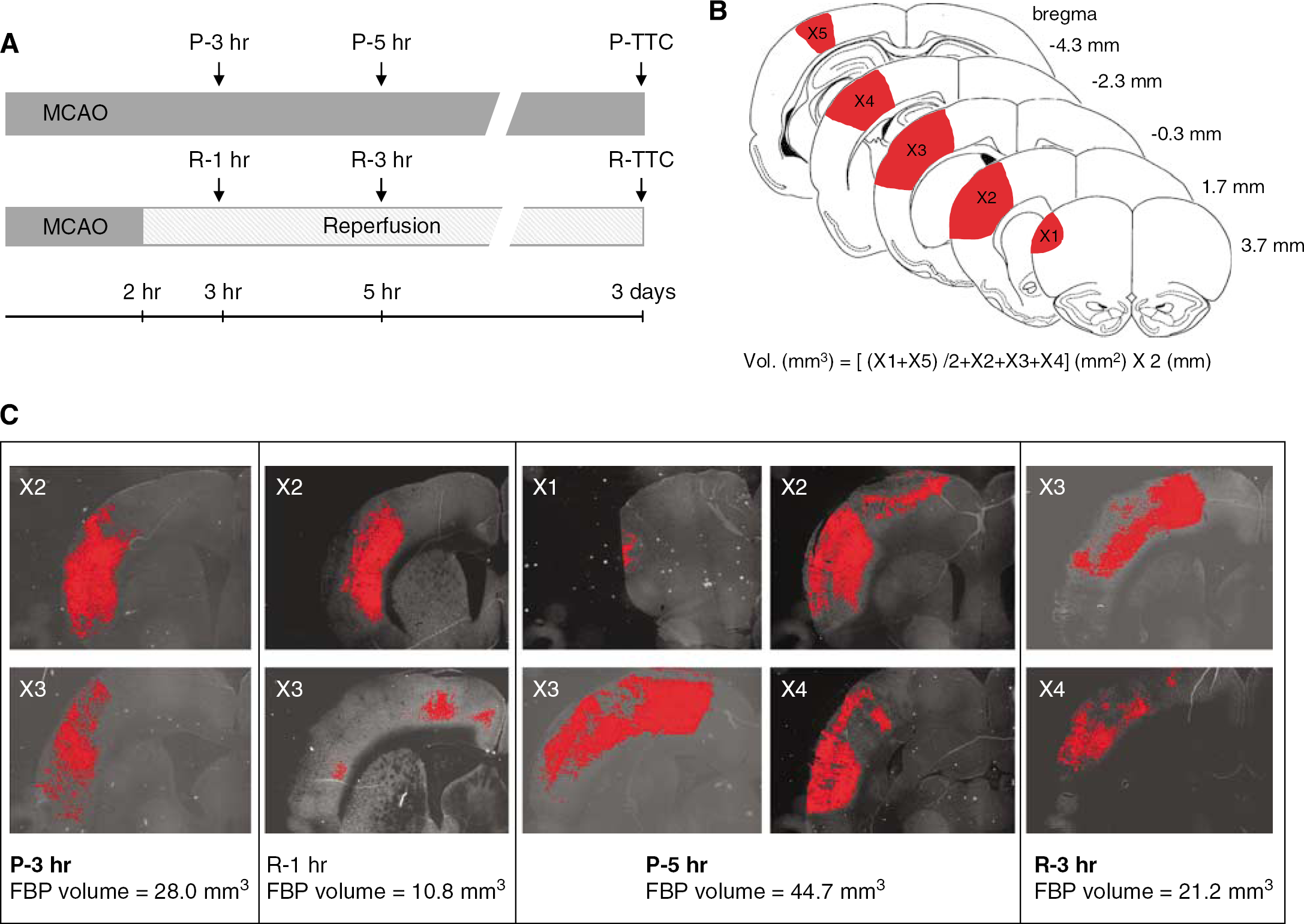
(
(
The fixed brains were coronally sectioned at 8 μm on cryostat at the five levels (3.7, 1.7, −0.3, −2.3, −4.3 mm from bregma) indicated as X1 to X5 in Figure 1. Each section was preincubated with 5% normal donkey serum and 0.1% Triton X-100 in phosphate-buffered saline for 30 mins at room temperature. The sections were then incubated with the primary affinity-purified antibody to the proteolyzed form of fodrin (Saido et al, 1993; Yokota et al, 1995) (1.25 μg/mL, a kind gift from Dr. TC Saido, RIKEN Brain Science Institute) for overnight at 4°C. After being rinsed, sections were incubated with fluorescein isothiocyanate-conjugated donkey anti-rabbit secondary antibody (1:50; Jackson Immunoresearch Laboratories) for 1 h at room temperature and then the sections were rinsed.
The sections were observed with a fluorescence microscope (Olympus, AX-80) and digital images were captured with a fluorescence microscope attached to a CCD camera (Olympus, DP70) using the image analysis software Image-Pro Plus ver. 4.5 (Media Cybernetics, Bethesda, MD, USA) (Figure 1). One of the authors (H.Y.) determined the immunoreactive areas corresponding to the proteolyzed fodrin with NIH Image software (version 1.56) in a masked fashion. The volume of fodrin breakdown zone was calculated by the following formula (trapezoidal rule): V=d(1/2(X1+X5)+X2+X3+X4), where V indicates volume; d, distance between sections; and X1 to X5, cross-sectional area of first to fifth section.
Infarct Volume
Three days after ischemic insult, rats were decapitated under amobarbital anesthesia (100 mg/kg, i.p.), and brains were rapidly removed. The brain was cooled in ice-cold saline for 10 min and was cut into 2 mm-thick coronal sections in a cutting block. Then the brain slices were stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC) (Wako Pure Chemical Industries Ltd) at 37°C for 30 min in the dark. We confirmed that the size of infarction was similar between TTC-stained- and hematoxylin and eosin-stained sections, and linear regression analysis revealed a good correlation between the two methods (Yao et al, 2003). Thus, TTC staining is a reliable indicator of 3-day-old infarction. The infarct volume was calculated by the trapezoidal rule with NIH Image software as shown in Figure 1B.
Experiment 2: Na+, K+, and Water Content
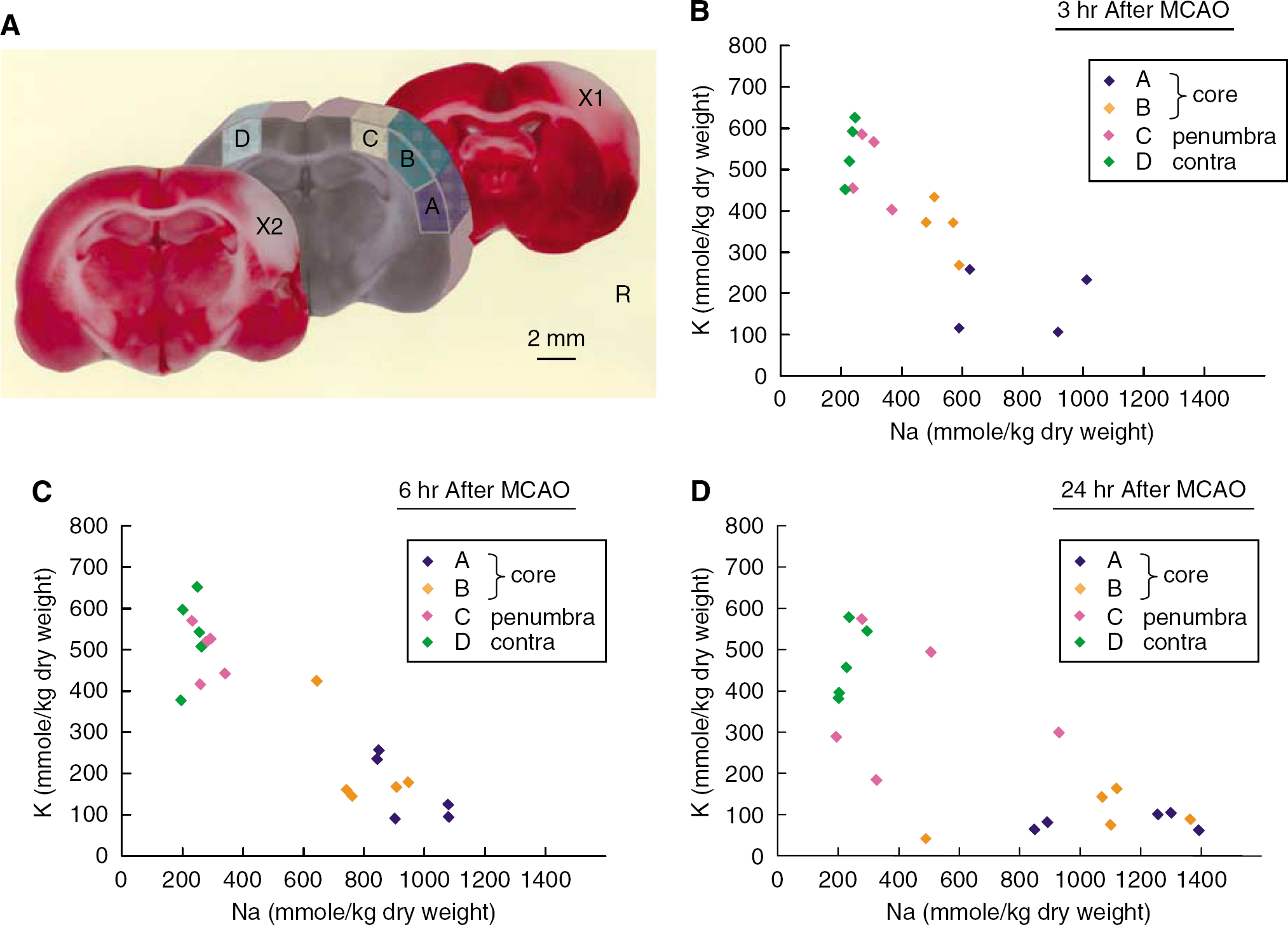
At 3, 6, or 24 h after MCAO with or without reperfusion, the rat was decapitated, and the brain was rapidly removed. The sampling procedure was described previously (Yao et al, 2001; Sadanaga-Akiyoshi et al, 2003). Briefly, the brain was cut into a 2-mm-thick coronal block, and four samples (A, B, C, and D) were dissected from the block at −20°C on a cooling plate under a stereomicroscope (Figure 2A). Because the beneficial effects by reperfusion or the therapeutic efficacies had been seen in the C zone, this fundamentally reversible region was considered to be the penumbral zone (Yao et al, 2002; Sadanaga-Akiyoshi et al, 2003). Our previous experiment showed that apoptotic DNA fragmentation was found only in this C zone at 6 h after MCA occlusion (Yao et al, 2001). The A and B zones were considered to be ischemic core. The slices anterior and posterior to the sampling block were stained with TTC. The samples (A, B, C, and D) were weighed on a balance (BP210D; Sartorius AG, Goettingen, Germany) with 0.01-mg precision to obtain wet weight (W). The tissue was then dried at 100°C for 24 h and reweighed to obtain dry weight (D). The water content was expressed as percent wet weight, calculated as (W−D)/W × 100 (%). The samples were then kept in a deep freezer at −80°C until use. These samples were again dehydrated at 100°C for 5 h, and reweighed on a microbalance (type 4125; Sartorius) with 0.001-mg precision. Tissue extracts were prepared according to Kuribayashi et al (1999). Brain tissue contents of Na+ and K+ were measured with atomic absorption spectroscopy (Z-9000; Hitachi Ltd., Tokyo, Japan) and expressed as mmol/kg dry weight.
Statistical Analysis
Values are mean±s.d. The statistical differences in volume estimations, Na+, K+, and water content were analyzed by analysis of variance (ANOVA) and Bonferroni's test.
Results
Physiologic Variables and CBF
Physiologic variables in Experiment 1 were maintained within the normal range, and there was no statistically significant difference in physiologic variables between the groups (Table 1). Physiologic variables in Experiment 2 were also maintained within the normal range (data not shown). After distal MCAO, mean CBF values determined in Experiment 1 decreased to 32% to 33% and 35% to 39% of the resting value in the permanent occlusion and reperfusion groups, respectively (Figure 3). YAG laser-induced reperfusion of occluded MCA increased CBF to 96±30% at 130 min and 81±23% at 180 min in the reperfusion group, which was significantly higher than 36±12% and 38±11% in the permanent occlusion group (P<0.05, ANOVA and Bonferroni's test).
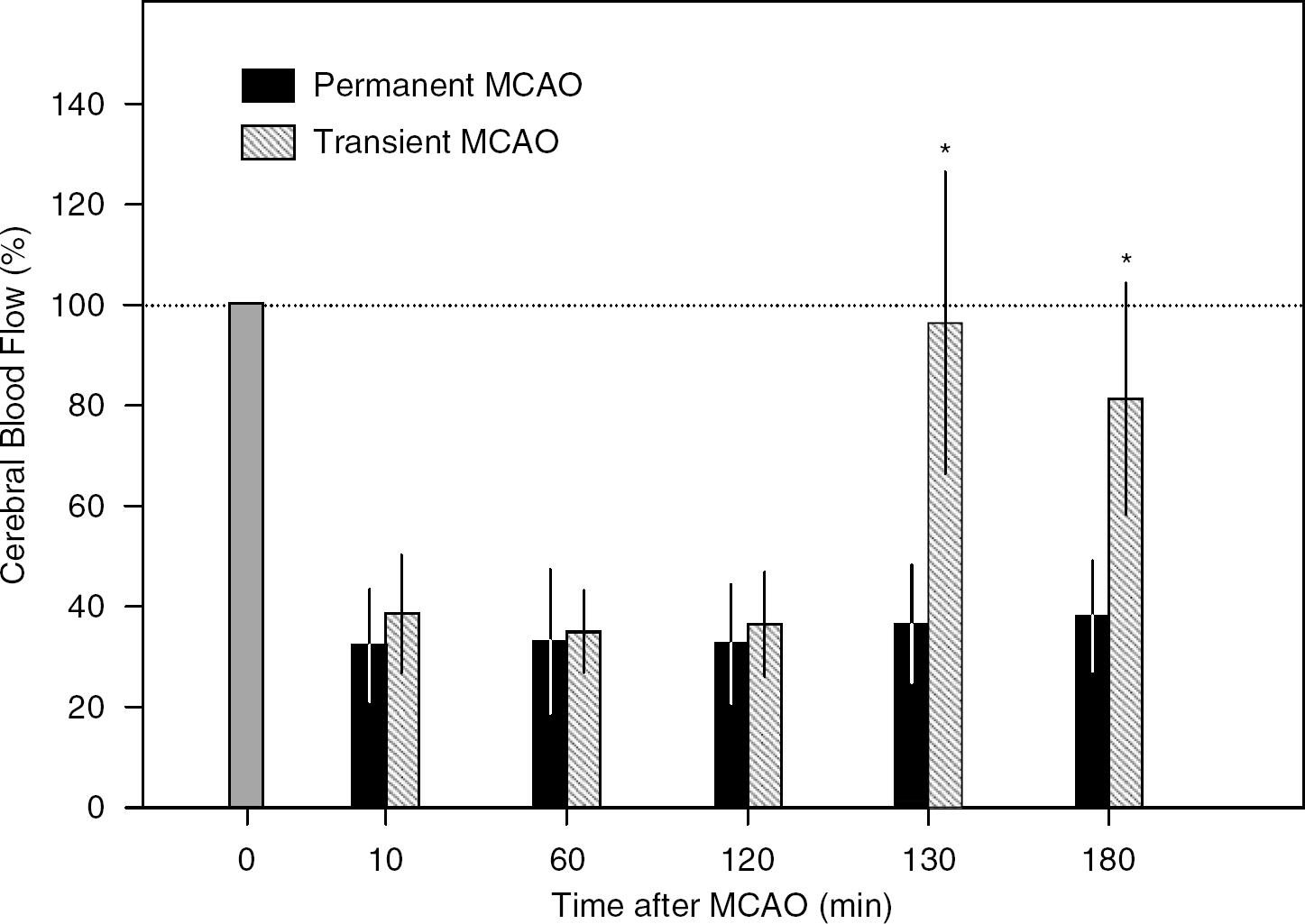
Regional CBF measured at 1 mm posterior and 4 mm lateral to the bregma with laser—Doppler flowmetry. ∗P<0.05 versus permanent MCAO, ANOVA and Bonferroni's test.
Physiological variables in the fodrin experiment
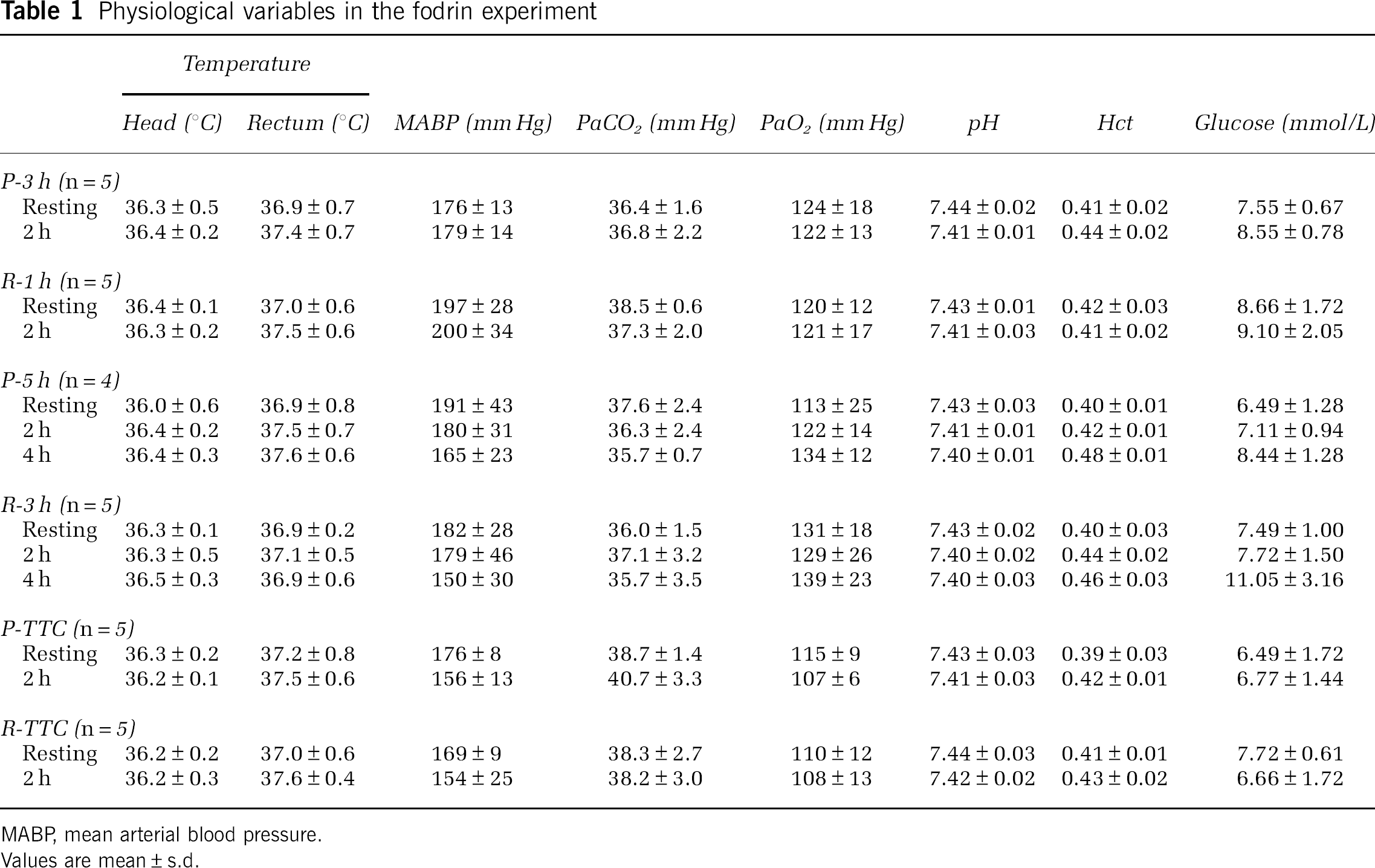
MABP, mean arterial blood pressure.
Values are mean±s.d.
Fodrin Breakdown Product Volume
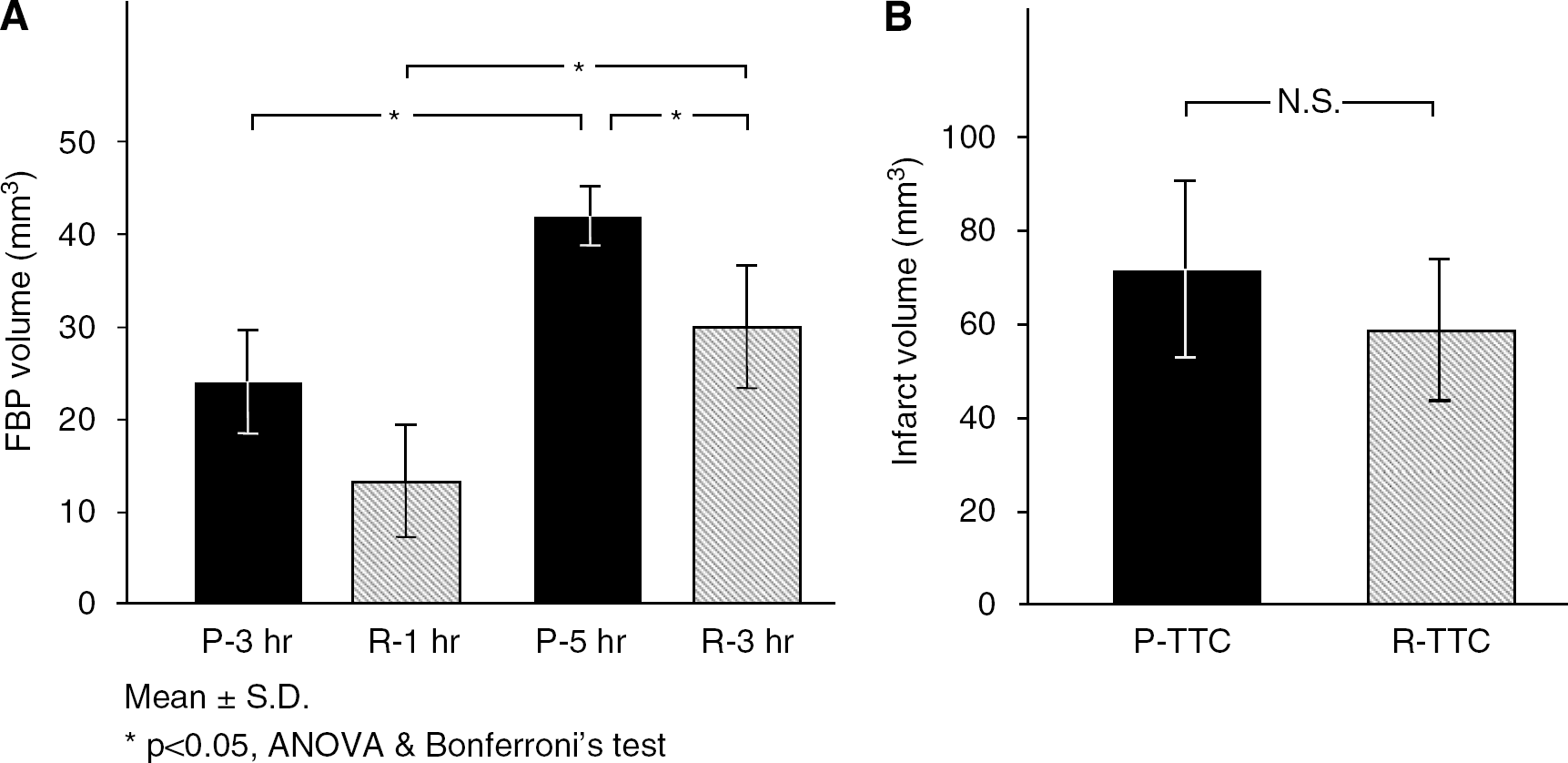
Because therapeutic efficacy had been consistently seen at the peripheral borderzone of distal MCA in our focal ischemia model as mentioned in the ‘Materials and methods’ section, this region was considered to be the penumbral zone. The immunoreactive areas with proteolyzed fodrin were restricted in the ischemic core at 3 h of ischemia, and expanded to penumbra at 5 h of occlusion (Figure 1). At 3 h after distal MCAO (1 h after reperfusion), volumes of fodrin breakdown product were 24±6 and 13±6 mm3 in the permanent occlusion and reperfusion groups, respectively (Figure 4A). The volume of fodrin breakdown product significantly increased at 5 h of occlusion (3 h after reperfusion) compared with that at 3 h. Reperfusion attenuated fodrin breakdown in the penumbra and the volume of fodrin breakdown product at 3 h after reperfusion (30±7 mm3) was significantly smaller than 42±3 mm3 of the permanent occlusion group (P<0.05, ANOVA and Bonferroni's test). There were no significant differences in infarct volume between the permanent occlusion group and the reperfusion group (Figure 4B).
(
Brain Tissue Electrolytes and Water Content
In the nonischemic side sample (D) of the sham-operation group, brain tissue Na+ and brain tissue K+ were nearly equal to previously reported values (Wang et al, 2000; Hu et al, 2000) at 240±31 mmol/kg dry weight and 557±81 mmol/kg dry weight, respectively (Table 2). In the ischemic core (A), Na+ increased and K+ decreased 3 to 6 h after onset of ischemia (Figures 2B and 2C). After 24 h, this tendency became even more prominent, particularly that of decreased brain tissue K+ in the ischemic core (A) which declined considerably to 79±20 mmol/kg dry weight. In the penumbra as well, K+ exhibited a slight tendency to decrease after 24 h (Figure 2D). However, the increased brain tissue Na+ and decreased brain tissue K+ in the acute phase of brain ischemia are thought to occur principally in the ischemic core. With regard to the effects of reperfusion after 2 h of MCAO on brain electrolytes and edema, significant improvements due to reperfusion were not observed (Table 2).
Brain water contents and electrolytes in focal ischemia and reperfusion
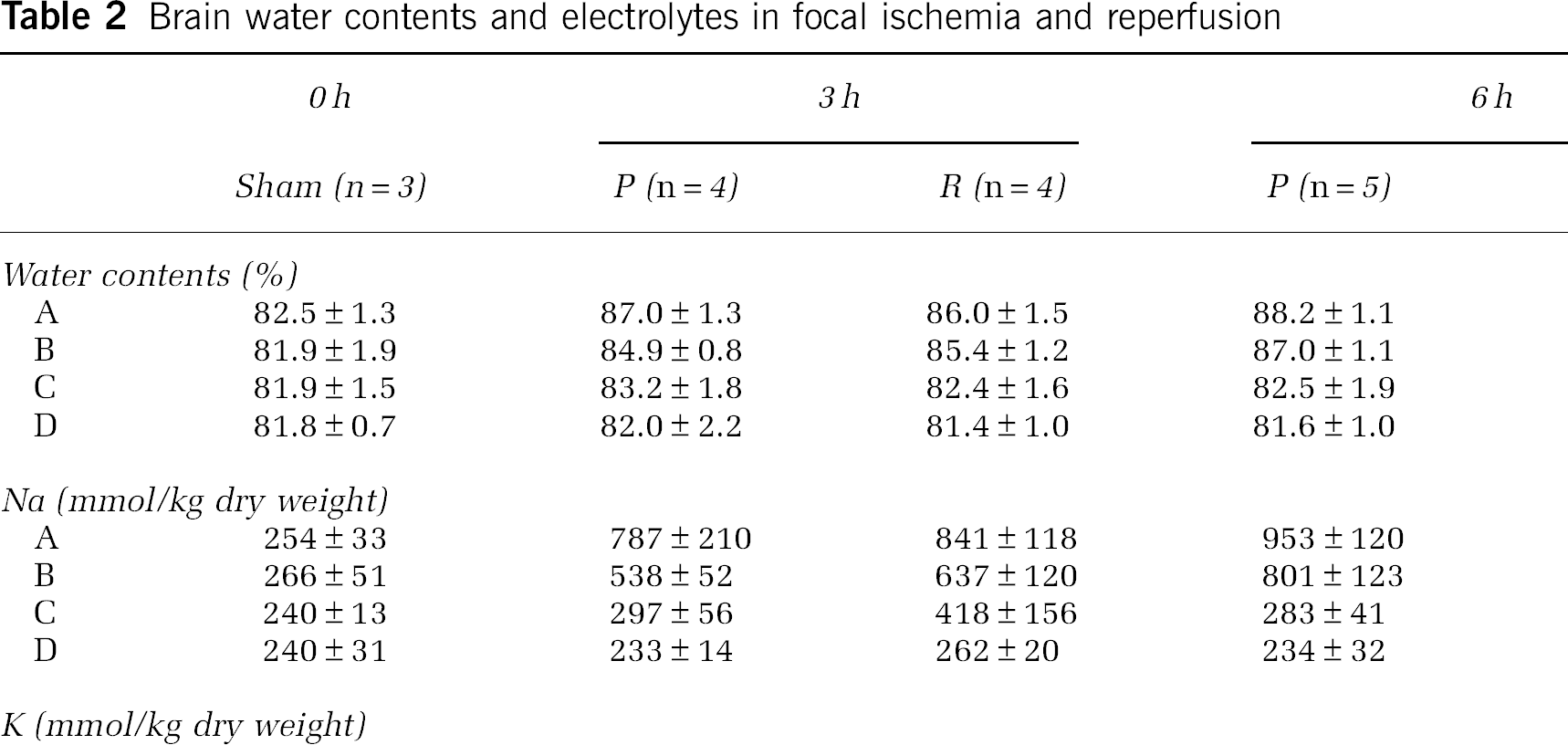
P, permanent occlusion; R, reperfusion.
Values are mean±s.d.
Discussion
Reperfusion after a relatively long period of ischemia (2 h) did not significantly decrease final infarct size in the photothrombotic distal MCAO model in this study, as we previously reported (Yao et al, 2002). In addition, not even a partial reversal was evident in brain tissue electrolytes or brain edema in the ischemic core. However, in the penumbra, fodrin breakdown as an index of evolving ischemic injury was attenuated by reperfusion, which may indicate the temporary appearance of a therapeutic window.
The photothrombotic distal MCAO model, originally established by Watson et al (1987) (for review, see Watson, 1998) and further developed by our own work using SHR, is highly reproducible and does not entail extensive surgery (Yao et al, 1996, 2003). Recently, the ultraviolet laser-induced recanalization technique (Watson et al, 2002) has been established, which confers reperfusion of occluded MCA in our model (Yao et al, 2002, 2003). Reperfusion injury can be defined as ‘ischemic injury aggravated by the act of reperfusion, more extensive than in the case where reperfusion does not occur (i.e., permanent occlusion)’ as shown by Aronowski et al (1997) and by Watson et al (2002) in normotenisve rats. It could be possible that reperfusion injury may aggravate ischemia to a degree less than does that of permanent occlusion. In the latter situation, one must verify the presence of injury mechanism in reperfused but not in nonreperfused brain tissue. In any case, in order to clarify reperfusion injury it is necessary to make a comparison with permanent occlusion. There are actually very few models, however, in which it is possible to test both permanent and transient MCAO. For example, although the most frequently used small animal focal ischemia model must be the intraluminal suture model, a critical view has been put forth that this model is a type of internal carotid artery occlusion model rather than a pure MCAO model (Kanemitsu et al, 2002). This model has a wide zone of severe ischemia, and because the mortality rate is high in the case of permanent occlusion, the permanent occlusion group and the reperfusion group that shared the same time lapse cannot be compared. In fact, of the reperfusion injuries seen in much of the research, those that exhibit real reperfusion injury are surprisingly rare. Our model has a mortality rate of essentially 0% even with permanent occlusion, hence it is possible to conduct tests under both permanent occlusion and reperfusion conditions in the same model, making it very useful in the pathophysiologic analysis of reperfusion injury.
Compelling evidence has accumulated over the past several years that stroke can be treated by means other than flow improvement at least under experimental conditions. Measures of these, which are not directly dependent on improvement of CBF, include blockade of ion channels and transporters (AMPA/kainate receptor (Yao et al, 1997), Na+/H+ exchanger (Kuribayashi et al, 1999; Kitayama et al, 2001) and 3Na+/Ca2+ exchanger (Matsuda et al, 2001)) where Na+ and Ca2+ flow into cells owing to brain ischemia, inhibition of protease (calpain and caspases) activated by a rise of intracellular Ca2+ (Markgraf et al, 1998; Sugawara et al, 2004), shutdown of caspase-dependent apoptosis or programmed cell death pathways (Chan, 2004). In contrast, caspase-independent cell death pathways have been identified such as poly(ADP-ribose)polymerase-1 activation mediated through apoptosis-inducing factor (Andrabi et al, 2006; Yu et al, 2006). The cascade of ischemic injury is essentially multiple, so it is unlikely that targeting a single aspect of any pathway will be efficacious. Furthermore, unless increased CBF of the ischemic penumbra occurs, the multiple pathways of the ischemic cascade will finally prevail, leading to failure of tissue salvage, which is a plausible scenario in clinical settings. However, even if reperfusion is successful, without the effective administration of neuroprotectants there is the possibility that any beneficial effects of reperfusion will be lost.
The intracellular concentration of Ca2+ in ischemic neurons will reach the micromolar range (Silver and Erecinska, 1990) required for activation of μ-calpain, which is expressed in neurons and some noted glial expression (Bevers and Neumar, 2008). Increased intracellular Ca2+ triggers detrimental cascades such as nitric oxide formation from iNOS (Sugawara et al, 2004), calpain activation, and caspase—endonuclease activation or apoptosis as mentioned above. The consequences of specific calpain substrate cleavage at various subcellular locations such as synapses, plasma membrane, endoplasmic reticulum, lysosomes, mitochondria, and the nucleus were reviewed by Bevers and Neumar (2008). However, the precise mechanisms by which calpains contribute to postischemic neuronal death have not been fully elucidated. Recently, Cao et al (2007) showed that calpain I (μ-calpain) of cytosolic and mitochondrial or cytosolic or mitochondrial origin translocated to the mitochondrial inner membrane, leading to the translocation and release of apoptosis-inducing factor, which causes large-scale DNA fragmentation (approximately 50 kb). Furthermore, inhibition of calpain and cathepsin B has been shown to provide neuroprotection by inhibiting the upregulation of MMP-9 (Tsubokawa et al, 2006). Ischemic injury of the brain is associated with calpain-induced cleavage of fodrin to generate a 150 kDa C-terminal fragment. We previously showed a milder and more variable degree of fodrin breakdown in the ischemic penumbra compared with the core (Yao et al, 1995). Fodrin breakdown products reflect intracellular Ca2+, which is important in the ischemic cascade. However, up until now the effects of reperfusion on fodrin breakdown have never been studied. Our results showed a delay in the progression of ischemic cascade can be effected by reperfusion. In other words, our study suggests the possibility that the therapeutic effect of the administration of neuroprotectants on recovery of penumbral tissue is more efficacious together with reperfusion not only due to the reperfusion-induced better delivery of injected neuroprotectants but also by an expansion of the time window.
The concentration of Na+ is typically 10 to 20 times higher outside cells than inside, whereas the reverse is true of K+. These gradients of electrolytes are maintained by a Na+–K+ pump (antiporter) or Na+–K+ ATPase. An important function of the Na+–K+ ATPase is to maintain osmotic balance and stabilize cell volume by pumping out three Na+ for every two K+ in, using ATP as driving force. Therefore, Na+–K+ ATPase is stopped under severe ischemia with energy failure (i.e., ischemic core), resulting in increased intracellular Na+ and decreased intracellular K+. Increase in the total amount of brain Na+ is explained by the fact that Na+ is supplied from the blood due to residual blood flow (Wang et al, 2000; Jones et al, 2006). In this way, brain tissue Na+, which gradually increases during the initial hours of experimental focal ischemia, linearly correlates with the time since the onset of ischemia and can be used to help estimate the age of the lesion (Wang et al, 2000; Jones et al, 2006; Thulborn et al, 1999). At 3 h of MCAO, brain Na+ increases and K+ decreases in the A region where ischemia is most severe. Tissue Na+ exhibited an inverse correlation with K+. In the penumbra, both Na+ and K+ exhibited nearly normal values. In the B region, values of Na+ and K+ were intermediate between the A region and the C region (penumbra) values (Figure 2B). Six hours later, the B region became almost equal to the A region in terms of deranged electrolytes, whereas tissue Na+ and K+ remained in the normal range in the penumbra. As was pointed out earlier (Wang et al, 2000), fluctuations of tissue Na+ and K+ takes place in the ischemic core. Although it was of particular interest whether the exacerbation of ischemic conditions such as these in the ischemic core would show even transient improvement or worsening reflecting reperfusion injury, neither improvement nor exacerbation was seen in this study. This is because Na+–K+ ATPase activity is not reversed by reperfusion even after 60 min of ischemia (Yang et al, 1992), whereas reperfusion within a moderate duration of ischemia restores energy metabolism (Selman et al, 1990; Hata et al, 2000).
Ischemic brain edema is believed to be cytotoxic initially, when the integrity of blood—brain barrier (BBB) is preserved within the first 3 h after MCA occlusion (Belayev et al, 1996). During this period, increase in intracellular Na+ first occurs, and according to osmotic gradient probably through the intermediary of water channels such as aquaporins, an increase in water occurs secondarily (Simard et al, 2007). Although the origin of cellular swelling in cytotoxic edema is unclear, it is attributed mainly to astrocytic swelling. Later, the barrier to plasma proteins breaks down, and a vasogenic phase of edema is recognized. In the photothrombotic MCAO model, photothrombosis of a feeding artery leads to the formation of blood-borne factors that acutely alter BBB permeability, and it should be noted that BBB disruption is more prominent compared with the mechanical occlusion model (Dietrich et al, 1988). Monocyte infiltration or trafficking toward areas of neuroinflammation was shown in the model of cortical photothrombotic stroke (Engberink et al, 2008). However, the effects of reperfusion on BBB integrity after photothrombotic MCAO have not been examined. In this study, 2 h of temporary MCAO did not exacerbate or attenuate brain edema in comparison to permanent MCAO.
In conclusion, even in reperfusion after relatively long periods of occlusion where brain infarction size was not significantly reduced and the precipitating indicators of the ischemic core (Na+, K+, water content) did not improve, temporary improvement or a delay in progression of ischemic cascade was discernible in the penumbra. Although the direct relationship of this temporary window to neuroprotection is unclear without specific interventions, reperfusion may be permissive to the effects of neuroprotection or may prolong the time window for neuroprotectants against stroke.
Footnotes
Acknowledgements
We thank Tetsuya Oogami PhD for excellent technical assistance, and Catherine Kawaharada for editing the paper. We also thank Brant D Watson PhD for the critical reading of the paper.
The authors declare that they have no conflict of interest.