
Abstract
Previously, we reported that transgenic mice overexpressing endothelin-1 in astrocytes showed more severe neurological deficits and increased infarct after transient focal ischemia. In those studies, we also observed increased level of aldose reductase (AR), the first and rate-limiting enzyme of the polyol pathway, which has been implicated in osmotic and oxidative stress. To further understand the involvement of the polyol pathway, the mice with deletion of enzymes in the polyol pathway, AR, and sorbitol dehydrogenase (SD), which is the second enzyme in this pathway, were challenged with similar cerebral ischemic injury. Deletion of AR-protected animals from severe neurological deficits and large infarct, whereas similar protection was not observed in mice with SD deficiency. Most interestingly, AR−/− brains showed lowered expression of transferrin and transferrin receptor with less iron deposition and nitrotyrosine accumulation. The protection against oxidative stress in AR−/− brain was also associated with less poly(adenosine diphosphate-ribose) polymerase (PARP) and caspase-3 activation. Pharmacological inhibition of AR by Fidarestat also protected animals against cerebral ischemic injury. These findings are the first to show that AR contributes to iron- and transferrin-related oxidative stress associated with cerebral ischemic injury, suggesting that inhibition of AR but not SD may have therapeutic potential against cerebral ischemic injury.
Introduction
Cerebral ischemia initiates a cascade of cellular and molecular events that eventually leads to brain damage (for review see Lo et al, 2003). Experimental studies have reported that there is a rapid increase in the production of oxidative stress markers after acute stroke due to the reperfusion event after ischemia, and the endogenous antioxidant defenses are rapidly depleted, permitting further tissue injury. There is also strong evidence that free radical production appears to be an important mechanism of brain injury after exposure to ischemia and reperfusion (Traystman et al, 1991). Animals are also vulnerable to oxidative stress in the brain after transient ischemia/reperfusion involving brain lipids (Yamamoto et al, 1983; Yoshida et al, 1984).
Another event is cerebral edema formation, which is influenced by numerous cellular and functional changes in activities, such as blood–brain barrier function and cell volume regulation (Unterberg et al, 2004). Because of the limited intracranial space, any expansion in brain volume as caused by edema formation increased intracranial pressure and contributes to additional brain injury that has a crucial impact on morbidity and mortality. A number of molecules have been implicated in the pathogenesis of cerebral edema after experimental stroke. Previously, we reported that transgenic mice with specific overexpression of endothelin-1 in astrocytes (GET-1 mice) displayed more severe neurological deficits and larger infarct associated with increased edema and aquaporin-4 expression after 2-h ischemia followed by 22-h reperfusion when compared with nontransgenic mice (Lo et al, 2005).
Aldose reductase (AR), the first and rate-limiting enzyme in the polyol pathway, is present in most tissues surveyed and has been implicated in a wide variety of physiological and pathological functions, such as diabetic vascular and neural complications (Chung and Chung, 2005). Under hyperglycemia, the exaggerated flux through AR, which reduces glucose to sorbitol using nicotinamide adenosine dinucleotide phosphate (NADPH) as a cofactor, increases sorbitol accumulation and osmotic stress because of the slower oxidation of sorbitol to fructose by sorbitol dehydrogenase (SD), the second enzyme in the polyol pathway, which uses nicotinamide adenine dinucleotide (NAD+) as a cofactor. Recently, this hypothesis has been challenged in the nervous tissue, which normally express high level of AR, since the exaggerated flux through AR may contribute to formation of free radicals by excessive use of nicotinamide adenosine dinucleotide phosphate, thereby depleting the pool of nicotinamide adenosine dinucleotide phosphate for other antioxidant enzymes, such as glutathione reductase, which is involved in generation of glutathione sulfhydryl (GSH) (Cheung et al, 2005; Chung and Chung, 2003). Moreover, the flux through SD can limit the amount of NAD+ available for glyceraldehydes-3-phosphate dehydrogenase (GAPDH), thus limiting glycolysis and glucose use. An increased nicotinamide adenine dinucleotide dehydrogenase (NADH)/NAD+ ratio can also lead to increased oxidative stress through the action of NADH oxidase (Chung and Chung, 2003). The expression of AR is regulated by a number of transcription factors, such as osmotic response element binding protein (OREBP) and nuclear factor (NF)-κB, by binding to the AR promoter (Iwata et al, 1999; Ko et al, 1997) possibly under osmotic or oxidative stress conditions associated with ischemia.
Iron is an essential element for normal oxidative mechanism within a cell, and therefore maintenance of homeostatic concentrations of iron is tightly controlled. It is the most abundant metal in the human body, and the brain contains a substantially high concentration of iron (Nakamichi et al, 2004), which is an essential cofactor for enzymes related to synthesis of neurotransmitters, such as tyrosine hydroxylase (Moos and Morgan, 2004). Disturbances in brain iron homeostasis have been linked to acute neuronal injury after cerebral ischemia (Selim and Ratan, 2004). Ferrous iron (Fe2+) is capable of forming hydroxyl radicals that are extremely toxic to the neuronal cell membrane, leading to lipid peroxidation and increased membrane fluidity. In diabetic condition, lipid peroxidation is thought to be contributed by activation of AR (Chung et al, 2003). Indeed, AR is also a major contributor to oxidative stress in diabetic neuropathy (Ho et al, 2006), and both inhibition of AR activity and transition metal chelator were able to completely prevent motor nerve conduction velocity (MNCV) and sensory nerve conduction velocity (SNCV) deficits (Nakamura et al, 2002).
To elucidate the role of the polyol pathway enzymes, AR and SD, in cerebral ischemia and related oxidative stress, we challenged AR- and SD-mutant mice (Ho et al, 2000; Ng et al, 1998) with ischemia/reperfusion injury. The findings that various agents can decrease cerebral infarct size after 2 h of middle cerebral artery occlusion (MCAO) with 22-h reperfusion (Ellsworth et al, 2003; Endres et al, 1998a) suggest that such ischemia-induced brain injury can still be reversed. Cerebral edema is also present 24 h after 2h of MCAO (Yang et al, 1992). Moreover, transient MCAO with reperfusion is proposed to more closely mimic the clinical situation (Hunter et al, 1995). Therefore, AR- and SD-mutant mice underwent 2-h MCAO and 22-h reperfusion (Lo et al, 2005), and their phenotypes were compared with those of the wild-type mice. Inhibitor for AR was also administered to normal mice to further understand the role of AR in cerebral ischemic injury. Our present findings indicated the involvement of AR in oxidative stress and cerebral ischemic injury.
Materials and methods
Animals
GFAP-ET-1 (GET-1) transgenic mice were generated by microinjection of a 15-kb SfiI fragment of pGFAP-ET1 (pGET-1) plasmid into pronuclei of F1 (CBAxC57BL/6N) mouse embryos as previously described (Lo et al, 2005). Age-matched nontransgenic littermates were used as controls in all experiments. Aldose reductase knockout (AR−/−) mice generated in our laboratory (Ho et al, 2000) were back-crossed to the C57BL/6N strain to the 11th generation (N11), which are considered to be congenic with C57BL/6N mice. Age-matched normal C57BL/6N mice were therefore used as control wild-type mice (AR+/+). C57BL/6N mice were also used in experiments with drug treatment. In addition, SD−/− (C57BL/LiA) mice and their age-matched wild-type controls (SD+/+) were generated by crossing of the SD heterozygous mice (Ng et al, 1998). All animals used were male mice and were housed under diurnal lighting condition and allowed access to food and water ad libitum. The protocol of this study was reviewed and approved by the Committee on the Use of Live Animals in Teaching and Research at The University of Hong Kong.
Cerebrovasculature and Water Content Measurement
Mice were euthanized with urethane and perfused with carbon black-ink solution (Huang et al, 1994). The Circle of Willis and its major branches on the ventral side of mouse brains were inspected with the Zeiss Stemi SV11 stereomicroscope (Thornwood, NY, USA). Water content in mouse brains was calculated from measured wet and dry weight of mouse brains collected immediately after killing without perfusion (Lo et al, 2005).
Transient Middle Cerebral Artery Occlusion
Procedure for MCAO is identical to that previously described (Lo et al, 2005). Adult mice (23 to 26 g) were anesthetized (2% halothane in 70% N2O/30% O2 for induction, and 1% halothane in 70% N2O/30% O2 for maintenance) and MCAO was induced by inserting an 8/0 nylon monofilament coated with vinyl polysiloxane impression material (3M Dental Products, St Paul, MN, USA) through the external carotid artery into the intracranial circulation to occlude the middle cerebral artery at its origin. An optic fiber was glued to the skull (2 mm posterior and 6 mm lateral to bregma) and connected to a laser Doppler flowmeter (Perimed, Järfälla, Sweden) for monitoring relative regional cerebral blood flow (CBF) in the middle cerebral artery territory until 10 mins after induction of ischemia. Throughout the experiment, the rectal temperature was monitored and maintained at 37°C ± 0.5°C with a homeothermic blanket system (FHC, Brunswick, ME, USA). One hour and 45 mins after ischemia, the mouse was re-anesthetized and the relative CBF was monitored again to ensure no displacement of the filament and maintenance of ischemia. Two hours after MCAO, the filament was pulled out to allow for reperfusion to the territory supplied by the middle cerebral artery. Cerebral blood flow was monitored for another 10 mins. Subsequently, anesthesia was removed and the mouse was kept in an incubator (ThermoCare® intensive care unit system, ThermoCare Inc., Incline Village, NV, USA) at 32°C for 4 h after ischemia. Sham-operated animals underwent the same anesthesia and surgical procedures without MCAO. For drug treatment, normal C57BL/6N mice were given Fidarestat (dissolved in H2O, i.e., a kind gift from Dr Chihiro Hibi at the Sanwa Kagaku Kenkyusho Co Ltd., Japan) either at 30 mins before ischemia (10 mg/kg body mass) or at 1 h and 45 mins after ischemia (2 mg/kg body mass), whereas vehicle (H2O)-treated mice were included as controls.
Neurological Deficits, Infarct Size, and Brain Swelling
Twenty-two hours after MCAO, the mouse was evaluated for neurological deficits (Lo et al, 2005). Briefly, mice were scored as follows: 0, no observable neurological deficits (normal); 1, failure to extend opposite forepaw (mild); (2) circling to the contralateral side (moderate); and 3, loss of walking and righting reflex (severe).
Immediately after scoring of neurological deficits, the brains were isolated and cut into six coronal slices of 2 mm thickness, stained with 2% 2,3,5-triphenyltetrazolium chloride (Sigma, St Louis, MO, USA) at 37°C in dark for 10 mins to detect lesion area and subsequently fixed in 10% buffered formalin overnight. The posterior surface of each brain slice was photographed and analyzed using a digital image analysis system (SigmaScan Pro, SPSS, Chicago, IL, USA). Infarct size was calculated using the indirect method in which the effects of edema and brain swelling were normalized (Endres et al, 1998b; Iadecola et al, 1997; Kokubo et al, 2002; Lin et al, 1993; Shimizu-Sasamata et al, 1998). Infarct area in each brain slice was calculated as the difference between the contralateral hemispheric area and the noninfarct ipsilateral hemispheric area and was expressed in square millimeters. Infarct volume was then calculated by multiplying the infarct areas with the distance between brain slices and was expressed in cubic millimeters. Hemispheric brain swelling was assessed by the following formula: hemispheric brain swelling = 100% × (ipsilateral volume–contralateral volume)/contralateral volume (Lo et al, 2005).
Immunohistochemistry
For immunohistochemical analyses, 7-μm sections were prepared from paraffin-embedded brain slices. Brain sections were then incubated with antibodies against cleaved caspase-3 (1:200; Cell Signaling, St Louis, MO, USA), nitrotyrosine (1:200, Santa Cruz Biotechnology, Santa Cruz, CA, USA), poly(poly(adenosine diphosphate-ribose) (PAR) (1:200; Alexis, Lausen, Switzerland), transferrin (1:500, sc-22599, Santa Cruz Biotechnology), or transferrin receptor (1:50, sc-7087, Santa Cruz Biotechnology). Immunoreactivity was detected and visualized by Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA) with 3,3′-diaminobenzidine tetrahydrochloride (Zymed, South San Francisco, CA, USA). Photomicrographs were taken with a Zeiss Axiophot microscope. For controls, primary antibody was omitted on adjacent sections. Slides were coded when examined and graded in a masked fashion. To semiquantitatively assess the extent of oxidative stress, cell counting was also conducted in the cortical periinfarct area at interaural line ∼ 1.74 mm using a Zeiss Axioplan microscope. Neurons with pyknotic nuclei that are immunoreactive for nitrotyrosine were counted and expressed as a percentage of the total number of neurons with pyknotic nuclei. For each animal, counting was performed in three separate fields and the values were averaged.
Iron Histochemistry
Brain iron was visualized by Perl's reaction with 3,3′-diaminobenzidine intensification with slight modifications (Kondo et al, 1995). No iron staining can be observed without intensifying the reaction. Briefly, brain sections were well rinsed in deionized water and incubated in Perl's solution (5% potassium ferrocyanide in 5% HC1) for 45 mins. For diaminobenzidine (DAB) intensification, sections were incubated in 0.5% DAB in 0.1 mol/L phosphate buffer for 30 mins followed by a 25-mins incubation in the same solution with the addition of 0.005% H2O2. Sections were then counterstained with methyl green.
Measurement of Bleomycin-Chelatable Iron and Nonheme Iron
In another set of experiment, mice were subjected to either sham operation or MCAO, and their cerebral hemispheres (with cerebellum and brainstem removed) were collected for iron determination. Bleomycin-chelatable iron was determined as previously described (Sohal et al, 1999). Briefly, 10% (w/v) tissue homogenate was prepared in 20 mmol/L Tris, pH 7.4. The following reagents were then mixed in sequence to yield a reaction mixture: 200μL herring testis DNA (1 mg/mL), 10 μL bleomycin (Calbiochem, La Jolla, CA, USA) (1.5 U/mL), 50 μL 50 mmol/L MgCl2, 50 μL 1 mol/L Tris (pH 7.4), 10 μL tissue homogenates or iron standard (2 to 10 μmol/L in 0.05 N HCl, atomic absorption standard, Sigma), and 25 μL 7.5 mmol/L ascorbic acid. The reaction mixture was incubated at 37°C and 50 μL 0.1 mol/L ethylenediaminetetraacetic acid, 250 μL thiobarbituric acid (1% w/v in 50 mmol/L NaOH), and 250 μL 3N HCl were subsequently added. After heating at 85°C for 20 mins, the mixture was centrifuged (10,000g, 10 mins) and the OD532 of supernatant (250 μL) was measured using the Spectra Max 340 microplate reader (GMI Inc., Ramsey, MN, USA).
Levels of nonheme iron were determined as previously described (Sohal et al, 1999). Hundred microliters of tissue homogenate was added to 100 μL 1.5 n HCl, heated at 85°C (30 mins) and then centrifuged (10.000g, 5 mins). Twenty microliters 40% trichloroacetic acid was added to 100 μL supernatant, and the mixture was then heated at 85°C (15 mins) and centrifuged (10.000g, 5 mins). The supernatant (20 μL) was subsequently mixed with 200 μL 1 mmol/L ferrozine solution 3-(2-pyridyl)-5,6-bis(4-phenyl-sulphonic acid)-1,2,4-triazene in 1.05 M sodium acetate, pH 4.8 and its absorbance at 562 nm was measured using the Spectra Max 340 microplate reader.
Western-Blot Analyses
Separate MCAO experiment was performed to collect protein samples. Cerebral hemispheres were homogenized in lysis buffer (50 mmol/L Tris-HCl, pH 6.8, 150 mmol/L NaCl, 5 mmol/L ethylenediaminetetraacetic acid, 0.5% sodium deoxycholate, 0.5% NP-40, and protease inhibitor cocktail) (Lo et al, 1994). The homogenate was then centrifuged (3000g, 5 mins, 4°C) and the resulting supernatant was used for Western-blot analyses. Blots were incubated with antibodies against mouse AR (1:1000, a kind gift from Professor KH Gabbay at Baylor College of Medicine) and transferrin (1:1000, sc-22599, Santa Cruz). Signals were visualized by enhanced chemiluminescence (Amersham, Piscataway, NJ, USA) and quantitated using PhosphoImager (Molecular Dynamics, Sunnyvale, CA, USA). Values for protein levels were given as percentage of AR+/+ sham-operated group after normalization with individual α-tubulin levels for equal loading.
Statistical Analyses
All experiments were performed in a masked fashion. Data are presented as mean±s.e.m., and statistical tests were performed using the GraphPad Prism software (San Diego, CA, USA) as specified. P < 0.05 was considered statistically significant.
Results
Upregulation of Aldose Reductase in GET-1 Mouse Brains after Middle Cerebral Artery Occlusion
A number of molecules, for example, AR, an osmoregulatory protein, have been implicated in the pathogenesis of cerebral edema after experimental stroke. We previously reported that GET-1 mice showed more severe neurological deficits as well as increased infarct and cerebral edema when subjected to transient MCAO (Lo et al, 2005). In light of the increased water content and therefore edema in GET-1 ipsilateral hemispheres after MCAO, the expression of AR was also examined by Western-blot analyses. In sham-operated mice, there was no significant difference between nontransgenic and GET-1 ipsilateral hemisphere (Figure 1). However, the amount of AR in GET-1 ipsilateral hemisphere was slightly increased, suggesting an upregulation of AR in GET-1 mouse brains with increased edema after MCAO (Figure 1).
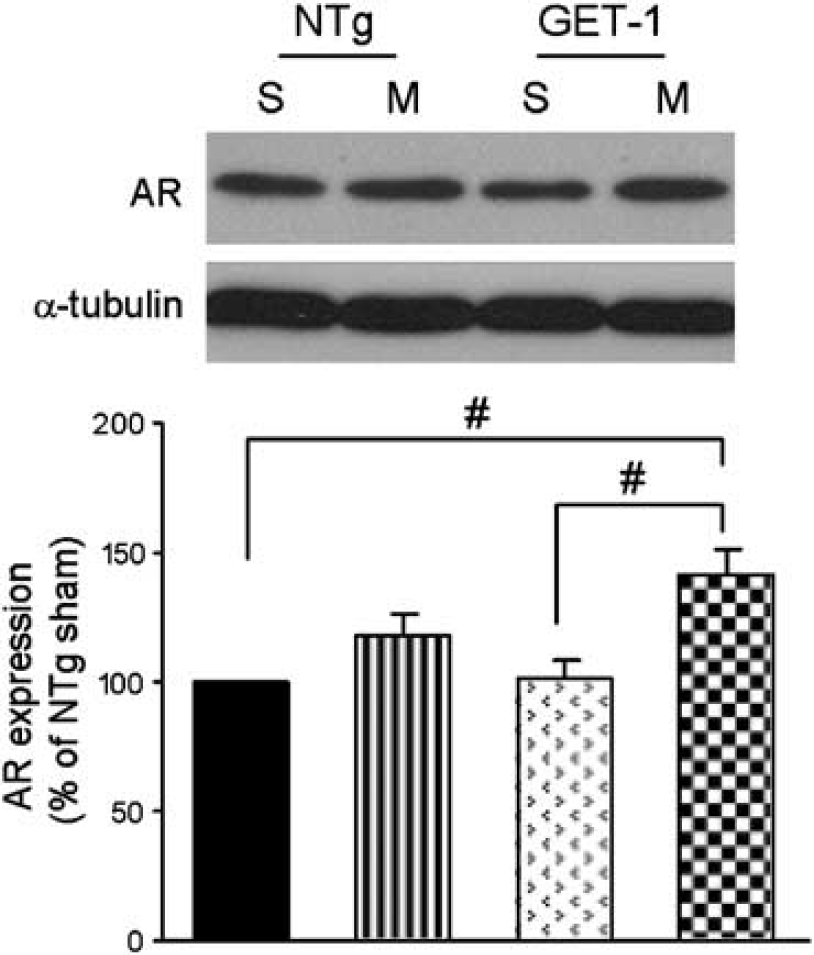
Transient ischemia-induced AR expression. Aldose reductase level, when analyzed by Western blots, was upregulated in both Ntg and GET-1 mouse cerebral hemisphere after MCAO, but note the higher AR level in GET-1 brains after MCAO. S, sham-operated, M, MCAO, NTg, non-transgenic. n = 4 to 5 for each group. #P < 0.05, analysis of variance (ANOVA) followed by Tukeys multiple comparison posttest.
Aldose Reductase Knockout Mice have Normal Cerebrovasculature
The increase in AR levels after MCAO suggested an involvement of AR in ischemic injury. To determine if AR is involved in ischemic stroke, we have chosen to use a general knockout mouse model deficient in AR (AR−/−), which we previously generated (Ho et al, 2000). Under normal conditions, AR−/− mice showed no changes in their behavior, growth, reproductive ability (Ho et al, 2000), and mean arterial blood pressure (data not shown). In addition, the blood glucose level in AR−/− mice under normal and diabetic conditions was not different from those in wild-type mice (Ho et al, 2000, 2006). As reported, AR−/− mice showed no evidence of AR expression in the brain when analyzed by both Northern and Western blots (Ho et al, 2000).
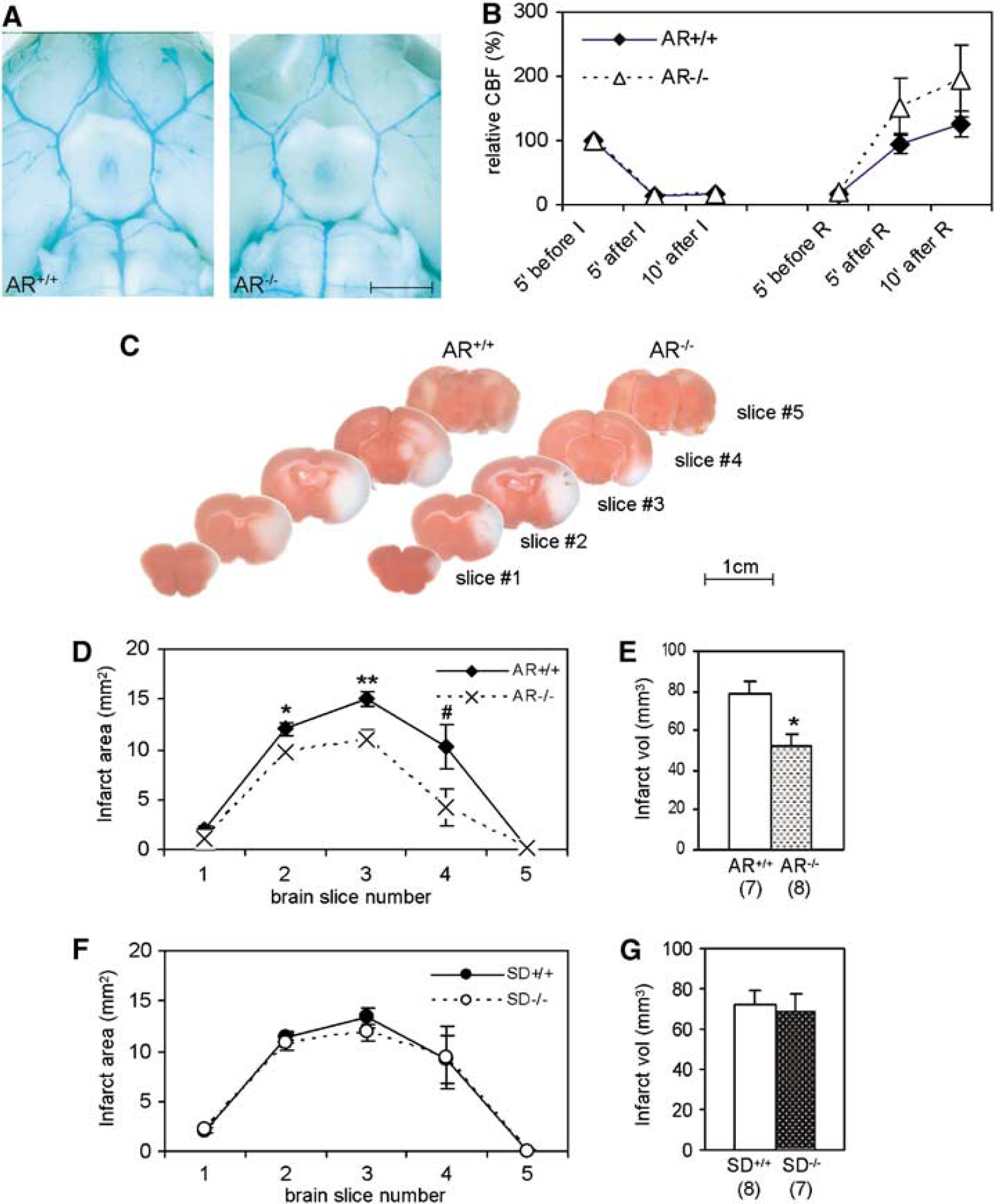
On careful examination in the central nervous system, AR deficiency did not affect the development of the central nervous system. AR-deficient mice displayed normal cerebrovasculature on inspection of the ink-filled Circle of Willis on the ventral surface of the brain (Figure 2A). Major branches were present and there were no missing vessels. Histological analyses of brain sections at the light microscopic level also revealed no major difference in the brain cytoarchitecture between AR+/+ and AR−/− mice (data not shown).
Decreased infarct size in AR−/− mice. (
Since AR is an osmoregulatory gene and its absence may lead to osmotic imbalance in the brain, cerebral water content was measured. Under normal conditions, AR−/− mice appeared to contain less water. The water content in AR−/− mouse brain was 78.04 ± 0.09% (n = 10), slightly decreased than that of the normal AR+/+ brains (78.24 ± 0.06%, n = 8, P < 0.05, Mann–Whitney test).
Ischemic Brain Injury and Neurological Deficits were Reduced in Aldose Reductase Knockout Mice after Middle Cerebral Artery Occlusion
Aldose reductase activation was observed in our model of ischemia/reperfusion (i.e., 2-h MCAO + 22-h reperfusion) in GET-1 mouse brains. To investigate the role of AR in ischemic brain injury, age-matched AR−/− and AR+/+ mice were challenged with the same experimental stroke conditions (2-h MCAO and 22-h reperfusion) and their phenotype was compared. During the experiment, core body temperature (data not shown) did not differ between the two groups. The percentage decrease in relative regional CBF during ischemia and the subsequent increase during reperfusion were similar in both groups (Figure 2B), indicating that the physiological state before and after ischemia in these two groups of mice were similar.
Twenty-two hours after reperfusion, improved neurological scores were observed in AR−/− mice. Most of the AR+/+ mice exhibited severe neurological deficits, having an average score of 2.4 ± 0.2 (Table 1). AR-deficient mice, however, showed significantly minor neurological deficits (1.4 ± 0.2, P < 0.01, Mann–Whitney test, Table 1). The sham-operated mice showed no neurological impairment (data not shown).
Less severe neurological deficits in AR−/− mice after MCAO
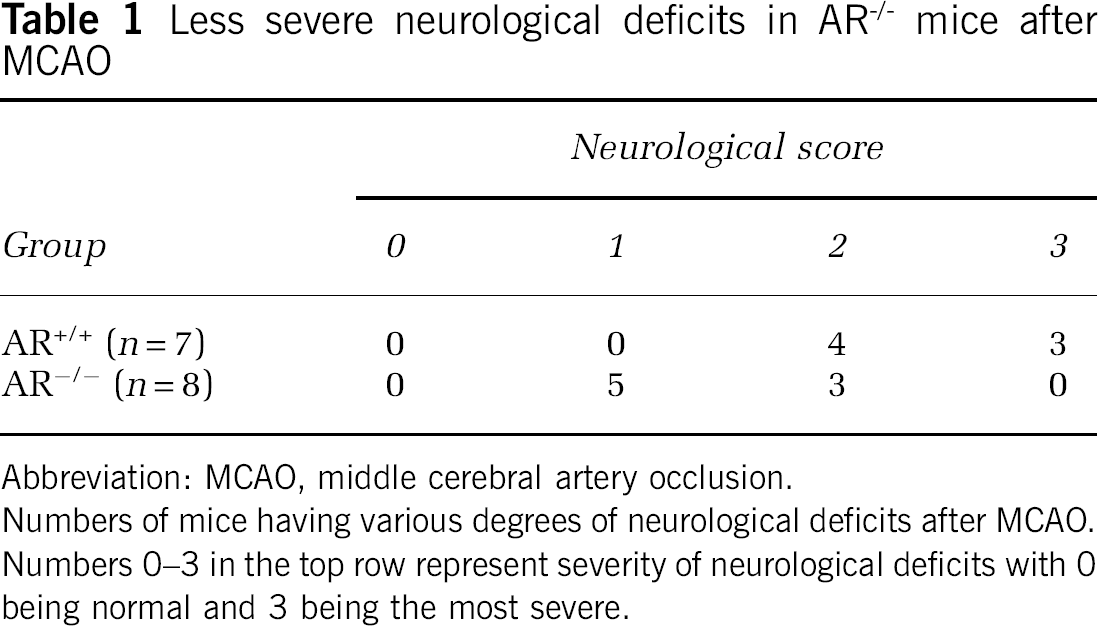
Abbreviation: MCAO, middle cerebral artery occlusion.
Numbers of mice having various degrees of neurological deficits after MCAO.
Numbers 0–3 in the top row represent severity of neurological deficits with 0 being normal and 3 being the most severe.
Moreover, significantly smaller infarct areas were evident in 2,3,5-triphenyltetrazolium chloride-stained brain slices of AR−/− mouse brains (Figure 2C). Infarct area in the brain slices #2, #3, and #4 were significantly smaller in AR−/− mice (9.8 ± 0.5, 11.0 ± 1.0, and 4.3 ± 1.8 mm2 respectively) than in AR+/+ mice (12.0 ± 0.7, 15.0 ± 0.7, and 10.3 ± 2.1 mm2 respectively) (Figure 2D). Accordingly, infarct volume in AR−/− mice was also significantly smaller (52.0 ± 5.8 mm3 versus 78.4 ± 6.1 mm3 Figure 2E) after correction for brain swelling. However, sham-operated mice did not display any infarct (data not shown). In addition, ipsilateral brain swelling in AR−/− brains showed a trend of decrease (12.8% ± 1.2% for AR+/+ mice and 10.7% ± 1.4% for AR−/− brains).
Mutation of Sorbitol Dehydrogenase Gene has no Effect on Cerebral Infarct Size in Mice after Middle Cerebral Artery Occlusion
We showed that the inhibition of AR, the first enzyme of the polyol pathway, is beneficial in reducing infarct size in cerebral ischemic injury. To further understand the involvement of the polyol pathway in cerebral ischemic injury, the importance of SD, the second enzyme in the polyol pathway, was investigated. SD-deficient mice carry a point mutation at the exon 8/intron 8 junction of the SD gene, leading to aberrant mRNA splicing, and therefore prematurely truncated proteins (Lee et al, 1997) resulting in total absence of SD enzyme activity (Ng et al, 1998). Here, SD−/− mice and their wild-type littermates were subjected to MCAO. Surprisingly, SD deficiency did not alleviate or worsen neurological deficits (1.4 ± 0.2, n = 7 versus 1.3 ± 0.2, n = 7 observed in wild-type littermates). Moreover, there was no reduction in infarct area among all brain slices (Figure 2F), infarct volume (Figure 2G), and hemispheric swelling (data not shown).
Aldose Reductase Deficiency Reduced Ischemia-Associated Oxidative Stress and Apoptosis
Because the polyol pathway is implicated in oxidative stress, the level of nitrotyrosine was assessed in brain sections after MCAO. Peroxynitrite is a compound formed by reaction of superoxide with nitric oxide. It is well known that peroxynitrite reacts with tyrosine residues in proteins to form nitrotyrosine in a nitration reaction as a peroxynitrite-specific reaction. Here, formation of nitrotyrosine, an oxidative–nitrosative stress marker (Cheung et al, 2005), was present at a low level in sham-operated mice. Immunoreactivity for nitrotyrosine greatly increased on ischemic damage after MCAO in the AR+/+ mouse brains. In AR+/+ brains, numerous neurons that showed nuclear disruption (e.g., in the CA1 area and periinfarct cortical region) displayed intense immunoreactivity for nitrotyrosine (Figures 3A-1 and 3A-2). However, nitrotyrosine immunoreactivity was much reduced in neurons in ipsilateral hippocampus and periinfarct cortical region of AR−/− brains after MCAO (Figures 3A-3 and 3A-4). Much less nitrotyrosine accumulation appeared in neurons in AR−/− brains when compared with that of the AR+/+ mice (Figures 3A-1 and 3A-3, arrows). After semiquantitative analysis, cell-counting results showed that the percentage of neurons with pyknotic nuclei that were immunoreactive for nitrotyrosine in AR+/+ brains was 89.4% ± 1.2% (n = 3). Similar to the qualitative analysis, the AR−/− brains displayed a significantly decreased number of neurons that were immunoreactive for nitrotyrosine (68.7% ± 0.2%, n = 3, P < 0.0001, t-test), further confirming our observation that nitrotyrosine accumulation was much less in AR−/− brains after MCAO.
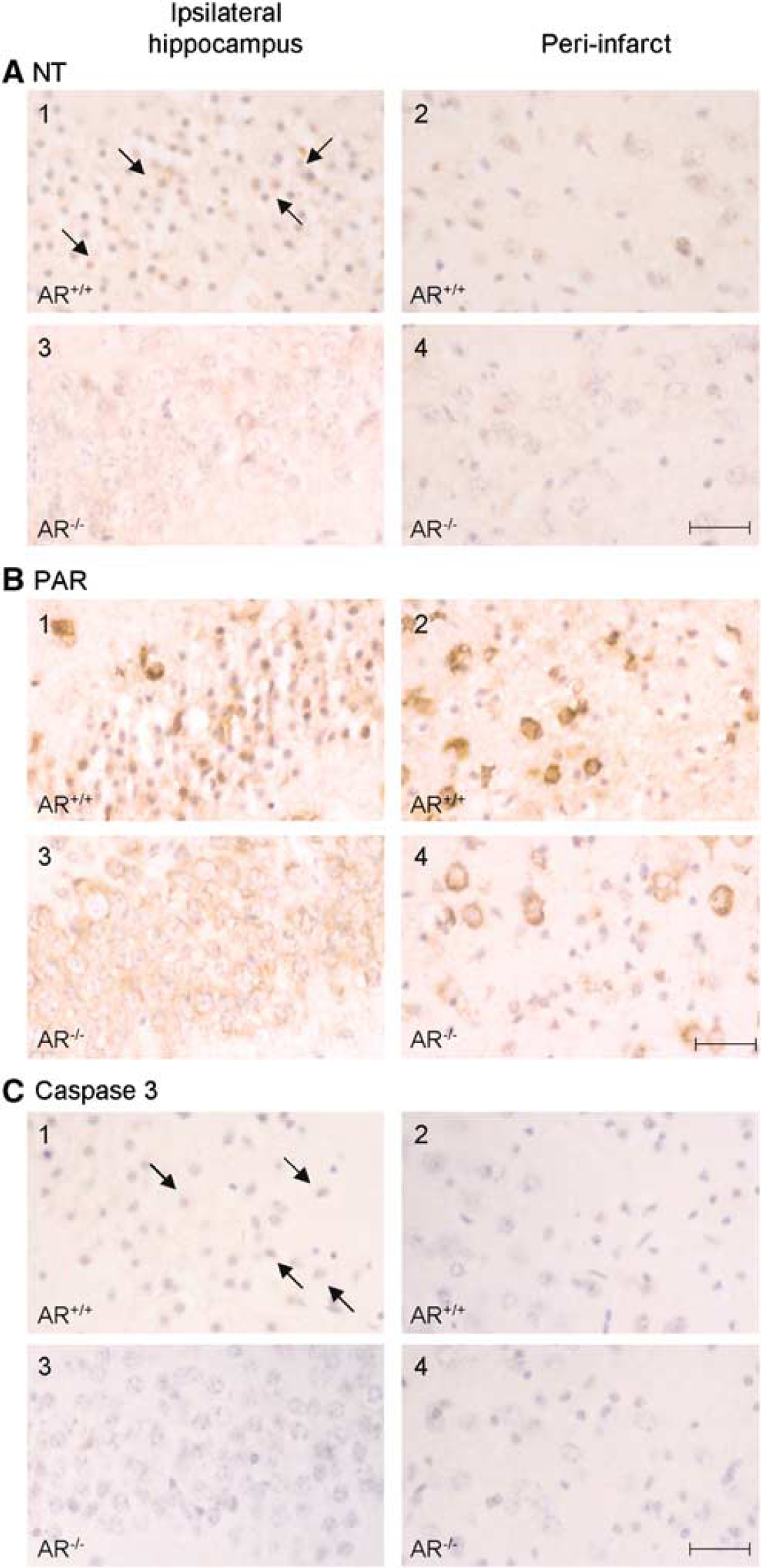
Decreased immunoreactivity for nitrotyrosine, PAR and cleaved caspase-3 indicating decreased oxidative stress in AR−/− mouse brains. Representative photomicrographs from IHC studies using antibodies against (
Since increased nitrosative stress may lead to DNA damage, the level of PAR, the product of PAR polymerase (PARP) that is activated by DNA strand breaks (Burkart et al, 1999), was also assayed. Again, in AR+/+ brains after MCAO, expression of PAR was highly increased in neurons with shrunken nuclei and was concentrated in the perikarya (Figures 3B-1 and 3B-2). However, PAR expression was significantly less intense in the neurons in the AR−/− brains (Figures 3B-3 and 3B-4). The difference in staining intensity became more apparent in the periinfarct region in the cortex, where robust immunoreactivity could be seen in both dead and living neurons.
Apoptotic neurons, as determined by immunohistochemical staining of cleaved caspase-3, were found in the neurons from AR+/+ brains after MCAO (Figures 3C-1 and 3C-2, arrows). However, both the immunoreactivity of cleaved caspase-3 per cell and the number of cleaved caspase-3-positive cells in these regions were less in AR−/− brains (Figures 3C-3 and 3C-4), possibly because of the smaller infarct observed.
Much Less Iron Deposit in AR−/− Brains after Middle Cerebral Artery Occlusion by Iron Histochemistry
Recent evidence has shown that deregulation of brain iron homeostasis plays a role in mediating neuronal damage after ischemic stroke. To determine the extent of iron deposition in AR+/+ and AR−/− mouse brains, iron-related pathology in the brain was investigated histochemically. Brain sections were treated with Perl's solution plus DAB intensification to assay for iron deposits in the brain after MCAO. Under normal conditions, little or no iron deposits were observed in brain of either AR−/− or AR+/+ mice at the level of hippocampus (data not shown) as previously reported (Danielisova et al, 2002; Kondo et al, 1995). After 2-h MCAO and 22-h reperfusion, increased positive, brown staining by Perl's DAB reaction was found. Numerous coarse, dark brown granular deposits were found in neurons with shrunken nuclei in the CA1 area of AR+/+ brains after MCAO (Figure 4A). However, very low amount of very fine, brown deposits were seen in the relatively normal neurons in the CA1 area of AR−/− brains after MCAO (Figure 4B, arrow heads).
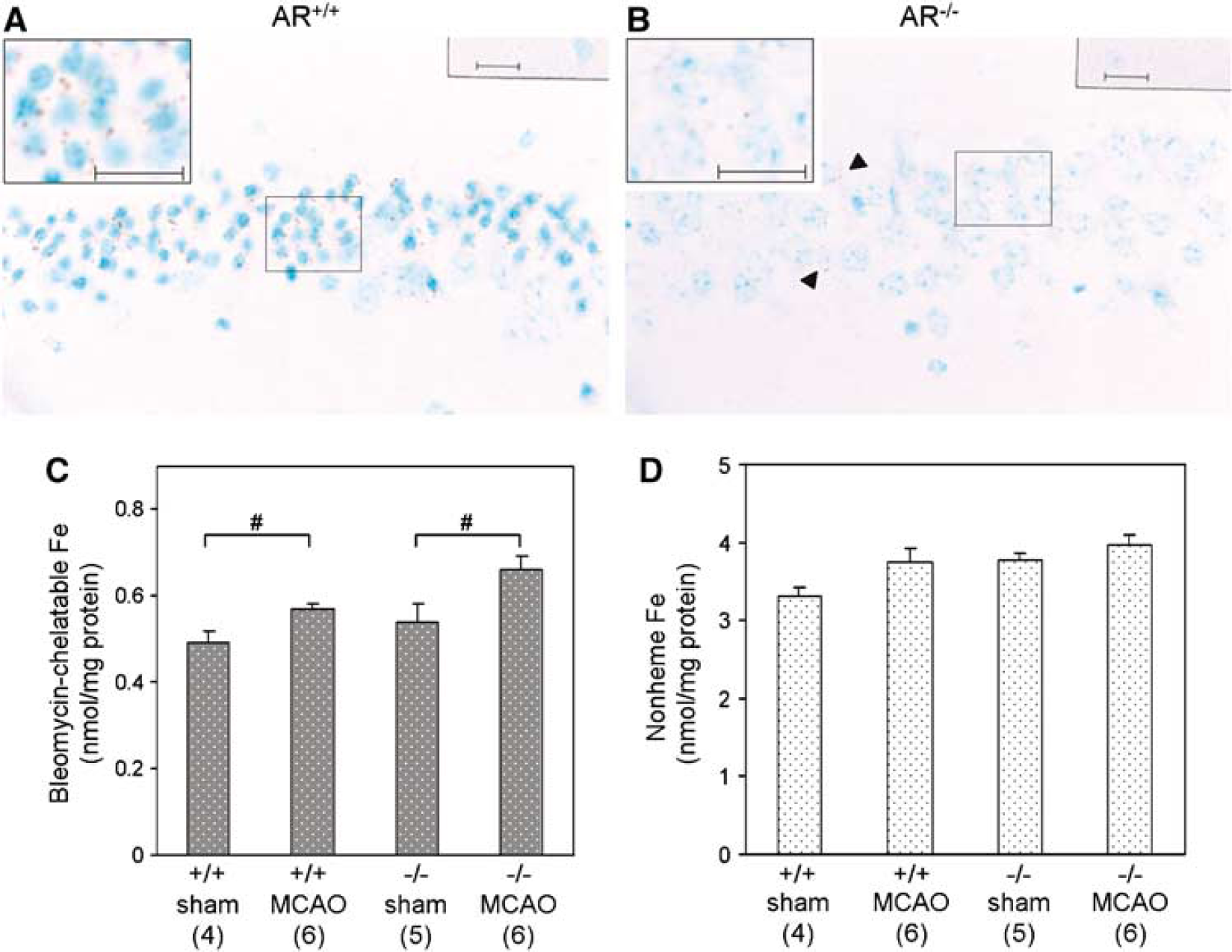
Iron deposits and iron content in AR−/− mouse brains after MCAO. Representative photomicrographs showing iron deposits in (
Higher Bleomycin-Chelatable Iron Levels in AR−/− Mouse Brains after Middle Cerebral Artery Occlusion
Iron levels in mouse brains after MCAO were also quantitatively determined and compared with sham controls. Here, both bleomycin-chelatable and nonheme iron levels were measured. Bleomycin-chelatable iron is widely believed to represent the redoxactive iron pool, whereas the nonheme iron consists of intracellular iron bound to ferritin, hemosiderin, and other proteins such as those in the electron transport chain. Our assay showed that both AR+/+ and AR−/− brains increased their bleomycin-chelatable Fe content after MCAO when compared with their respective sham controls (Figure 4C). Most surprisingly, bleomycin-chelatable iron is higher in AR−/− brains after MCAO (Figure 4C). However, no significant changes could be observed in the nonheme iron pool among all four experimental groups (Figure 4D).
AR−/− Mouse Brains had Lower Cerebral Transferrin and Transferrin Receptor Levels after Middle Cerebral Artery Occlusion
Since transferrin (Tf) levels are also associated with the amount of iron present, Western-blot analyses were used to determine the level of Tf in mouse brains after 2-h MCAO and 22-h reperfusion. There was a very low level of Tf in sham-operated ipsilateral hemispheres (Figure 5A). Transferrin level was then highly induced after MCAO in AR+/+ mice. Similar to AR+/+ ipsilateral hemisphere, the level of Tf was fairly low in sham-operated AR−/− mice but was upregulated after MCAO (Figure 5A). Most importantly, the level of Tf induction in ipsilateral hemisphere of AR−/− mice was significantly decreased than that in AR+/+ mice.
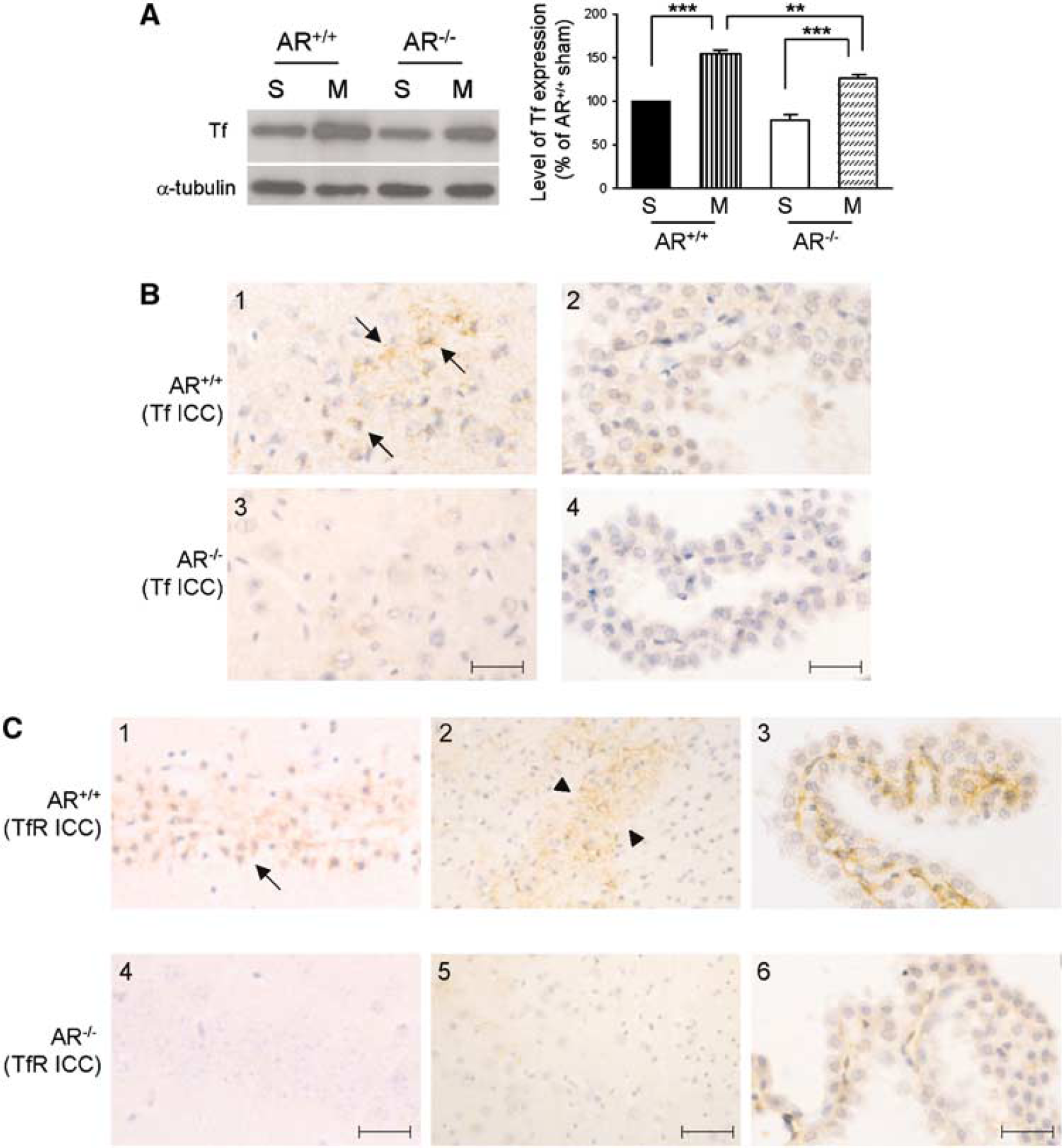
Decreased transferrin and transferrin receptor levels in AR−/− mouse brains after MCAO. (
Immunohistochemistry results for Tf also correlated with the Western-blot findings. Generally, Tf immunoreactivity was stronger in AR+/+ brains after cerebral ischemia induced by MCAO. As described above, shrinkage of neuronal nuclei was more common and abundant in AR+/+ mouse brains, reflecting a more severe infarct in these brains. In particular, Tf immunoreactivity was found in neurons with shrunken nuclei and in neuropil where vacuolation could be seen (Figure 5B-1, arrows). However, the intensity of immunostaining in similar area in AR−/− brains was significantly weaker (compare Figures 5B-1 and 5B-3). Besides, some signals were also found in the choroid plexus in both groups of mice, particularly in the cytoplasm of epithelial cells (Figures 5B-2 and 5B-4). Again, weaker Tf immunostaining was observed in the choroids plexus of AR−/− brain compared with that of AR+/+ brains (Figure 5B-4).
Since transferrin receptor (TfR) is required for Tf-mediated iron transport, the cellular distribution of TfR in both AR+/+ and AR−/− mouse brains after MCAO was also examined. Immunohistochemistry studies using antibody against TfR revealed that increased immunoreactivity was seen in shrunken neurons in the infarct areas. One example was the dead/dying neurons with shrunken nuclei in the CA1 hippocampal region (Figure 5C-1, arrows), and the periinfarct region in the cortical area where there was vacuolation of the neuropil (Figure 5C-2, arrow heads). In AR−/− brains, a similar staining pattern was found except that much fewer neurons were seen with TfR immunoreactivity. Also the staining intensity was much weaker when compared with that observed in AR+/+ brains. Figure 5C-4 showed the neurons in the CA1 area of AR−/− mouse brains with normal morphology that displayed little/no TfR immunoreactivity after MCAO. Moreover, little TfR immunoreactivity could be observed in periinfarct area in these brains (Figure 5C-5). In addition, signals for TfR also resided in endothelial cells inside the choroids plexus in AR+/+ brains after MCAO (Figure 5C-3). Again, the intensity of TfR immunoreactivity in these cells was of much lower intensity in the AR−/− brains (Figure 5C-6).
Administration of Aldose Reductase Inhibitor Reduced Infarct Size
To further confirm our result that deletion of AR activity is beneficial to the animal after cerebral ischemia, an AR inhibitor, Fidarestat, was administered to normal C57BL/6N mice that underwent MCAO. Similar to our results with AR−/− mice, administration of Fidarestat to normal C57BL/6N mice improved the postischemic outcome. When Fidarestat was given at 30 mins before induction of ischemia, the neurological score was better (1.2 ± 0.1, n = 9, compared with 1.8 ± 0.1 in vehicle-treated mice, n = 9, P < 0.05, Mann–Whitney test). Similarly, when Fidarestat was administered at 1 h and 45 mins after ischemia, neurological deficits were diminished in Fidarestat-treated mice (1.2 ± 0.2, n = 9, compared with 2.0 ± 0.2 in vehicle-treated mice, n = 9, P < 0.02, Mann–Whitney test). In parallel, infarct size was significantly reduced (Figure 6) after Fidarestat administration at both time points. Fidarestat treatment decreased the infarct area of brain slice #4 in both experiments and that of brain slice #3 when administered 1 h and 45 mins after ischemia (Figures 6A and 6C), while reducing the infarct volume at the same time (Figures 6B and 6D). However, hemispheric swelling was not significantly reduced (data not shown).
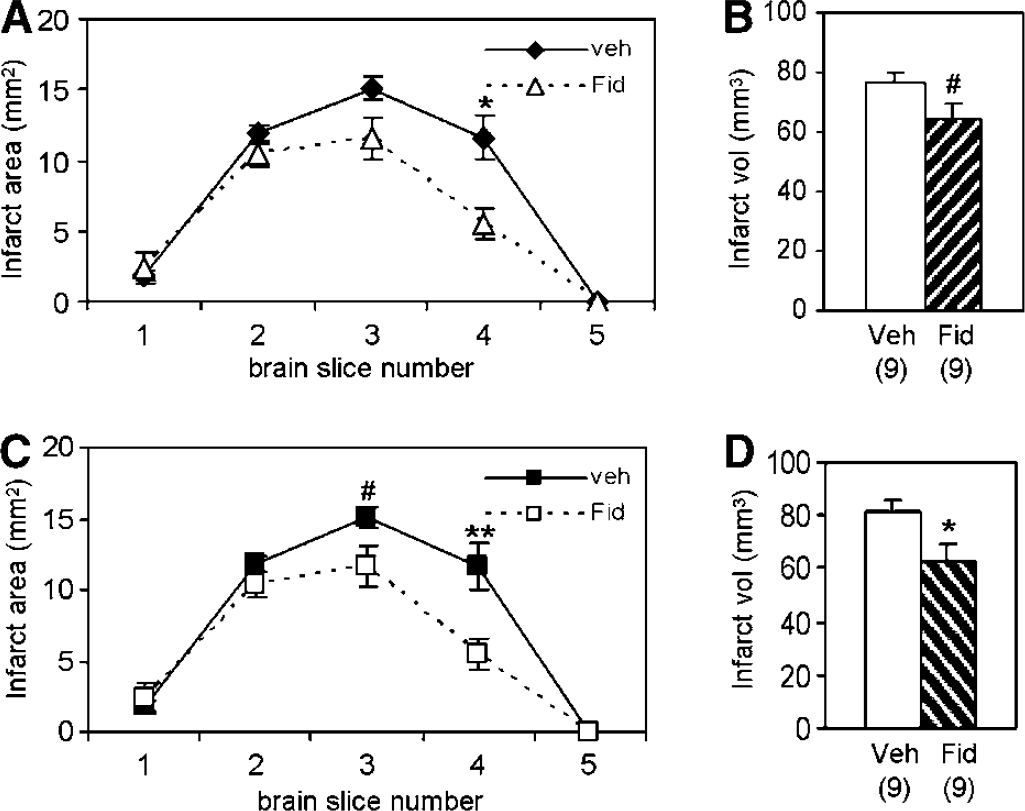
Mice treated with Fidarestat displayed smaller infarct size. Fidarestat was administered at 30 mins before ischemia (
Discussion
As described above, AR protein level was elevated in GET-1 brains when compared with nontransgenic brains after MCAO, suggesting that this increased AR level was related to astrocytic ET-1 overexpression, which led to more severe neurological deficit with increased cerebral edema and infarct. Previous studies showed that the transcription of AR is regulated by osmotic response element binding protein (TonEBP/NFAT5/OREBP), a transcription factor, which also regulates transcription of several ischemic injury-related genes, such as HSP70, SMIT, and TNFα (Lopez-Rodriguez et al, 2001; Rim et al, 1998; Woo et al, 2002). The fact that TonEBP/NFAT5/OREBP is significantly expressed in neurons and some glial cells suggests that nerve cells can accumulate compatible osmolytes (Loyher et al, 2004) by changing their expression level or translocation. It is possible that the flux through the polyol pathway can lead to elevation of intracellular sorbitol and therefore increasing intracellular hyperosmotic pressure, drawing more water into neurons, and resulting in an increased cytotoxic edema; thereby contributing to larger infarct in GET-1 brains after MCAO. It is also possible that ET-1-mediated activation of AR under ischemic condition and the consequent flux through SD can further increase oxidative stress by altering the redox state and activation of NADH oxidase (Chung and Chung, 2005), which may lead to accumulation of more oxygen free radicals contributing to ischemic injury. In agreement with such notion that AR activation in brain of wild-type mice after MCAO is detrimental to brain, the deletion of AR in AR−/− mice improved neurological score and smaller infarct after MCAO. Previously, AR is also shown to be implicated in ischemic myocardial injury. Aldose reductase activity was increased in ischemic heart (Hwang et al, 2002), whereas AR inhibition was cardioprotective both in vitro and in vivo (Hwang et al, 2002, 2004; Tracey et al, 2000). Moreover, the result that absence of AR is beneficial to the brain after ischemia is further confirmed by administration of Fidarestat, an AR inhibitor, to C57BL/6N mice in our experiments. Blocking AR activity at either 30 mins before ischemia or 1 h and 45 mins after ischemia by Fidarestat can reduce neurological deficits and infarct size, in agreement with the results observed in AR−/− brains. However, the finding that ipsilateral hemispheric swelling did not significantly decrease after MCAO on AR deficiency suggested that the activation of AR in GET-1 brains after MCAO was not a major contribution to increased water content and therefore swelling was observed in GET-1 brains.
Interestingly, deletion of SD did not protect the mice from ischemic injury, suggesting that the flux through AR and its related pathway, such as depletion of nicotinamide adenosine dinucleotide phosphate is responsible for ischemic injury. It has been proposed that the conversion of sorbitol to fructose in the second step of the polyol pathway leads to a reduction in the level of NAD+ the cofactor for SD, thereby creating a pseudohypoxic state by increasing the NADH-to-NAD+ ratio (Williamson et al, 1993). Moreover, increased sorbitol pathway activity has been shown to generate oxidative stress in diabetic complications (Ho et al, 2006). Sorbitol dehydrogenase can increase cytosolic NADH/NAD+ ratio, triggering excess production of reactive oxygen species such as superoxide radical (O•−2) through the reaction of NADH with NADH oxidase, and thereby resulting in oxidative stress. However, the lack of reduction in cerebral infarct size after MCAO in SD−/− mice indicates that this second step of the polyol pathway and its associated biochemical consequences, such as lowering of NAD+ levels, appears to contribute less to cerebral ischemic injury. This is somewhat surprising since it has been shown that SD inhibition using an SD inhibitor, CP470711, can protect ischemic myocardium (Hwang et al, 2003). However, our previous studies on SD−/− mice in experimental diabetes suggested that SD is not a major culprit in the pathogenesis of diabetic neuropathy (Ng et al, 1998).
It appears that AR itself contributes to oxidative stress after ischemic/reperfusions since the expression of PAR and nitrotyrosine, indicators of increased oxidative or nitrosative stress, is decreased in AR−/− brains after MCAO. Increased reactive free radicals would cause DNA damage, leading to the activation of PARP to synthesize more PAR. Deletion or inhibition of AR has been shown to attenuate the diabetes-induced increase in PAR and nitrotyrosine (Cheung et al, 2005). Recently, we also showed that AR−/− db/db mice accumulated less PAR and nitrotyrosine than AR+/+ db/db mice with fewer cleaved caspase-3-positive cells in the retinal ganglion cell and inner nuclear layer (Cheung et al, 2005). Similarly, our data on the AR−/− brains showed that there is less accumulation of PAR and nitrotyrosine. Moreover, fewer cleaved caspase-3-positive cells are present, suggesting less apoptotic cells in AR−/− mice. Increased AR activity can also contribute to oxidative stress by promoting glycation and protein kinase C activation (Chung and Chung, 2003). Furthermore, AR-mediated oxidative stress can lead to detrimental consequences, such as mitogen-activated protein kinase activation and PARP activation (Ho et al, 2006). Our results also showed that less Jun NH2-terminal kinase activation and reduction in nerve conduction were observed in AR−/− diabetic nerve (Ho et al, 2006), whereas inhibition of Jun NH2-terminal kinase reduced the infarct volume and improved the functional outcome (Hirt et al, 2004).
Injury was observed in the hippocampus, which is not part of the middle cerebral artery territory, of AR wild-type mouse brains after MCAO. One of the explanations is that occlusion of the internal carotid artery during MCAO may reduce the blood supply to the hippocampus, as the blood supply to the hippocampus partially originates from the internal carotid artery via the posterior-communicating artery in mice (Barone et al, 1993). In fact, it has been shown that C57BL/6 mice have hypoplastic posterior-communicating arteries, and therefore incomplete Circle of Willis making them more susceptible to transient cerebral ischemia (Fujii et al, 1997). In our AR studies, all mice were of C57BL/6 background as the AR−/− mice were backcrossed to the C57BL/6N strain to the 11th generation (N11), and therefore C57BL/6N mice were used as controls. Secondly, it is also possible that breakdown of the blood–brain barrier in the ipsilateral side contributed to a widespread brain damage after transient ischemia (States et al, 1996).
Our results also showed that the choroids plexus was affected as well after MCAO. The anterior choroidal artery, which supplies the choroids plexus in the lateral ventricle, arises from the internal carotid artery, and as a variation may also arise from the posterior-communicating artery (Baskaya et al, 2004). In either case, blood supply in the anterior choroidal artery may be reduced by occlusion of the internal carotid artery or anomaly of the posterior communicating artery.
In addition, our studies showed that upregulation of cerebral Tf and TfR after transient ischemia as seen in AR+/+ mice was smaller in AR−/− brain, suggesting an involvement of AR in brain iron homeostasis associated with oxidative damage in cerebral ischemia. Previous studies showed that a period of brain ischemia leads to increased TfR expression (Omori et al, 2003), probably as a result of increased expression of hypoxia inducible factor-1α that can increase TfR expression (Lok and Ponka, 1999). Indeed, hypoxia inducible factor-1α may also increase TfR expression on neurons, thereby worsening the problem (Moos and Morgan, 2004). Here, our data showed that TfR upregulation is less in AR−/− brain. Whether hypoxia inducible factor-1α plays a role in the less severe phenotype seen in AR−/− mice after MCAO remains to be determined.
During ischemia, lactate acidosis occurs leading to decreased pH. Ferric iron (Fe3+) become unbound from Tf resulting in extracellular Fe3+ accumulation, which may upregulate expression of Tf and TfR. This extracellular iron can in turn be taken up by neurons, and it is reflected in the observed increased iron deposits in dead neurons of the AR+/+ brains after MCAO. In addition, low pH can facilitate the reduction of intracellular ferritinbound Fe3+ to Fe2+ on its release, causing increased production of hydroxyl radicals (OH•) by the Fenton reaction (Fe2+ + H2O2 → Fe3+ + OH + OH−) (Selim and Ratan, 2004). OH• is extremely toxic to neuronal cell membrane and can lead to oxidative stress and subsequent cell death.
In AR−/− brains after MCAO, Perl's staining results indicate that less iron deposits (in the form of Fe3+) are present, suggesting that the extent of the Fenton reaction is smaller, and in turn a lower production of hydroxyl radicals (OH•) indicative of less oxidative stress. This is also reflected in our IHC experiments on expression of nitrotyrosine and PAR, in which AR−/− brains showed much less intense immunoreactivity. Because the extent of the Fenton reaction is less, we anticipate that a smaller amount of Fe2+ is converted to Fe3+ leading to increased Fe2+ as shown in our bleomycin experiment, again suggesting less oxidative stress in AR−/− brain after MCAO. Similarly, bleomycin-chelatable iron is actually higher in mouse tissues under calorie restriction (Sohal et al, 1999), a condition that can reduce oxidative stress and stimulate mitochondrial proliferation at the same time (Lopez-Lluch et al, 2006) and is found to ameliorate the oxidative and inflammatory effects of diabetes in the brain (Ugochukwu et al, 2006). It is also possible that H2O2 can be shuttled and metabolized to nontoxic products (H2O and O2) by glutathione sulfhydryl peroxidase under AR deficiency where there is more glutathione sulfhydryl supply. Indeed, our previous studies showed that in AR−/− mice, diabetes did not lead to any decrease in the nerve glutathione sulfhydryl level (Chung and Chung, 2003; Ho et al, 2006).
Taken together, our novel finding that cerebral ischemia induced AR level in the brain and deletion of AR protects the animals from oxidative stress and more severe neurological deficit and larger infarct suggest that increased AR observed in our GET-1 mice may contribute to more severe neurological deficit and larger infarct via oxidative stress. The fact that SD did not protect the animals against cerebral ischemia/reperfusion suggest that AR is a key culprit in ischemic brain injury, and provide a basis for evaluating pharmacological interventions of AR as novel therapeutic approach for treating stroke patients.
Footnotes
Acknowledgements
The author Justin Leung for his help in drug treatment, Oi Fung Cheung, and James Lau for their technical help in histology work, Alfred Chan for providing SD−/− mice as well as Yuan Zhou for her excellent assistance in 2-DE.