
Abstract
We evaluated cerebral blood flow by laser Doppler during 30 secs of hypoxia (0.10 FiO2) in anesthetized, ventilated adenosine 2a receptor knockout (A2aR KO) and wild-type (WT) mice to test the hypothesis that cerebral hypoxic hyperemia in KO mice would be attenuated. We also studied the effects of selective and nonselective A2aR antagonists. During 30 secs of hypoxia, PaO2 decreased significantly (P < 0.05) and to a similar degree in both types of mice, whereas PaCO2 remained relatively stable. However, mean arterial blood pressure (MABP) decreased to a greater extent (P < 0.05) during hypoxia in KO mice (58.6 ± 1.5 mm Hg) than in WT animals (76.1 ± 3.2 mm Hg). Consequently, in a separate group of mice, we stabilized and matched MABP during hypoxia. Hypoxic hyperemia was attenuated by 38% (P < 0.05) in KO animals whose MABP was uncontrolled, and by 81% (P < 0.05) in KO animals whose MABP changes were matched to the MABP in the hypoxic WT mice. In animals treated with adenosine antagonists, hypoxic hyperemia was decreased by 44% to 48% (P < 0.05) in WT mice, but was without effect in KO mice. We conclude that adenosine via A2aR is responsible for a significant proportion of the hyperemia during hypoxia.
Keywords
Introduction
A variety of potential factors have been suggested as being the chemical substance regulating cerebral blood flow (CBF) during hypoxia. Adenosine, a breakdown product of adenine nucleotides, has emerged as a candidate for this chemical link because of its potency as a cerebral vasodilator (Berne et al, 1974; Ibayashi et al, 1991; Ngai and Winn, 1993; Ngai et al, 2001). Furthermore, concentrations of adenosine in brain, interstitial fluid, and cerebral spinal fluid are increased during acute and prolonged hypoxic hypoxia (Meno et al, 1993; Rubio et al, 1975; Winn et al, 1981b; Zetterstrom et al, 1982). Recently, Martin et al (2007) have shown that adenosine is produced by hypoxic astrocytes. The latter, which links the neuron and the cerebral vasculature, are a critical component of the neurovascular unit. Although brain adenosine concentrations during hypoxia have been studied in multiple species, there is limited information regarding the changes in brain adenosine concentrations during hypoxia in mice (Aden et al, 2003; Ohta et al, 2006), and none in studies of CBF using knockout (KO) animals.
The actions of adenosine are mediated through extracellular receptors, of which four subtypes (A-1 receptor (R), A2aR, A2bR, and A-3R) have been characterized (Fredholm et al, 2001). Out of these four subtypes, the high-affinity A2aR appears to be the primary receptor responsible for cerebrovascular dilatation, with a possible lesser contribution from the low-affinity A2bR. Stimulation of the abluminal, but not the luminal (Ngai and Winn, 1993), A2R results in vasodilatation, both in isolated, perfused, penetrating cerebral arterioles in vitro (Ngai et al, 2001) and in perfused pial arterioles in vivo (Ibayashi et al, 1991).
Many investigators have used adenosine receptor agonists and antagonists in an attempt to define the role of adenosine in the regulation of CBF. The most widely used drugs are the methylxanthines, such as caffeine, theophylline, and aminophylline (theophylline dissolved in ethylenediamine, 2:1), which are nonspecific (A1 and A2) adenosine receptor antagonists and appear to minimally affect A2bR and A3R subtypes (Fredholm et al, 2001). Earlier studies have shown that during acute (∼30 secs) (Morii et al, 1987) and prolonged (Emerson and Raymond, 1981; Morii et al, 1987; Phillis et al, 1984) hypoxia, methylxanthines attenuate hypoxic hyperemia. Recently, more specific adenosine receptor drugs (i.e., A2a) have become available and have been used to further define the physiology of adenosine related vasodilatation. However, the absence of effective A2b agonists precludes precise definition of the contribution of A2bR. In contrast, direct A-1 and A-3 stimulation of in vitro cerebral arterioles is without effect (Ngai et al, 2001). Irrespective of the pharmacologic specificity of adenosine antagonists, the in vivo application of both nonspecific and specific adenosine drugs may be problematic because of dose-related effects, unrecognized actions, and/or confounding influences on the noncerebral circulation. The use of adenosine receptor KO animals may overcome these pitfalls.
Consequently, we evaluated the in vivo response of the cerebral circulation during transient (30 secs) systemic hypoxia in A2aR KO mice and compared this response with that in wild-type (WT) animals. We hypothesized that in KO animals, hypoxic hyperemia would be attenuated. We also studied the effects of the specific A2aR antagonist ZM-241385 (4-(2-(7-amino-2-(2-furyl)(3,2,4)triazolol(2,3-a)(1,3,5)triazin-5-yl-amino) ethyl)phenol), and the nonspecific antagonist aminophylline, in both WT and KO mice.
Materials and methods
Animals
Adenosine 2aR C57Bl/6 KO mice were genetically engineered by Chen et al (1999). Homozygous A2aR KO mice and C57Bl/6 WT mice were generated by homozygous interbreeding using breeding pairs from University of Virginia (Joel Linden). This study was approved by the Animal Care Committee of the Mount Sinai School of Medicine.
Surgical Procedures
Mice weighing 22 to 31 g were initially anesthetized in a chamber with 5% isoflurane, intubated with 20 G intravenous catheter, and ventilated. Anesthesia was maintained during the procedure with 1.1% to 1.7% isoflurane in ∼35% O2 balanced with nitrogen; end-tidal CO2 (etCO2) was continuously monitored. Because the small intravascular volume (2 to 3 mL) in mice precluded multiple arterial sampling (100 μL) for measurement of arterial blood gases, we performed preliminary studies in a separate group (n = 15) of anesthetized, ventilated mice, in which we simultaneously measured PaCO2 and etCO2 and established that PaCO2 = 1.48 (etCO2) + 3.8. Therefore, our targeted baseline etCO2 value was ∼25 mm Hg.
All surgical sites were infiltrated with 1% lidocaine. The temperature was monitored by a rectal probe and maintained at 37°C. In all animals, the right femoral artery was cannulated with PE-10 (i.d., 0.28 mm) tube for continuous blood pressure monitoring and for sampling blood for arterial blood gases and pH. The right femoral vein was also cannulated for administration of drugs. Throughout the experiment, pancuronium bromide was administered intraperitoneally at 0.4 mg/kg q45 mins.
After securing the arterial line, the animal's head was fixed to a stereotactic frame and the scalp was incised along the midline to expose the bone. The bone over the right parietal bone was thinned with a high-speed drill. Then, a laser-Doppler probe (Periflux 5000; Perimed, Järfälla, Sweden) was placed over the thinned skull to allow measurement of CBF. Alpha-chloralose was administered intraperitoneally (75 mg/kg, q1 h) and isoflurane was gradually discontinued. The level of anesthesia was repetitively assessed by tail flick, paw withdrawal, and analysis of physiologic parameters, and anesthesia was supplemented to assure that the animal was not in pain.
In a separate group of animals (groups B to F, see below), we altered the blood pressure by adjusting the intravascular volume before (groups E and F, see below) or during hypoxia (groups B and C) to match the mean arterial blood pressure (MABP) in the comparison animals by either withdrawal or transfusion of blood of varying volumes (100 to 300 μL).
Experimental Protocol
One hour after isoflurane was discontinued, each animal was ventilated with 7% CO2 (in 40% O2 balanced with nitrogen) for 60 secs and then allowed to recover for 5 mins during which all physiologic parameters returned to baseline. The hypercarbic challenge was then repeated. Animals having greater than 10% variation between their CO2 challenges, as determined by laser-Doppler flowmetry, were discarded. Then each animal was ventilated with 10% O2 (balanced with nitrogen) for 30 secs and CBF response assessed by laser-Doppler flowmetry. The animals were then allowed to recover for 5 mins during which all physiologic parameters returned to baseline. The hypoxic challenge was then repeated. Animals having greater than 10% variation in their CBF response, as determined by laser-Doppler flowmetry, were discarded. In animals used to assess the effect of adenosine antagonists (ZM-241385, intravenously, or aminophylline, intraperitoneally) during hypoxia, the drugs, diluent, or saline were administered, and after 20 mins, the hypercarbic and hypoxic challenges repeated (x 2) as described previously.
Our initial comparison of the CBF response during hypoxia in the WT and A2aR KO mice (group A, see below) was done with the experimentalist blinded to the genotype of the mouse. Consequently, at the end of the four steps outlined above, we administered CGS-21680 (0.1 mg/kg), an A2aR agonist, to uncover the genotype and to confirm the presence/absence of functional A2aR.
During hypoxia, the WT animals maintained their MABP within the autoregulatory range, whereas the KO mice decreased their MABP significantly. Consequently, in a separate group of animals (group B), we matched the MABP in the KO and WT mice during hypoxia by augmentation or withdrawal of intravascular volume. In addition, because of alterations in MABP in the animals treated with adenosine antagonists, we also created four additional groups (groups C to F; see Table 2), in which we matched the MABP before (groups E and F) or during hypoxia (group C). In total, we analyzed the following six groups (see Table 2):
Physiological parameters before, during and after hypoxia
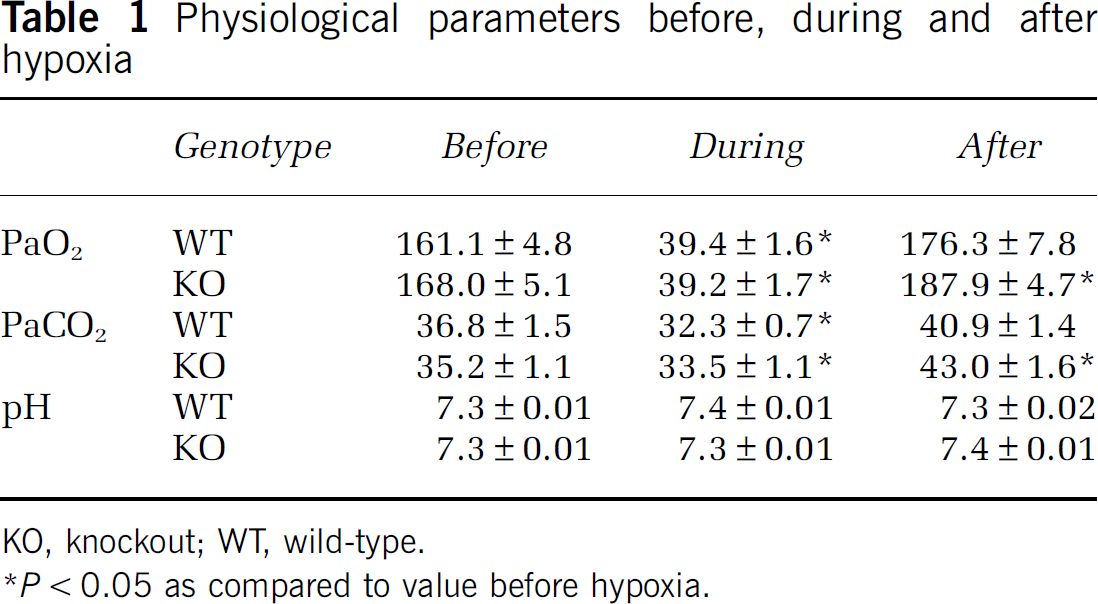
KO, knockout; WT, wild-type.
P < 0.05 as compared to value before hypoxia.
MABP, before and during hypoxia; CBF, % change during hypoxia
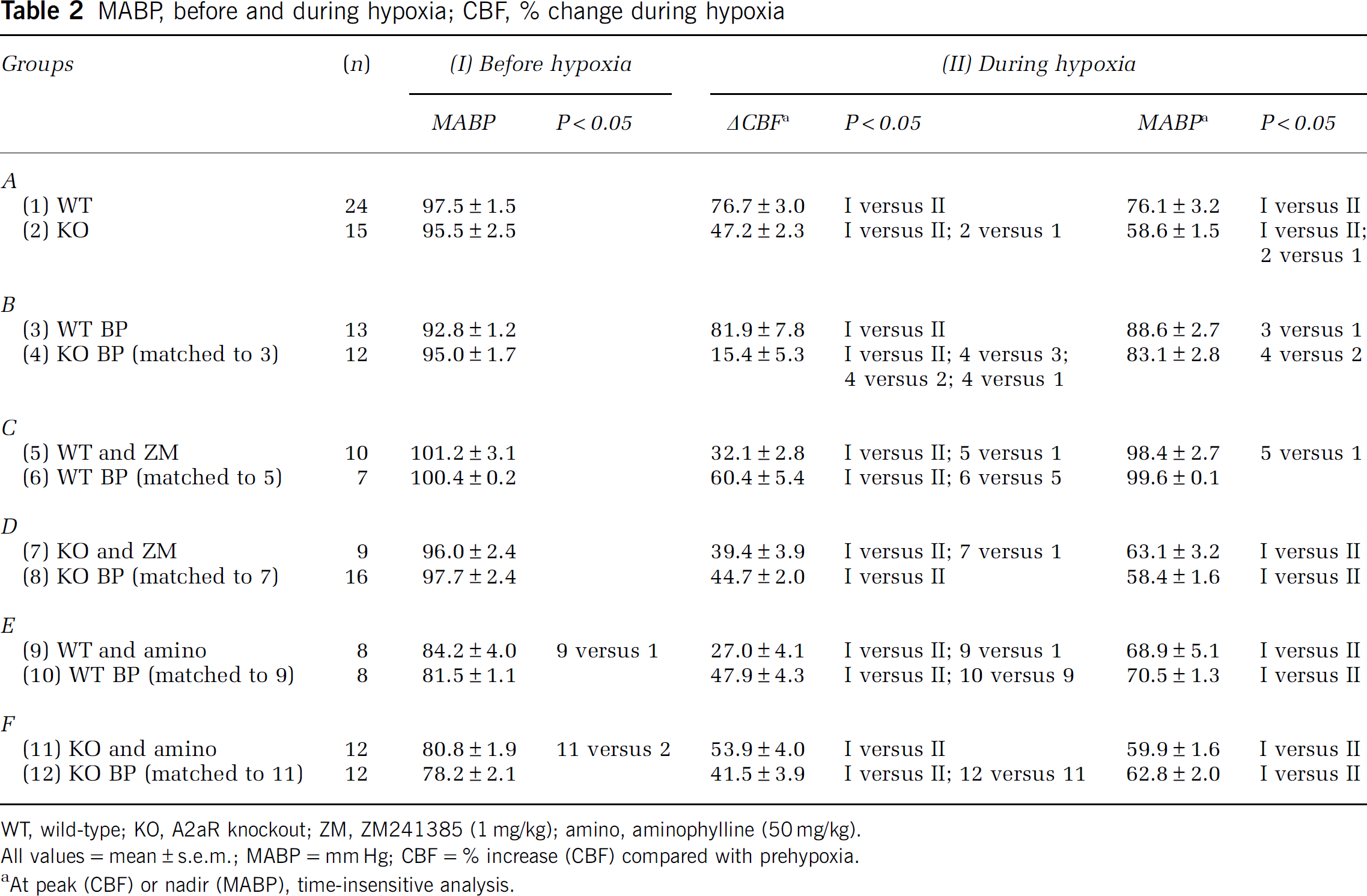
WT, wild-type; KO, A2aR knockout; ZM, ZM241385 (1 mg/kg); amino, aminophylline (50 mg/kg).
All values = mean ± s.e.m.; MABP = mm Hg; CBF = % increase (CBF) compared with prehypoxia.
At peak (CBF) or nadir (MABP), time-insensitive analysis.
Group A (row 1 versus 2): WT versus A2aR KO; no MABP manipulation.
Group B (row 3 versus 4): During hypoxia, we attenuated the decrease in MABP in KO mice to match the MABP changes in the WT animals.
Group C (row 5 versus 6): ZM-241385; during hypoxia, we maintained a stable MABP (i.e., prevented hypotension) in WT mice to match the stable MABP in the ZM group.
Group D (row 11 versus 12): ZM-241385; the MABPs in KO-ZM and KO animals before and during hypoxia were not different. Consequently, no MABP interventions were performed in this group of mice.
Group E (row 5 versus 6): Aminophylline; during the prehypoxic period, we lowered MABP in the WT mice to match the prehypoxic MABP in the WT-aminophylline animals.
Group F (row 7 versus 8): Aminophylline; during the prehypoxic period, we lowered MABP in the KO mice to match the prehypoxic MABP in the KO-aminophylline animals.
Preliminary Studies
We also performed the following preliminary studies:
Time controls: In WT and KO mice, we documented that CBF responses to repetitive periods of hypoxia were stable despite the hypoxic challenges being separated by hours.
Adenosine antagonists—preparation and dosage: ZM-241385. We based our preparation and dose (1 mg/kg, intravenously) on our earlier studies in rats, in which we found that systemic application of ZM-241385 attenuated pial arteriolar dilatation evoked by topical application of adenosine (Meno et al, 2001) or glutamate (Iliff et al, 2003), or during sensory cortical activation caused by contralateral sciatic nerve stimulation (Meno et al, 2001). ZM-241385 had no effect on resting MABP in either WT or KO animals. Previous studies with a vehicle for ZM-241385 did not report any alteration in CBF (Meno et al, 2001).
To show the biologic effectiveness of ZM-241385, we administered 0.1 mg/kg of CGS-21680 (A2a agonist) in the absence (equal volume of saline, intravenously) and presence of ZM-241385 (1 mg/kg). In the absence of ZM-241385, CGS-21680 caused the WT mice to become rapidly hypotensive and tachycardic. With prior administration of ZM-241385, this cardiovascular response to CGS-21680 was blocked. In contrast, CGS-21680 had no cardiovascular effect in the KO mice. Further evidence of the biologic activity of our ZM-241385 preparation was the attenuation of hypotension during hypoxia in WT animals, a previously described activity related to cardiac A2aR (Koos and Maeda, 2001).
Theophylline. Because the solubility of theophylline in saline is not high and the intravascular volume of the mouse is small, we chose to use the more soluble preparation of aminophylline (theophylline and ethylenediamine in a 2:1 ratio). We used 50 mg/kg of aminophylline (or 37.5 mg of theophylline or 0.2 μmol/g) based on our earlier studies of theophylline in rats, which revealed that this dose functioned as an adenosine antagonist in the brain, had no effect on the brain concentrations of cyclic AMP, and attenuated hypoxic hyperemia (Morii et al, 1987). Aminophylline (50 mg/kg) in both the WT and KO mice caused a slight, but statistically significant, decrease in MABP (Figure 1). This decrease in MABP was similar to the downward trend observed in the rat after an equivalent dose (0.2 μmol/g) of theophylline (Morii et al, 1987). Higher doses of aminophylline decreased MABP further in a dose-dependent manner (Figure 1). When we plotted MABP versus the dose of aminophylline, we noted a similar relationship in the WT and KO mice. Administration in increasing concentrations of the diluent contained in aminophylline (ethylenediamine) did not result in a decrease in MABP.
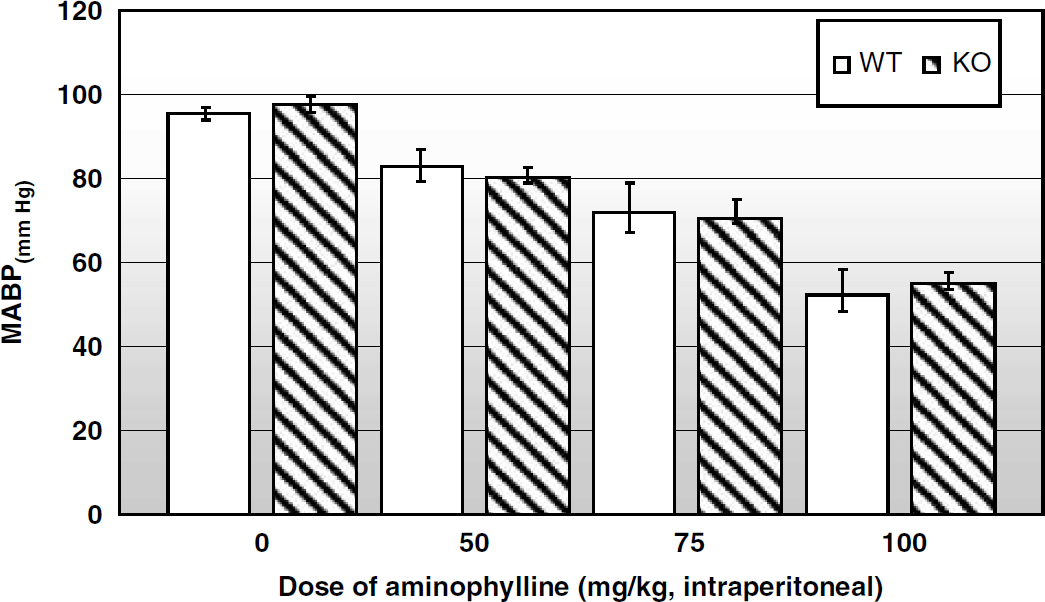
Effect of increasing dose of aminophylline (mg/kg, intraperitoneally) on mean arterial blood pressure (MABP). WT: n = 15; *P < 0.0.05 compared with 0 mg/kg (Bonferroni correction); KO: n = 13; *P < 0.0.05 compared with 0 mg/kg (Bonferroni correction).
Changes in PaO2, PaCO2, and pH before, during, and after 30 secs of hypoxia: Because the small intravascular volumes in mice precluded repetitive and rapid sampling of arterial blood gases during hypoxia, we performed a series of preliminary studies to document the changes in physiologic parameters in the WT and KO mice. As illustrated in Table 1, there were no differences in the response of the WT (n = 12) and the KO (n = 13) mice to transient hypoxia.
Data Analysis
The data were acquired with a Digidata 1322A digitizer (Axon CNS, Union City, CA, USA) and analyzed with the SPSS 14.0 data analysis package (SPSS Inc., Chicago, IL, USA). All values are expressed as means ± s.e.m. We analyzed CBF with two methods: peak flow (time insensitive or ti) and averaged peak flow (time sensitive or ts). In the former method, we measured the maximal flow in each subgroup irrespective of the time of onset of hypoxia, whereas in the latter method, we averaged flow by time after we changed the inspired O2. There was a delay between switching the source gases and when the mouse was actually exposed to the inspired gases. This delay was related to a number of factors, including the length of the respiratory tubing and the respiratory rate. In general, the CBF (ti) was slightly higher than CBF (ts). The duration of hypoxia was maintained at a constant value of 30 secs. Independent t-tests and analysis of covariance were used to make statistical comparisons. When multiple simultaneous comparisons were made, a Bonferroni correction was applied. A P-value of < 0.05 was used to indicate statistically significant differences.
Drugs
Aminophylline (Abbott Laboratories Inc., Abbott Park, IL, USA); ZM-241385 (Sigma Aldrich, St Louis, MO, USA); CGS-21680 (Sigma Aldrich); pancuronium bromide (Gensia Sicor Pharmaceuticals, Irvine, CA, USA); Lidocaine (Hospira Inc., Lake Forest, IL, USA); Isoflurane (Baxter Corp., Deerfield, IL, USA); and Alpha-chloralose (Fischer Scientific, St Louis, MO, USA).
Results
Hypoxia
Physiologic parameters: Initial MABP measurements in our A2aR KO mice were comparable to those noted by Chen et al (1999).
Prehypoxia period. There were no differences in MABP in WT and KO mice (Table 2) without (groups A and B) or with treatment of adenosine receptor antagonists ZM-241385 (group C or D). However, as previously noted (Figure 1), aminophylline treatment (groups E and F; Table 2) resulted in a significant (P < 0.05) decrease in MABP in both WT and KO mice.
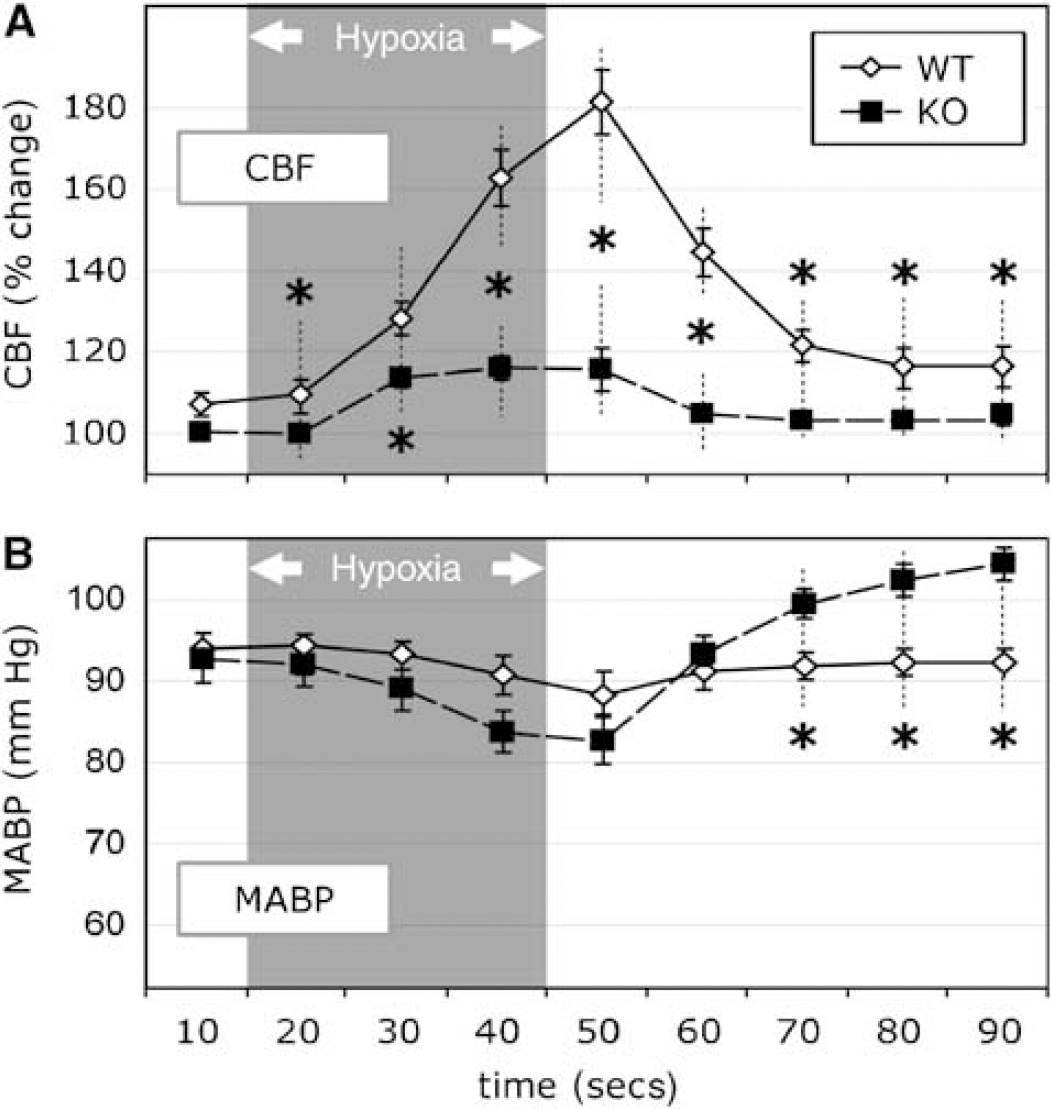
Hypoxic and posthypoxic period. Group A (Table 2, rows 1 and 2): In both WT and KO mice, MABP decreased significantly (P < 0.05) during hypoxia, reaching a nadir at termination. However, as noted earlier, MABP decreased to a greater extent (P < 0.05) during hypoxia in KO mice (58.6 ± 1.5 mm Hg) than in WT animals (76.1 ± 3.2 mm Hg) and remained lower than WT mice in the posthypoxic period (Figure 2).
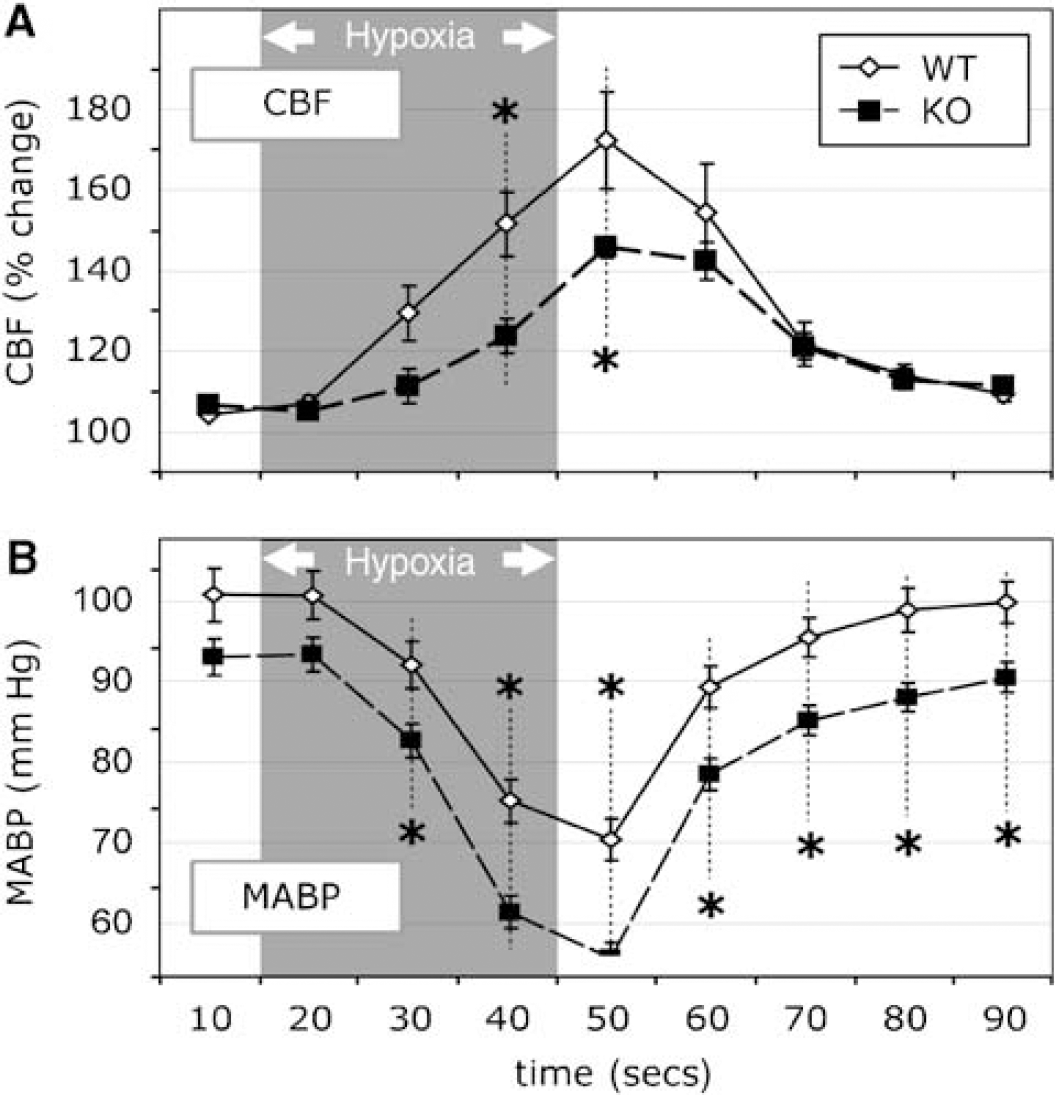
Results from group A. Changes in (
Group B: We were successful in matching the MABP changes during hypoxia, but less so in the posthypoxic period (≥ 25 secs after termination of hypoxia; see Figure 3).
Results from group B. Changes in (
Group C (WT-ZM): We did not observe any change in MABP during hypoxia in WT mice treated with ZM-241385. The MABPs in this group were well matched (see Table 2 and Figure 4).
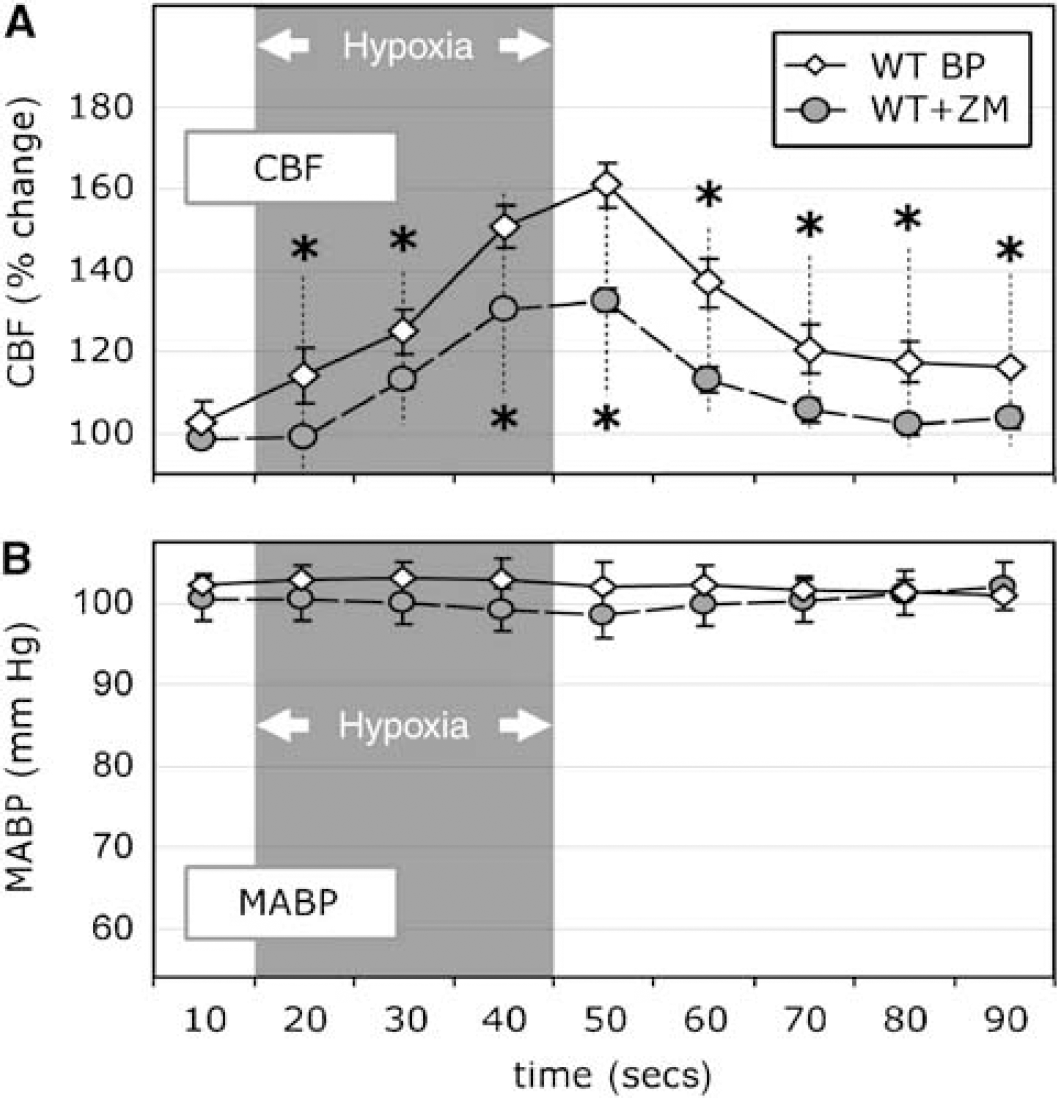
Results from group C. Changes in (
Group D (KO-ZM): The decreases in MABP during hypoxia were similar in the KO mice treated with ZM-241385 and the KO mice receiving vehicle alone. The decrease in MABP during hypoxia in the latter group was identical to KO mice receiving saline (Table 2, row 2).
Group E (WT-aminophylline): MABP decreased during hypoxia to 68.9 ± 5.1 mm Hg (or a decrease of 20.8% ± 3.7%) and promptly returned to prehypoxic levels. In our matched group, the changes in MABP were similar during the hypoxic (Table 2, rows 9 and 10) and posthypoxic periods.
Group F (KO-aminophylline): MABP decreased to 59.9 ± 1.6 mm Hg (or decrease of 19.3% ± 3.8%), a similar change in MABP as observed in the WT mice treated with aminophylline. Our attempts to match blood pressures were successful without any differences (Table 2, rows 11 and 12) in MABP responses in this group.
Cerebral blood flow response to transient hypoxia Wild-type compared with A2aR knockout: Uncontrolled blood pressure (group A-blinded): CBF in the WT (n = 24) by peak analysis(ti) increased maximally to 76% ± 3.0% (P < 0.05) as compared with prehypoxia (see Table 2, row 1, prehypoxia versus posthypoxia or I versus II), whereas in the KO animals (n = 15), CBF only increased to 47.2% ± 2.3% (see Table 2, row 2). This increase in CBF in the KO mice was 38% less than that measured in the WT mice, a significant difference (P < 0.05). The differences in CBF were evident during hypoxia and posthypoxia (Figure 2).
Blood pressure matched (group B): With MABP matched during hypoxia in the WT and KO mice, the differences (P < 0.05) in the CBF response(ti) (Table 2, rows 3 and 4) between WT and KO animals (81.9% ± 7.8% and 15.4% ± 5.3%, respectively, or an 81% decrease in response) became more evident. Figure 3 indicates the response(ts) in CBF and MABP in group B animals.
Effects of adenosine antagonists on hypoxic CBF response.
ZM-241385
WT (group C): During hypoxia in the WT mice (see Table 2, row 5 versus 6; Figure 4), CBF increased significantly (P < 0.05) by 60.4% ± 5.4%. In contrast, in mice treated with ZM-241385, CBF increased by only 32.1% ± 2.8%, which represents a 47% reduction (P < 0.001) in CBF. KO (group D): The CBF response during hypoxia in the A2aR KO mice (39.4% ± 3.9%) treated with ZM-241385 (Table 2, row 7 versus 8) was similar to that observed in the untreated KO animals (44.7% ± 2.0%). Aminophylline
WT mice (group E): As indicated in Table 2 (row 9 versus 10), aminophylline significantly (P < 0.05) attenuated the CBF response in WT animals. For example, CBF in WT mice treated with aminophylline increased only by 27.0% ± 4.0%, whereas in the blood pressure-matched WT animals treated with saline, CBF increased by 47.9% ± 4.3%, that is, a 44% reduction in flow. KO mice (group F): We observed no attenuation in the CBF increases during hypoxia as compared with the MABP-matched, untreated animals. In fact, the aminophylline-treated mice had a slightly greater increase in CBF (53.9% ± 4.0% versus 41.5% ± 3.9%, P < 0.05; Table 2, lines 11 versus 12).
Hypercarbia
The changes in CBF during 60 secs of ventilation with 7% CO2 in WT and KO mice are illustrated in Figure 5. The increases in CBF and the temporal profile were not different. Repeat measurements after hypoxia and in the presence of adenosine antagonists were similar to those shown in Figure 5.
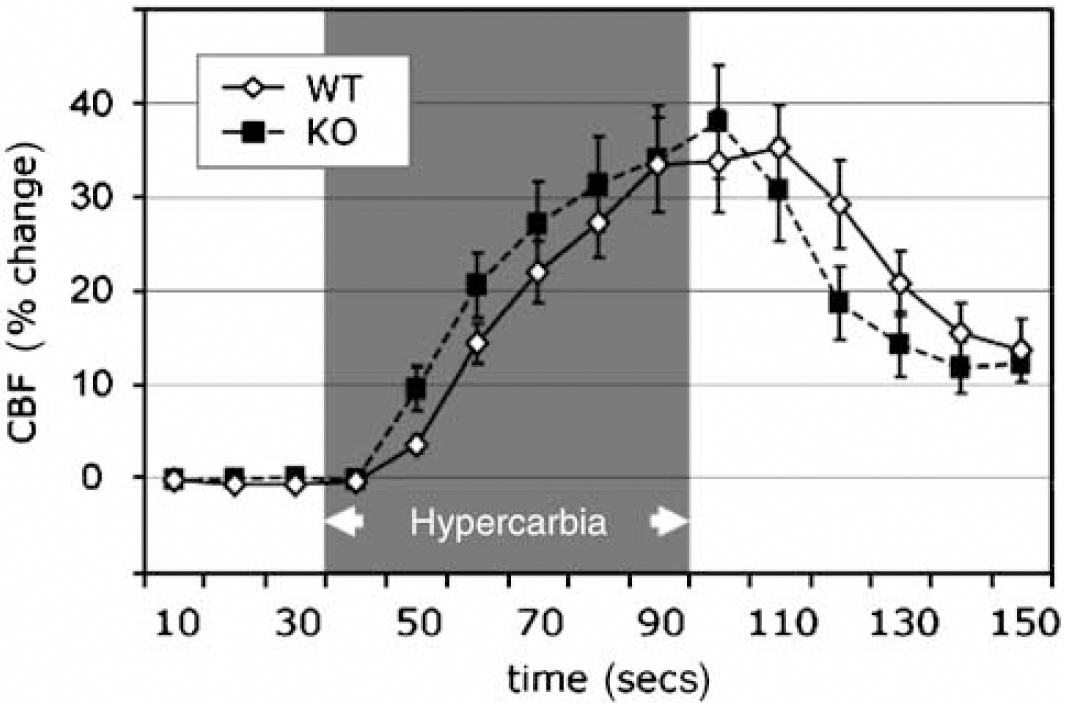
Changes in CBF with hypercapnia. WT and KO mice had a similar response to 60 secs ventilation with 7% CO2. The MABP was stable in both groups and unchanged from prehypercarbic period. n ≥ 15 in each group.
Physiologic Response to CGS-21680
At the end of the experiments with the experimentalist blinded to the genotype of the animal, mice were administered CGS-21680, an A2a agonist. As illustrated in Figure 6, we observed two distinct patterns of responses. In one group, we observed a rapid and significant (P < 0.05) decrease in MABP (Figure 6A) and an increase in heart rate (Figure 6B) as compared with pre-CGS injection values. We presumed that these animals were WT. In contrast, the second group, presumably the KO mice, had no change in MABP or heart rate with injection of CGS-21680. Based on these cardiovascular responses, we characterized the groups as either WT or KO animals.
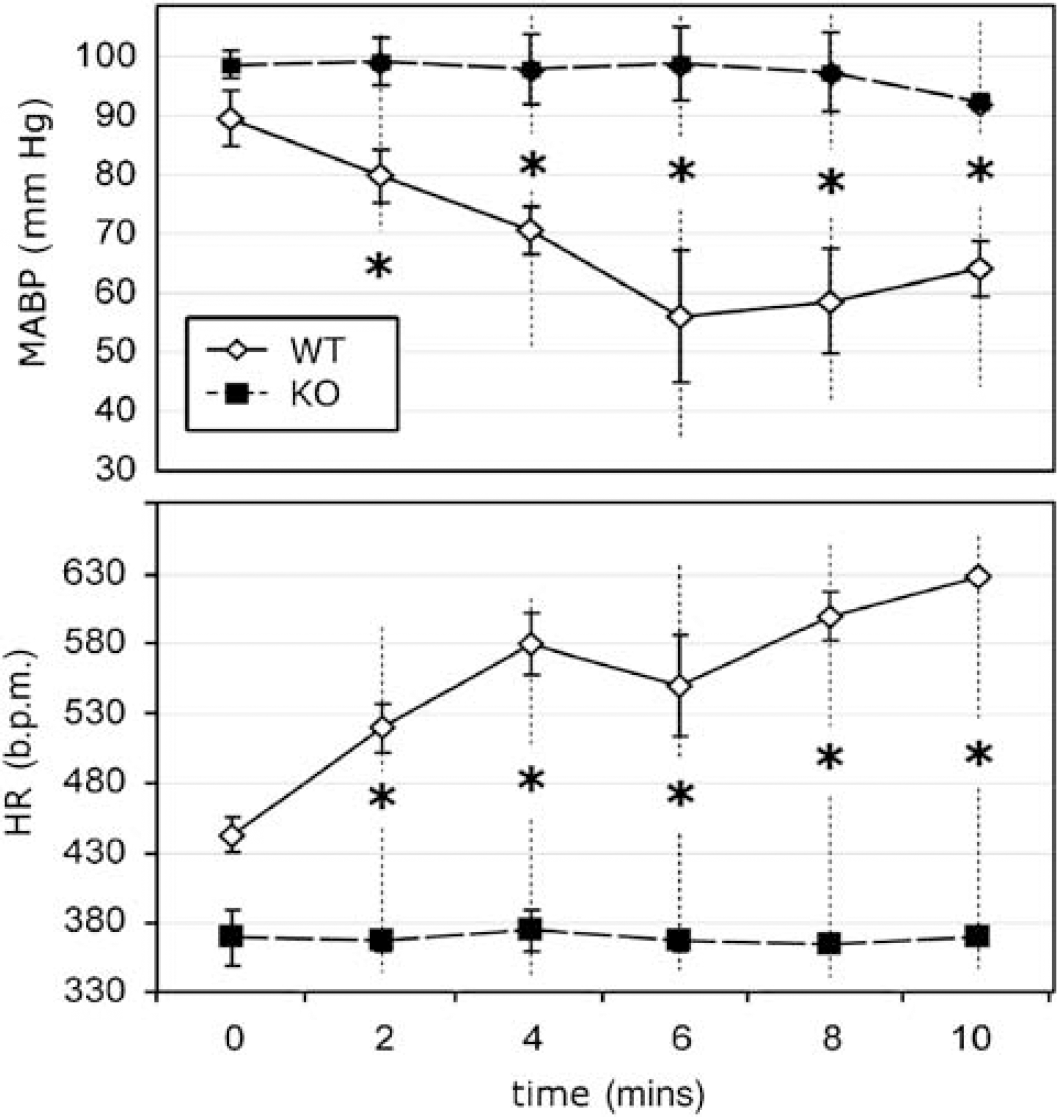
Changes in mean arterial blood pressure (MABP, upper panel) and heart rate (HR, lower panel) before and after administration of CGS-21680, an A2a agonist. The experimentalists were blinded as to genotype (group A). Two patterns were observed: in one group (n = 12), hypotension and tachycardia occurred presumably because of the presence of functional A2a receptors. In the other group (n = 13), MABP and HR were stable, presumably related to nonfunctional A2a receptors. Based on these cardiovascular responses, the mice were characterized as either WT (hypotension and tachycardia) or KO (stable cardiovascular parameters) animals. Pretreatment with ZM-241385 (1 mg/kg) in WT mice blocked the cardiovascular effects of CGS-21680. *P < 0.01 (with Bonferroni correction) before versus after administration of CGS-21680.
Discussion
The present study was designed to evaluate CBF responses to short duration (30 secs), moderate hypoxic hypoxia in WT and adenosine 2aR KO mice. We sought to test the hypothesis that hypoxic hyperemia would be attenuated in KO in comparison with WT mice. We found that CBF during hypoxia was attenuated by 38% in KO animals whose MABP was not controlled and by 81% in KO animals whose MABP was matched to the MABP in WT mice. The latter finding strongly suggests that CBF during hypoxia is significantly regulated by means of A2aR. Furthermore, in the WT animals, administration of adenosine receptor antagonists diminished hypoxic hyperemia by 44% to 48% whether the antagonist was A2a-specific (ZM-241583) or nonspecific (aminophylline) affecting both A1R and A2R. We conclude that adenosine via A2R, and most probably through A2aR, is responsible for a significant proportion of the hyperemia during hypoxia.
Our study is the first investigation of the physiology of CBF during hypoxia using adenosine receptor KO mice. However, Chen et al (1999), who developed and described the KO mouse used in the present study, documented the protective effect of A2aR KO with prolonged (2 h) focal ischemia and reperfusion using the middle cerebral artery occlusion model.
There are a number of methodological, physiologic, and pharmacologic factors that should be considered while evaluating our results and before accepting our conclusion that adenosine is significantly involved in the regulation of CBF during hypoxia.
Methodological Considerations
We chose a relatively short period of hypoxia to avoid hypoxic patho-physiologic changes in the brain tissue and alterations in the cardiovascular responses during subsequent periods of hypoxia, which precluded repetitive challenges to our animals (Pelligrino et al, 1995). In addition, the short exposure to hypoxia circumvented significant changes in PaCO2, which might have confounded the interpretation of CBF responses. Nevertheless, the short period of hypoxia elicited a consistent CBF response, which was similar to that in rat under comparable conditions (Morii et al, 1987; Ngai et al, 1995). In addition, we based our hypoxic challenge on our previous studies in rat (Winn et al, 1981a, b ) in which we showed an increase in brain tissue adenosine concentrations within 30 secs of the onset of hypoxia, and on our more recent study (Kung et al, 2007) showing a rise in extracellular adenosine concentration within the initial seconds of the onset of hypoxia. Lastly, our preliminary time-controlled studies showed that neither these short periods of hypoxia nor the length of the experiments altered our animals' physiologic status or ability to respond to a hypoxic challenge.
The CBF response to hypercarbia observed in KO mice indicates that augmented flow in KO animals is not impaired and is similar to the response in WT mice. Moreover, the hyperemic response to ventilation with increased PaCO2 did not deteriorate over time or after the administration of adenosine antagonists.
The differences in CBF in WT and KO mice might be attributed to differences in physiologic parameters, but the changes in these measurements were similar. Alternatively, the differences in CBF response might be attributed to the pulmonary response to hypoxia, as lung adenosine concentrations are elevated during hypoxia and adenosine is a pulmonary circulation dilator (Mentzer et al, 1975). However, as already noted, the decrease in PaO2 was similar in WT and KO mice. We conclude that differences in the ability to augment CBF in KO mice, duration of our experiments, disparity in physiologic changes or pulmonary function cannot account for the observed difference in CBF responses in WT and KO mice during hypoxia.
Physiologic Considerations
During hypoxia, MABP decreased, more so in the KO animals than in the WT mice. The status of the autoregulatory response during hypoxia is unclear, with some investigators reporting persistence (Craigen and Jennett, 1981) and others suggesting loss (Haggendal and Johansson, 1965) of autoregulation. The dominant influence appears to be the state of the vascular tone (Heistad and Kontos, 1983), which may account for the varied response to hypoxia in the WT mice (groups A, B, C, and E). Irrespective of its functional status, the autoregulatory response is not instantaneous, with the onset occurring within 3 to 7 secs and steady-state responses being achieved within ∼ 60 secs. (Kontos et al, 1978). Thus, even if autoregulation is intact, the short duration of hypoxia in the present study may lead to a varied response. To obviate the interplay between the cerebrovascular response to systemic hypoxia and the cerebrovascular response to systemic hypotension, we created matched MABP groups.
Thus, the MABP-matched groups are the optimal conditions to determine the contribution of adenosine to CBF regulation during transient hypoxia. When MABP was matched, CBF was reduced by ∼80% in the KO mice as compared with the WT animals, whereas in the MABP-uncontrolled animals, CBF in the KO mice was reduced by 38%. Surprisingly, CBF was higher in KO mice with low MABP than in KO mice in which MABP was stabilized during hypoxia. Perhaps, the hypotension (MABP = 58.6 ± 1.6 mm Hg) in KO animals evoked additional adenosine release (Winn et al, 1979, 1980). This increase in adenosine concentrations could activate the less-sensitive A2bR, leading to additional dilatation of the cerebral arterioles. Alternatively, other nonadenosine mechanisms such a nitric oxide may be involved (Pelligrino et al, 1995).
Pharmacological Consideration
The present study using adenosine antagonists documented a 44% to 48% reduction in hypoxic hyperemia. These results are similar to those found in earlier studies with other species, but are the first in mice (Armstead, 1997; Emerson and Raymond, 1981; Hoffman et al, 1984; Morii et al, 1987; Pelligrino et al, 1995; Phillis et al, 1984; Winn et al, 1981b).
The present study used both specific A2a antagonist (ZM-241385) and nonselective (A1R and A2R) antagonist (aminophylline). We chose ZM-241385 because an earlier in vitro study by Ngai et al (2001) showed that adenosine-induced vasodilatation was predominantly related to stimulation of the A2aR, but found residual vasodilatation of cerebral arterioles with high concentrations of adenosine in the presence of an A2aR antagonist. Unfortunately, there are no effective A2bR antagonists and thus the role of A2bR activity in cerebral vasodilatation cannot be studied directly. As compared with rat, penetrating cerebral arterioles in mouse appear to possess similar dose responses to adenosine (Coyne et al, 2002).
The ZM-241385 dose used in the present study (1 mg/kg, intravenously) was comparable to that used previously by Meno et al (2001), which decreased pial arteriolar vasodilatation by 54% during sensory cortical activation. Although there are no studies documenting the degree of blood—brain barrier penetration by ZM-241385, the study by Meno et al (2001) showed that pial arteriolar vasodilatation by topical adenosine (10−5 mol/L) was attenuated by ∼50% by intravenous ZM-241385 (1 mg/kg), the same dose that we used in the present study. In addition, in the present study, 1 mg/kg of ZM-241385 was highly effective in blocking the cardiovascular effect of the A2aR agonist, CGS-21680. Potentially, use of a higher dose of ZM-241385, which could affect both A2aR and A2bR, might have diminished hypoxic hyperemia to a greater extent.
We also chose to use aminophylline, a nonspecific (both an A1 and A2) receptor antagonist. The range of depression in CBF (44% to 66%) in the WT mice, with and without MABP control, is similar to that observed in our earlier studies in the rat (Morii et al, 1987) as well as comparable with those noted by other investigators (Armstead, 1997; Emerson and Raymond, 1981; Hoffman et al, 1984; Pelligrino et al, 1995; Phillis et al, 1984; Pinard et al, 1989). We used aminophylline rather than the less-soluble theophylline to avoid volume overloading in mice. The dose of aminophylline was also based on earlier studies, which indicated that at 0.2 μmol/g administered intraperitoneally, theophylline easily penetrated the blood—brain barrier, persisted for hours in the cerebral spinal fluid, and did not inhibit phosphodiesterase activity (Morii et al, 1987).
The mechanisms that could account for the 19% residual CBF in the KO and ∼40% to 60% residual CBF in the aminophylline and ZM-241385-treated animals stated below. Multiple nonadenosine mechanisms, such as nitric oxide, potassium, and so on, have been suggested as being involved in CBF increases during hypoxia (Armstead, 1997; Pelligrino et al, 1995) and could be responsible for the residual CBF response during hypoxia. In addition, in the A2aR KO animals, A2bR activation could account for persistent cerebral vasodilatation. As noted earlier, at high concentrations of adenosine, Ngai et al (2001) showed vasodilatation in penetrating cerebral arterioles treated with ZM-241385. Conceivably, in the A2aR KO animals, CBF during hypoxia is insufficient, thereby increasing the brain concentration of adenosine, which in turn activates the less-sensitive A2bR. Lastly, lateral ‘coadaptation’ could have developed in the A2aR KO mice. In the WT mice treated with aminophylline or ZM-141385, residual CBF increase during hypoxia might be related to incomplete receptor blockade and/or the intrinsic pharmacological properties and/or pharmacokinetic limitations of these agents. However, taken together, the results of the present study strongly supports the hypothesis that adenosine is involved in cerebrovascular regulation to hypoxia and that such regulations occur primarily by means A2aR subtype receptors.
Conclusion
Using WT and A2aR KO mice, we have shown that hypoxic hyperemia is attenuated by nonfunctioning A2aR and by adenosine receptor blockade using receptor antagonists. These results are consonant with the hypothesis that adenosine is involved in CBF regulation during hypoxic hypoxia.
Footnotes
The authors state no conflict of interest.