
Abstract
The nuclear enzyme poly(ADP-ribose) polymerase (PARP) is activated by oxidative stress and plays a significant role in postischemic brain injury. We assessed the contribution of PARP activation to the blood–brain barrier (BBB) disruption and edema formation after ischemia–reperfusion. In male Wistar rats, global cerebral ischemia was achieved by occluding the carotid arteries and lowering arterial blood pressure for 20 mins. The animals were treated with saline or with the PARP inhibitor N-(6-oxo-5,6-dihydrophenanthridin-2-yl)-N, N-dimethylacetamide.HCl (PJ34); (10 mg/kg, i.v.) before ischemia. After 40 mins, 24, and 48 h of reperfusion, the permeability of the cortical BBB was determined after Evans Blue (EB) and Na-fluorescein (NaF) administration. The water content of the brain was also measured. The permeability of the BBB for EB increased after ischemia–reperfusion compared with the nonischemic animals after 24 and 48 h reperfusion but PARP inhibition attenuated this increase at 48 h (nonischemic: 170 ± 9, saline: 760 ± 95, PJ34: 472 ± 61 ng/mg tissue). The extravasation of NaF showed similar changes and PJ34 post-treatment attenuated the permeability increase even at 24 h. PARP inhibition decreased the brain edema seen at 48 h. Because PARP has proinflammatory properties, the neutrophil infiltration of the cortex was determined, which showed lower values after PJ34 treatment. Furthermore, PJ34 treatment decreased the loss of the tight junction protein occludin at 24 and 48 h. The inhibition of PARP activity accompanied by reduced post-ischemic BBB disturbance and decreased edema formation suggests a significant role of this enzyme in the development of cerebral vascular malfunction.
Introduction
Cerebral endothelial cells are characterized by the formation of tight junctions, low numbers of pinocytotic vesicles, and the presence of specialized transport systems, which together comprise the blood–brain barrier (BBB). Normal functioning of the BBB is essential for the maintenance of optimal ionic and metabolic homeostasis of the brain parenchyma (Joo, 1996). During pathophysiologic conditions, damage to cerebrovascular endothelial cells causes alterations of BBB function which can exacerbate neuronal cell injury and death. The mechanisms involved in BBB function alterations following brain insults are incompletely understood but several recent studies on endothelial cells from peripheral circulations suggest that the nuclear enzyme poly(ADP-ribose) polymerase (PARP) may play a prominent role. Poly(ADP-ribose) polymerase is activated secondarily after reactive oxygen species-mediated breaks in DNA strands during conditions such as ischemia–reperfusion. The contribution of PARP activation to BBB damage has been shown in meningitis and allergic encephalitis (Koedel et al, 2002; Scott et al, 2004). However, we are unaware of previous studies that have directly assessed the role of PARP activation in disruption of the BBB after ischemia–reperfusion.
Activation of PARP can lead to endothelial dysfunction and cell death via a variety of pathways (Pacher and Szabo, 2005). When activated, PARP catalyzes the attachment of ADP-ribose units from its substrate, NAD+, onto various nuclear proteins. NAD+ serves as a cofactor for glycolysis and the tricarboxylic acid cycle, thus providing ATP for most cellular processes. Thus, one cause of cellular injury and necrosis after PARP activation is the depletion of cellular NAD+ and ATP (Szabo, 2006). An additional consequence of PARP activation is cell death through the activation of the apoptosis-inducing factor (Komjati et al, 2004). Finally, augmented development of the proinflammatory phenotype by polyADP-ribosylation of histones, transcription factors, and signaling molecules can indirectly affect endothelial cell function (Chiarugi, 2005).
The purpose of our study was to examine the contribution of PARP activation to the increased permeability of the BBB observed after reperfusion. We examined both functional and structural changes related to BBB disturbance after transient global cerebral ischemia in rats and the effect of the administration of the PARP inhibitor, N-(6-oxo-5,6-dihydrophenanthridin-2-yl)-N, N-dimethylacetamide. HCl (PJ34) on the observed changes.
Materials and methods
Surgery and Experimental Groups
Experiments were carried out on male Wistar rats; weighing 270 to 320 g. Before surgical procedures, food was deprived for 3 h with free access to water. Animals were maintained and used in compliance with the principles set forth by the Animal Care and Use Committee of Wake Forest University Health Sciences. Anesthesia was induced with 3.5% halothane in a mixture of 72% nitrous oxide and 28% oxygen and maintained at 1.0% to 1.2% halothane in spontaneously breathing animals. The right femoral artery and vein were cannulated for physiologic monitoring, infusion of drugs, and blood withdrawal. Using a midline cervical incision, both common carotid arteries were isolated from adhering tissue and nerves, and silk threads (3 to 0) were placed loosely around them. Arterial blood pressure was monitored continuously, and blood gases and pH were measured before and during ischemia.
The animals were exposed to global cerebral ischemia using combined bilateral common carotid artery occlusion and arterial hypotension. We used this transient global ischemia model because we wanted to use a widely accepted procedure that affected most of the cortex so that regional sampling problems associated with focal ischemia would not be a problem. In addition, the model that we used will mimic some forms of human brain pathology such as those seen after cardiogen shock. After the administration of heparin (450 IU/kg, i.v.), mean arterial blood pressure was lowered to 45 to 50 mm Hg by blood withdrawal (1.5 to 2.2 mL/100 g body weight) through the femoral vein, and both common carotid arteries were occluded by placing microaneurysm clips around the vessels for 20 mins. Reperfusion was achieved by removing the clips and giving back the shed blood with a speed of 1.2 mL/min. Body temperature was measured and maintained at 37.5°C ± 0.2°C. After ischemia, reperfusion was allowed for different durations (40 mins, 24 h, 48 h). In each reperfusion group, animals were treated with saline or PJ34 (Sigma-Aldrich, Inc., St Louis, MO, USA) (10 mg/kg, i.v.) 40 mins before the start of ischemia. One set of animals was treated with the drug at the start of reperfusion and again 6 h later (10 mg/kg, i.p.). The effect of the post-treatment was checked only after 24 h of reperfusion. The pre-treatment dose of PJ34 that was chosen has been found to be effective against cerebral ischemia in rats (Abdelkarim et al, 2001; Park et al, 2004). The dual administration protocol for the delayed PJ34 treatment was designed to counteract the lack of access of this drug to the cerebral circulation and brain before ischemia as occurred in the pre-treatment group. In sham animals, the carotid arteries were exposed but no ischemia was made. Mortality was 5/17 in the saline-treated and 3/15 in the PJ34 pretreated rats when 48 h reperfusion was allowed. There were no mortalities in the other groups.
Tissue Sampling and Determination of Blood–Brain Barrier Permeability
Blood–brain barrier permeability was assessed by measuring the Evans Blue (EB) and Na-fluorescein (NaF) content in the brain. Normal saline containing EB (2%) and NaF (2%) (4 mL/kg) were administered intravenously 30 mins before the end of the reperfusion period. In the 24 and 48 h reperfusion groups, the rats were reanesthetized and the left femoral artery and vein were cannulated for physiologic monitoring and infusion of the dye. At the end of the reperfusion period, the animals were transcardially perfused with ice-cold, heparinized phosphate-buffered saline at 100 mm Hg pressure for 4 mins. The brains were removed and rinsed with phosphate-buffered saline, and two 4-mm wide coronal slices were made, starting at bregma levels + 1.80 mm and −2.20 mm. After removing the meninges, the cerebral cortex above the rhinal fissure from both slices were dissected and weighed. The weighed tissue samples were maintained at −80°C for further processing. Later, the samples from the rostral coronal slice were homogenized in 5 times volume of phosphate-buffered saline compared with the tissues and divided into two parts. The first part was returned to −80°C after the addition of proteinase inhibitors (1 μg/mL aprotinin, 50 μg/mL phenylmethylsulfonyl fluoride, and 1 μg/mL leupeptin) for protein or total RNA isolation.
The other half of the cortex was used for the EB and NaF measurement. Trichloroacetic acid (80%) was added to the homogenate in the same volume to precipitate the proteins, followed by centrifugation for 10 mins at 13,600g. For the EB measurement, the supernatant was diluted with ethanol (1:4), and its fluorescence was determined (excitation at 620 nm and emission at 680 nm). For the NaF measurement, the supernatant was diluted with 5 mol/L NaOH (1:0.8), and its fluorescence was determined (excitation 440 nm and emission at 525 nm). A microplate spectrophotometer (FLUOstar Optima, BMG, Durham, NC, USA) was used for the fluorescence measurements. Calculations were based on external standards in the same solvent (10 to 200 ng/mL). The tissue content was quantified from linear standard curves derived for each of the dyes and expressed per gram of tissue.
Determination of Brain Water Content
Brain water content was determined by the dry–wet weight method on other set of animals. At the end of the experiments, the brains were immediately removed, and weighed to obtain the wet weight (without the brainstem and cerebellum). The tissue was then dried in an oven at 100°C for 24 h and reweighed to obtain the dry weight. Brain water content was calculated as (wet weight-dry weight)/wet weight × 100%.
Myeloperoxidase Activity Assay
Inflammatory cell infiltration was determined using an assay for myeloperoxidase (MPO), an enzyme found within the azurophilic granules of neutrophil leukocytes (Bradley et al, 1982). The MPO activity assay has been found to be a reproducible and objective method to reliably estimate neutrophil infiltration and it correlates well with other estimations of neutrophil movement into inflamed tissues (Bradley et al, 1982; Matsuo et al, 1994a). The cortical samples from the second, caudal lying coronal sections were used for the biochemical determination of MPO activity. Each sample was homogenized in 5 mmol/L potassium phosphate buffer (pH 6.0, 4°C) using a glass homogenizer and centrifuged at 30,000g (30 mins, 4°C). The supernatant was discarded and the pellet was suspended in 0.5% hexadecyltrimethylammonium bromide in 50 mmol/L potassium phosphate buffer (pH 6.0, 4°C) at an original tissue wet weight-to-volume ratio of 1:5. The samples were frozen immediately and subjected to three freeze–thaw cycles, after which sonication was repeated between cycles. After the last sonication, the samples were incubated at 4°C for 20 mins and centrifuged at 12,500g (15 mins, 4°C). Supernatant MPO activity was assayed as described by Bradley et al (1982). In brief, 20 μL of supernatant was mixed with 180 μL of 50 mmol/L potassium phosphate buffer, pH 6.0, containing 167 mg/L o-dianisidine dihydrochloride and 0.0005% hydrogen peroxide. The rate at which a colored product formed during the MPO-dependent reaction of o-dianisidine dihydrochloride was measured. The change in absorbance at 460 nm was recorded at 15 secs intervals over 2 mins using a spectrophotometer (FLUOstar Optima, BMG, Durham, NC, USA). The MPO activity of each sample was expressed as a relative value compared with the average MPO activity of the sham-operated control animals.
Real-Time Polymerase Chain Reaction
Total cellular RNA was extracted from the cortical homogenates using an SVtotal RNA isolation system (Promega, Madison, WI, USA). Real-time reverse transcription-polymerase chain reaction (RT-PCR) was performed using an ABI Prism 7700 Sequence Detection System and Taqman probe and primer set for intracellular adhesion molecule-1 (ICAM-1) (Applied Biosystems, Foster City, CA, USA). Polymerase chain reaction products were detected using probes labeled with reporter dye FAM (6-carboxy-fluorescein) at the 5′ end and quencher dye TAMRA (6-carboxy-tetramethyl-rhodamine) at the 3′ end. Specificity of the polymerase chain reaction products was verified by agarose gel electrophoresis. The 2−ΔΔCt method was used to analyze the results. In brief, the Ct (threshold cycle) value of a gene was subtracted from the Ct value of a reference housekeeping gene (β-actin) to standardize for the amounts of RNA template and efficiencies of reverse transcription. The resulting change in Ct values was then converted to a linear form using 2−ΔCt and used in subsequent statistical analysis.
Western Blotting
Cortical samples were first homogenized in 5 × volume of phosphate-buffered saline compared with tissue weight and protease inhibitors were added as mentioned above. Next, sodium dodecyl sulfate buffer (1% sodium dodecyl sulfate, 10 mmol/L Tris, pH 7.6) was added in the amount of the homogenate and sonicated. Equal amounts of protein for each sample were separated by 4% to 20% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred onto a polyvinylidine difluoride membrane and blocked for 1 h at room temperature with Tris-buffered saline containing 5% skimmed milk powder, 0.1% Tween 20. Blots were incubated overnight at 4°C with a polyclonal anti-occludin antibody (1:1,000, Zymed Laboratories, San Francisco, CA, USA). The membranes were then washed three times in Tris-buffered saline containing 0.1% Tween 20 and incubated for 1 h in the blocking buffer with anti-rabbit IgG conjugated to horseradish peroxidase (1:50,000; Jackson Immuno-Research, West Grove, PA, USA). The bound antibodies were visualized using enhanced chemiluminescence. (Super-Signal West Pico; Pierce, Rockford, IL, USA) and recorded on X-ray film. The bands were scanned and the densities of the bands were quantitated by using FOTO/Analyst PC Image and Image J (Fotodyne Inc., Hartland, WI, USA).
Drugs
All chemicals were obtained from Sigma (Sigma-Aldrich Chemical Co., St Louis, MO, USA) if not mentioned otherwise. PJ34 was dissolved in saline (6 mg/mL).
Data Analysis
Data are presented as means ± s.e.m. Group differences were determined by one-way analysis of variance followed by pair-wise comparisons using the Student–Newman–Keuls test or by one-way analysis of variance on ranks if the normality test failed. We used Pearson's correlation analysis to determine how the different findings independently related to each other. In all cases, a P-value of < 0.05 was considered statistically significant.
Results
Physiologic Variables
The values of the physiologic variables measured approximately 10 mins before the induction of ischemia and during the ischemic period are shown in Table 1. There were no significant differences in these values among the different subgroups within the same treatment (saline; PJ34 pre-treatment; PJ34 post-treatment), so these values were pooled. PJ34 treatment 40 mins before ischemia caused a slight decrease in the basal heart rate compared with saline treatment and a very small increase in the arterial blood pressure during ischemia. However, the observed difference is close to the limit of accuracy of our blood pressure registration system and probably has no biologically significant effect on the level of ischemia. This small increase probably is a side effect of the observed resistance of the blood pressure to blood withdrawal in the PJ34 pretreated animals (Table 1). During ischemia, the blood pressure was initially reduced to 45 mm Hg by blood removal. Blood pressure was allowed to increase to 50 mm Hg, and then it was reduced to 45 mm Hg again by further blood removal. The PJ34-treated animals required 15% more blood to be removed because of more frequent pressure increases from 45 to 50 mm Hg during the ischemic period. This could account for the somewhat higher average pressure. There were no statistically significant differences among the treatment groups in other parameters.
Physiological variables, arterial blood gases, and acid–base parameters of saline, PJ34 pre- and post-treated animals before and during ischemia
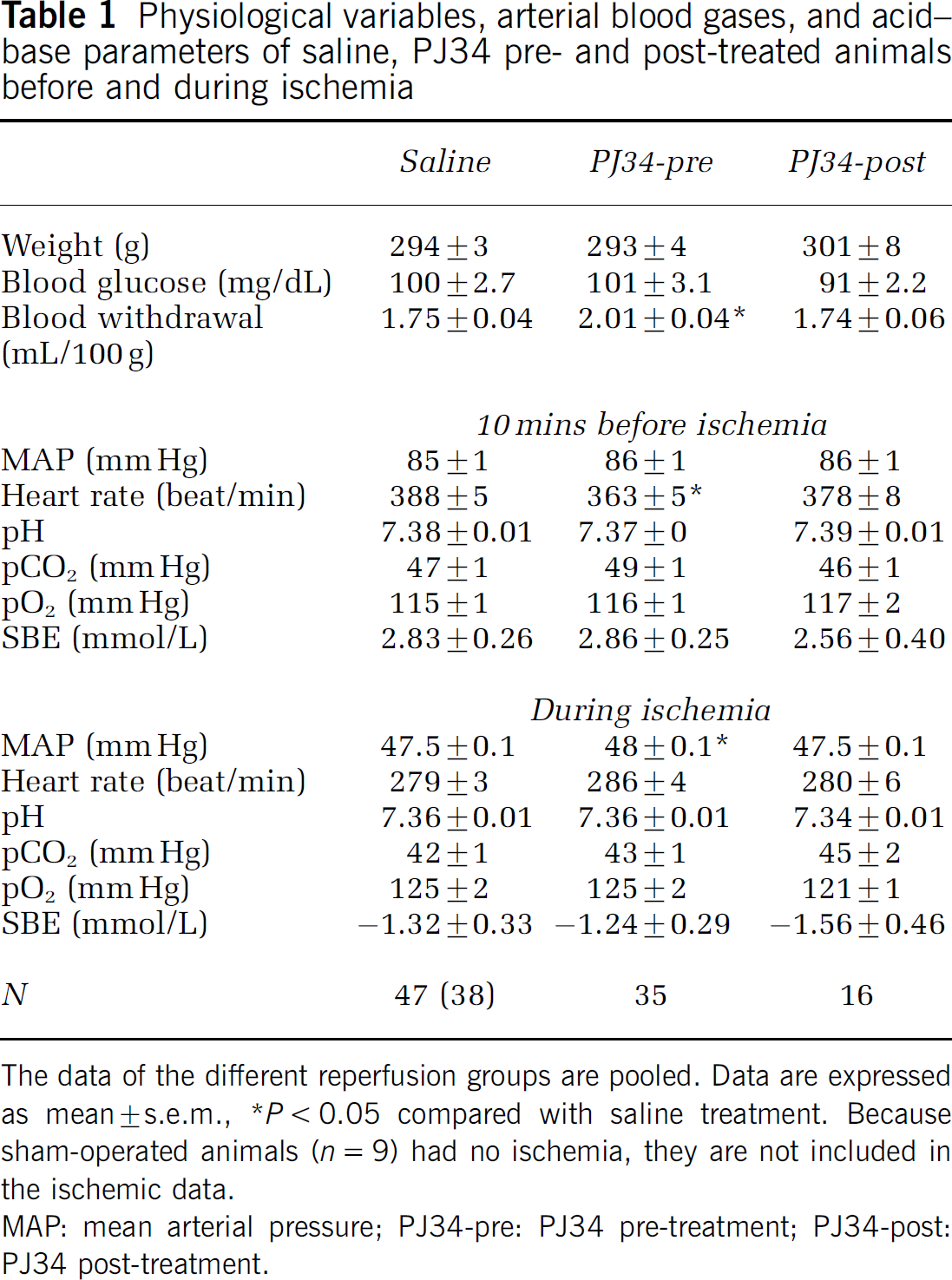
The data of the different reperfusion groups are pooled. Data are expressed as mean ± s.e.m.,
P < 0.05 compared with saline treatment. Because sham-operated animals (n = 9) had no ischemia, they are not included in the ischemic data.
MAP: mean arterial pressure; PJ34-pre: PJ34 pre-treatment; PJ34-post: PJ34 post-treatment.
Blood–Brain Barrier Permeability
Blood–brain barrier permeability in the cortex was assessed by the amount of extravasated EB and NaF dyes during a 30-min period at different reperfusion times. Extravasation of EB dramatically increased after ischemia measured at 24 and 48 h reperfusion compared with the sham-operated animals. PJ34 treatment significantly lowered EB extravasation in the 48 h group (Figure 1). Na-fluorescein extravasation was higher in all examined reperfusion time points and showed an increase with the elapsed time. PJ34 pre-treatment significantly decreased NaF permeability at 48 h reperfusion and our post-treatment protocol caused protection against the NaF permeability increase even at the examined 24 h time point (Figure 2).
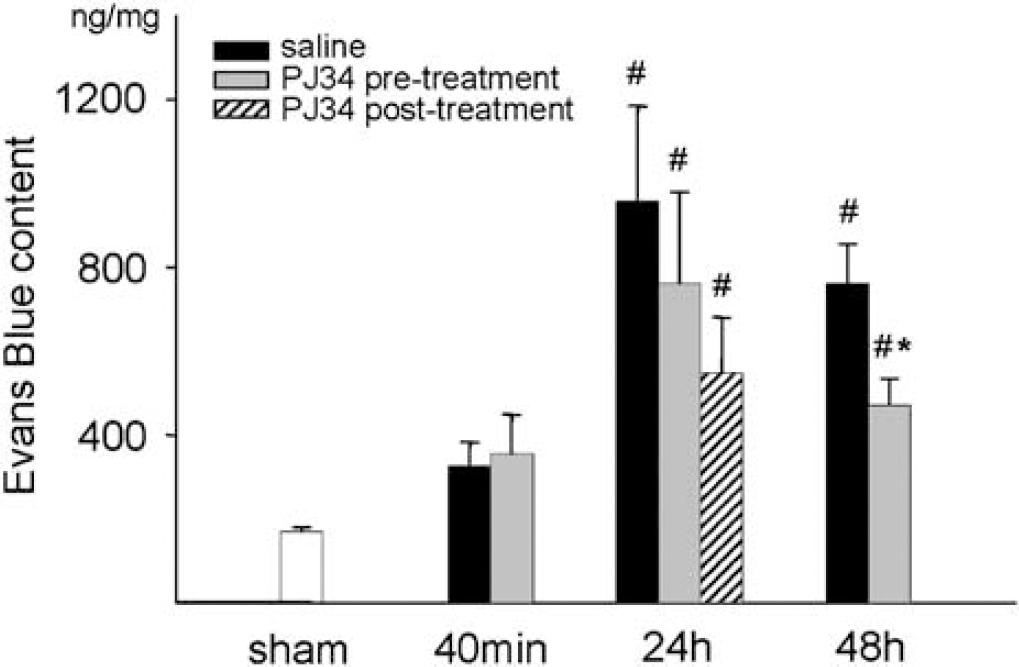
Evans Blue content in the cerebral cortical tissue after sham operation, and after 40 mins, 24 h, and 48 h reperfusion after global cerebral ischemia in saline and PJ34 pre- and post-treated animals. Data are shown as mean ± s.e.m., *P < 0.05 compared with saline treatment in the same reperfusion group; *P < 0.05 compared with sham. Sham (n = 9), 40 mins (n = 8 to 9), 24 h (n = 15 to 17), 48 h (n = 12 to 12).
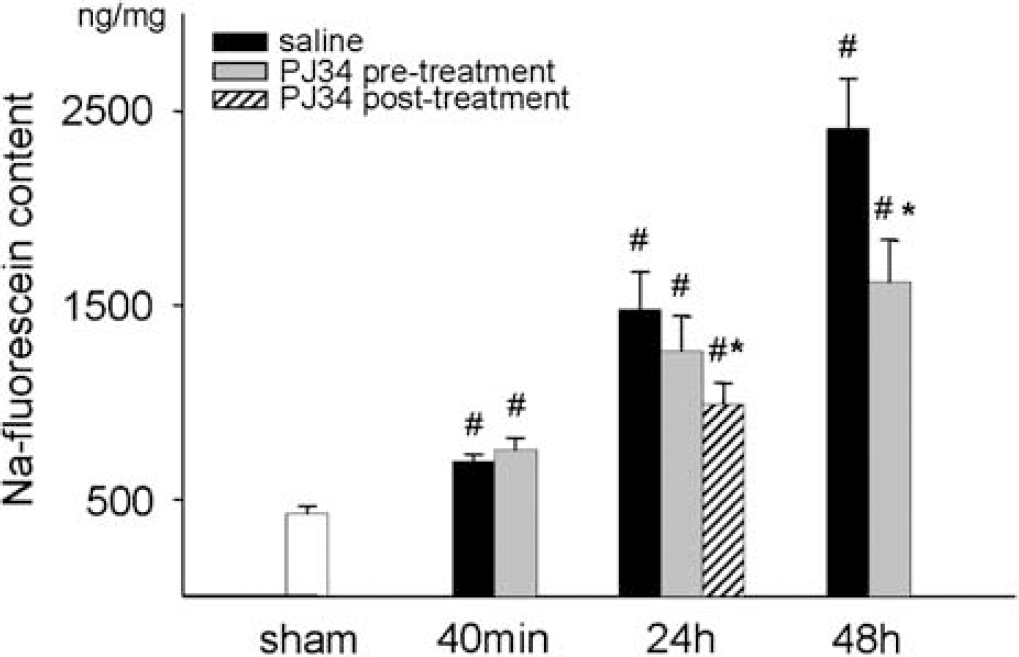
Na-fluorescein content in the cerebral cortical tissue after sham operation, and after 40 mins, 24 h, and 48 h reperfusion after global cerebral ischemia in saline and PJ34 pre- and post-treated animals. Data are shown as mean ± s.e.m., *P < 0.05 compared with saline treatment in the same reperfusion group; #P < 0.05 compared with sham. Sham (n = 9), 40 mins (n = 8 to 9), 24 h (n = 15 to 17), 48 h (n = 12 to 12).
Although without statistically significant difference, EB extravasation showed a clear tendency towards higher values at 40 mins reperfusion, and PJ34 pretreatment to have some protection even at 24 h. At 40 mins, the permeability of the barrier in the cortex was almost identical in the saline and PJ34-treated groups.
Water Content
After ischemia–reperfusion, there was no statistically significant difference in the whole brain water content in either treatment group compared with the sham animals at 24 h. However, the water content of the brains was elevated after 48 h reperfusion and the increase was significantly smaller in the PJ34 pretreated animals (Figure 3).
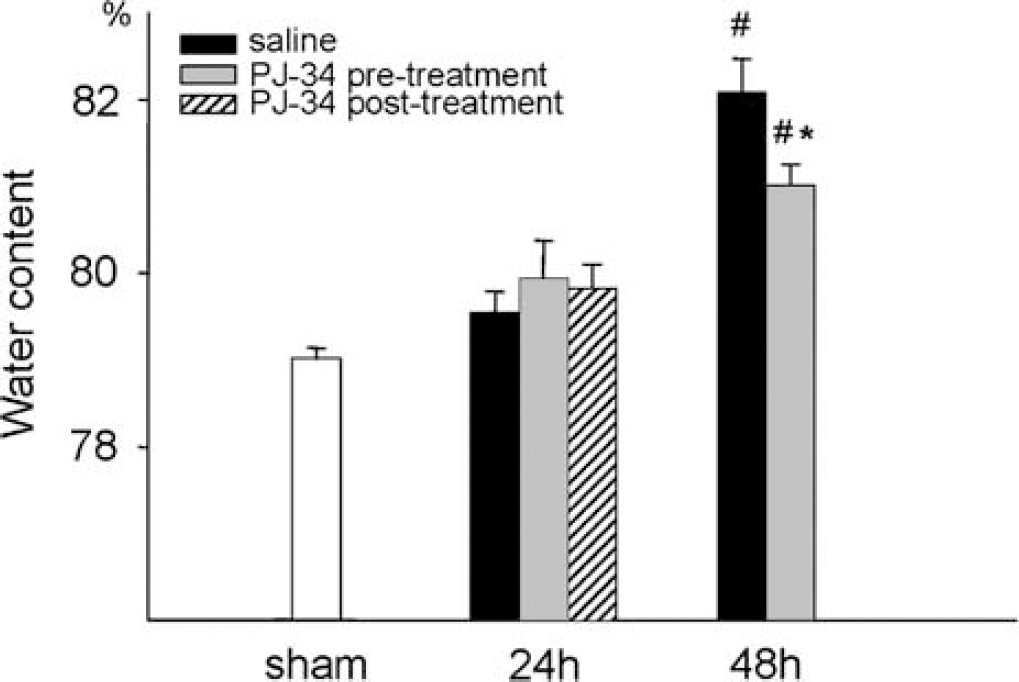
Water content of the brains in the sham, 24 h, and 48 h reperfusion groups. Water content given as percentage of the wet weight. Data are shown as mean ± s.e.m., *P < 0.05 compared with saline treatment in the same reperfusion group; *P < 0.05 compared with sham. Sham (n = 5), 24 h (n = 5 to 6), 48 h (n = 6 to 8).
Myeloperoxidase Levels
At 40 mins reperfusion, there were no MPO activity changes. The MPO activity was significantly elevated in the saline-treated ischemic animals at 24 and 48 h of reperfusion. Although the reduction in MPO at 24 h for both pre- and post-ischemia treatment was not statistically significant, pre-treatment with PJ34 significantly reduced MPO to near baseline values at 48 h reperfusion (Figure 4). In saline-treated rats, MPO activity correlated well with the extravasation of EB and NaF (Figure 5) at both 24 h (r: 0.668, P = 0.003 and r: 0.650, P = 0.005) and 48 h reperfusion (r: 0.833, P: < 0.001 and r: 0.902, P < 0.001).
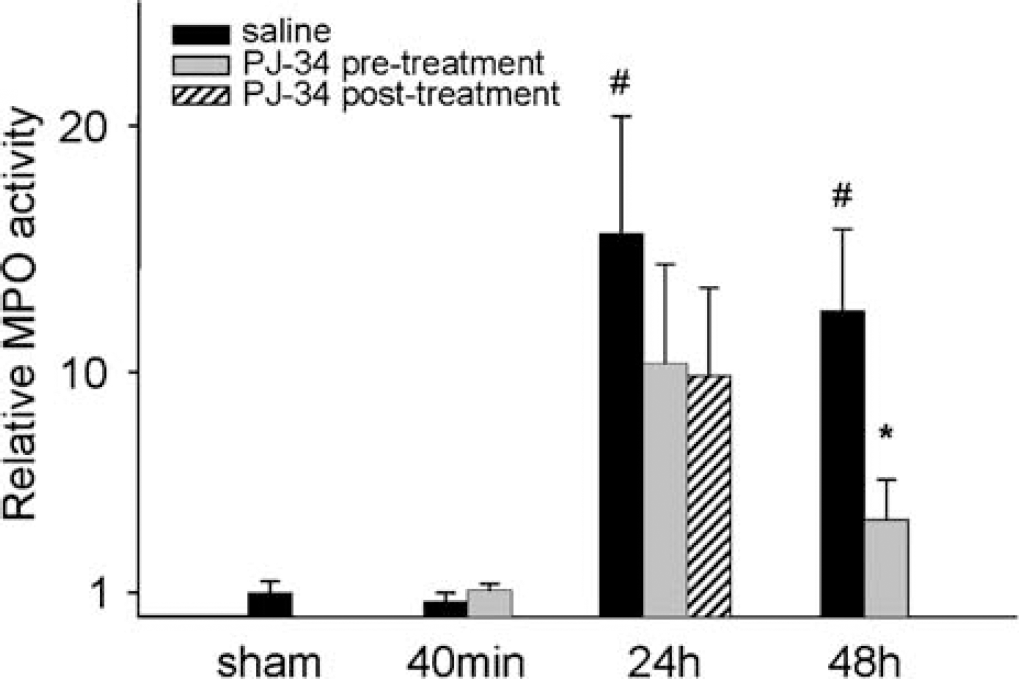
Myeloperoxidase activity in the cortices in relation to the average MPO activity measured in the sham-operated rats. The MPO activity was measured 40 mins, 24 h, and 48 h after the start of reperfusion in saline and PJ34 pre- and post-treated animals. Data are shown as mean ± s.e.m., *P < 0.05 compared with saline treatment in the same reperfusion group; *P < 0.05 compared with sham. Sham (n = 9), 40 mins (n = 8 to 9), 24 h (n = 15 to 17), 48 h (n = 12 to 12).
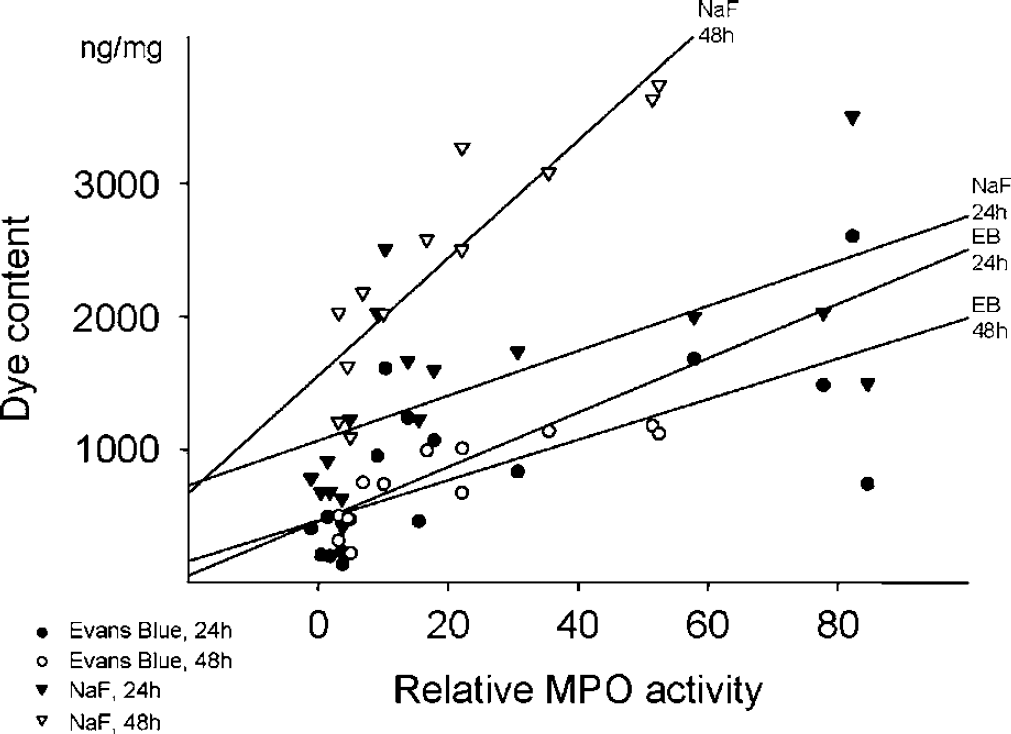
Correlation of EB and NaF content and relative MPO activity in the cortical samples of saline-treated rats after 24 and 48 h reperfusion. Evans Blue: r24 h = 0.668 and r48 h = 0.833. Na-fluorescein: r24 h = 0.650 and r48 h = 0.902, P < 0.05 in each cases.
Reverse Transcription-Polymerase Chain Reaction
The transcription of the endothelial adhesion molecule ICAM-1 markedly increased at 24 and 48 h after the start of reperfusion in the cortical samples compared with the sham-operated animals. PJ34 pre-treatment decreased the changes at both time points (Figure 6). Intracellular adhesion molecule-1 transcriptional activity correlated with MPO activity at 48 h reperfusion in the saline-treated rats (r: 0.596, P = 0.04) but not at 24 h.
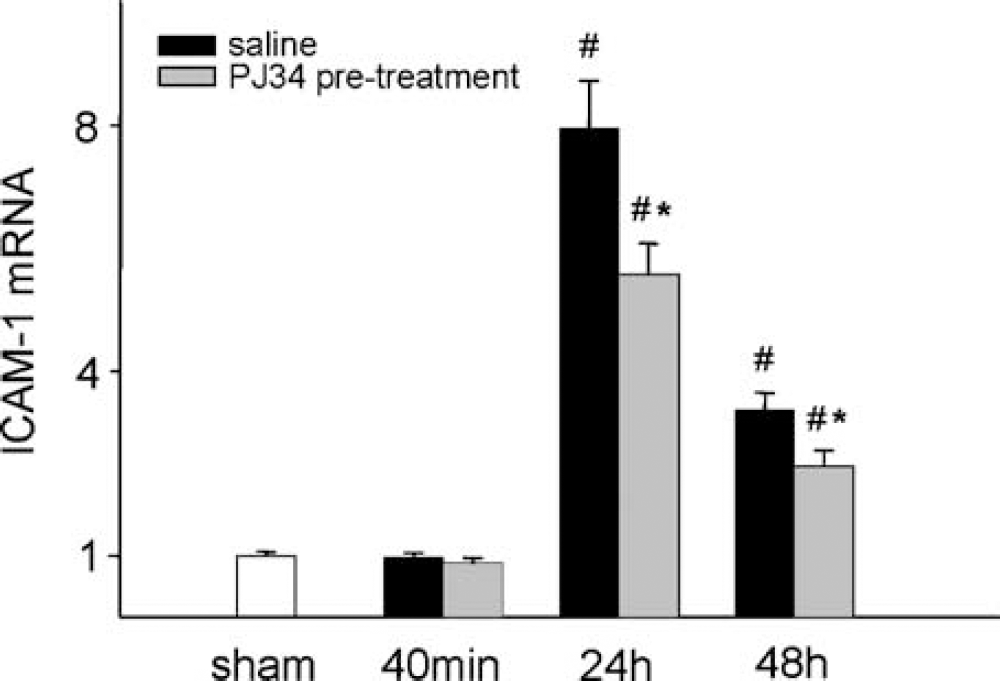
Amount of the mRNA of the endothelial adhesion molecule, ICAM-1 found in the cortices after 40 mins, 24 h, and 48 h reperfusion in saline and PJ34 pretreated animals. The RNA content is expressed in relation to the average RNA amount expressed in the sham cortices. Data are shown as mean ± s.e.m., *P < 0.05 compared with saline treatment in the same reperfusion group; #P < 0.05 compared with sham. Sham (n = 4), 40 mins (n = 6 to 6), 24 h (n = 12 to 12), 48 h (n = 12 to 12).
Western-Blotting
The amount of the endothelial tight junction protein occludin did not change at 40 mins reperfusion but significantly decreased to less than half of the original amount at 24 h reperfusion and was still lower at 48 h reperfusion in the cortices of the saline-treated animals. Pretreatment with PJ34 resulted in a trend for higher occludin content at 24 h (not significant), but on the second day the occludin content was significantly higher in the drug-treated animals. The post-treatment protocol protected against the loss of occludin at 24 h reperfusion (Figures 7A–7C). On the Western blots there was a prominent band detected at ∼25 kDa, which was inversely related to the occludin band located at ∼65 kDa (r: −0.537; P = 0.0022) (Figure 8). The amount of the 25 kDa protein was elevated only at 24 h reperfusion in the saline and PJ34 pretreated groups (3.1 ± 0.41 versus 3.0 ± 0.23 times increase in optical density compared with sham, P < 0.05), but the PJ34 post-treatment protocol significantly decreased its amount (1.8 ± 0.22, P < 0.05 compared with saline treatment).
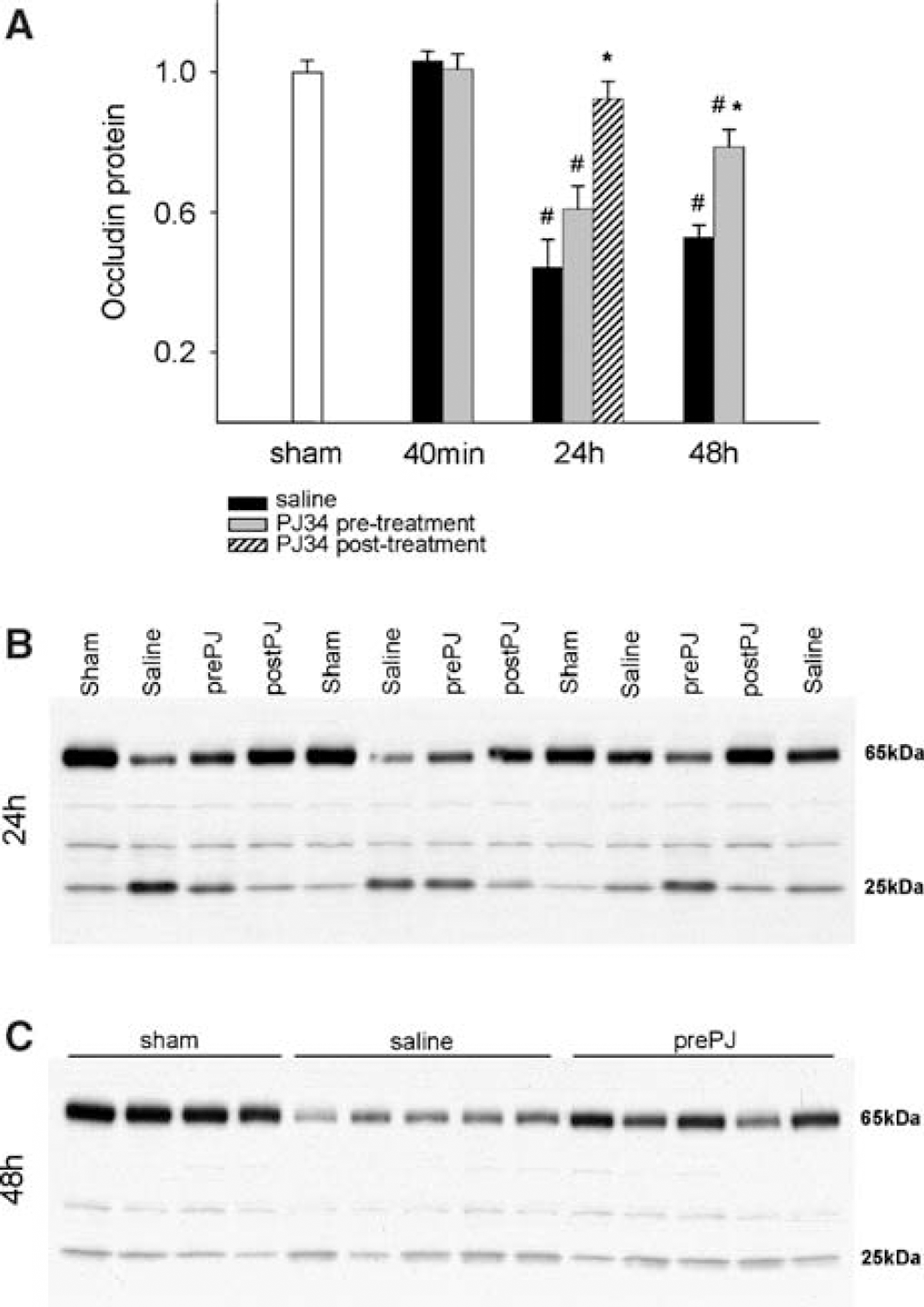
(
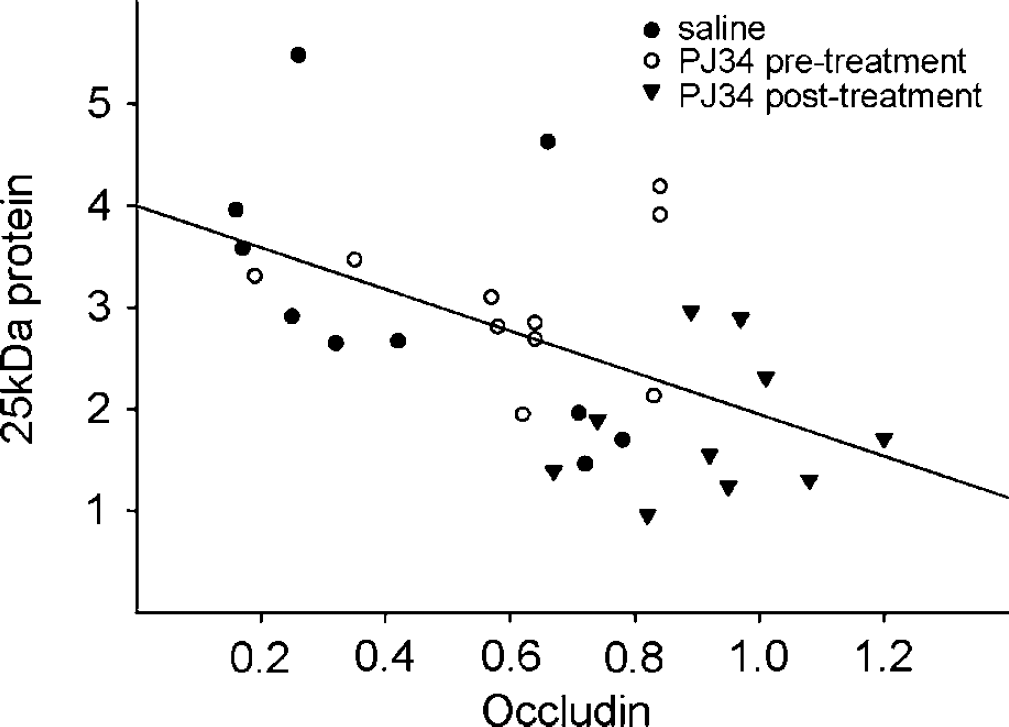
Relationship between the optical densities of the 65 and 25kDa bands on the Western blots for occludin in the 24 h reperfusion group. The densities are expressed as a relative value compared with the average density of the bands from the sham animals. Data are normalized to β-aktin for the individual animals. r = −0.537, P < 0.05.
The occludin content correlated well with EB and NaF extravasation at 48 h reperfusion in the untreated animals (r: −0.718, P = 0.02 and r: −0.689; P = 0.03), and although there was no significant correlation after 24 h, the dye contents agreed well with the amount of the 25 kDa band at this time point (r: 0.805; P = 0.005 and r: 0.67; P = 0.03). Occludin content was in inverse relation to MPO activity at 48 h as well (r: −0.656; P = 0.04).
Discussion
The new finding of our study is that PARP activation causes a major disruption of the BBB after transient ischemia. Thus, PARP inhibition by PJ34 either administered before or after global cerebral ischemia protects against delayed increases in BBB permeability to large and small molecules, reduces edema formation, preserves endothelial tight junction proteins, and decreases the leukocyte infiltration into the brain. PARP inhibition, therefore, represents an important therapeutic target for protection of the cerebral vasculature and brain against ischemic insult.
The BBB opens in a biphasic fashion after transient ischemia. There is an early opening of the BBB immediately after reperfusion and then a period of enhanced permeability of the BBB starting 6 to 24 h later (Haqqani et al, 2005; Pluta et al, 1994). Because the physical nature of the disruption of the BBB may vary over time, we used two dyes with different molecular weights for evaluating the permeability properties of the BBB. The dye EB binds to albumin in the blood. Large molecules such as EB-bound albumin are transported across the BBB mainly by pinocytotic vesicles through endothelial cells whereas small molecules such as NaF penetrate into the brain via a paracellular route involving passage through interendothelial clefts (Nakagawa et al, 1990; Pluta et al, 1994). We found that a modest increase in extravasation of NaF occurred at 40 mins of reperfusion and also observed a tendency for increased EB content at this time in the cortex as well. However, PARP inhibition did not reduce NaF and EB content at 40 mins of reperfusion. In the early phase of reperfusion, SOD and catalase have been reported to partially diminish the BBB opening (Fagan et al, 2004; Zuckerman et al, 1994), emphasizing the role of increased production of reactive oxygen species such as superoxide anion and hydrogen peroxide. The lack of effect of PARP inhibition against the early BBB opening is not contradictory to these findings because PARP activation is an event which is downstream of reactive oxygen species formation (Koedel et al, 2002).
There was a more pronounced BBB opening at 24 h of reperfusion for both NaF and EB, whereas NaF levels increased further from 24 to 48 h. EB levels did not change from 24 to 48 h, but still remained well above baseline values at this later time. These patterns of EB and NaF penetration into the brain suggest the differences in the permeability of the BBB for the two dyes and the progressive increase of the paracellular transport after ischemia. Pretreatment with PJ34 attenuated BBB permeability to both EB and NaF. Direct evidence for the speculation concerning paracellular transport is the reduction in the level of the tight junction protein, occludin, at 24 and 48 h, and its preservation with PJ34 treatment. Occludin, a ∼65kDa protein linking neighboring endothelial cells together via homotypic adhesion, is a marker of BBB integrity in vitro and in vivo (Koedel et al, 2002; Krizbai et al, 2005; Romero et al, 2003; Wachtel et al, 1999). During the 24 to 48 h reperfusion period, the levels of intact occludin decreased whereas the levels of a lower molecular weight (∼25kDa) fragment, possibly a degradation product of occludin, increased. A precursor–product relationship was further strengthened by the significant and inverse correlation between the two bands at 24 h reperfusion. It is possible that the activation of matrix metalloproteinases could be involved in occludin degradation, as it has been reported previously (Giebel et al, 2005; Wachtel et al, 1999).
An event also associated with the augmentation of BBB permeability was the dramatic increase in water content of the brain. PARP inhibition decreased EB content at 48 h and NaF content at both 24 and 48 h. One of the primary life-threatening consequences of the BBB opening after ischemia is the brain swelling caused by vasogenic edema. The development of serious brain edema between 24 and 48 h is a possible explanation for the mortality observed in some animals during this period. PARP inhibition was able to reduce brain water content at 48 h.
The increased BBB permeability and brain edema at 24 to 48 h were associated with other events which were indicative of adverse changes or damage to the cerebral endothelium. At these time periods, but not at 40 mins, we found that ICAM-1 expression and MPO activity increased. Both increased ICAM-1 expression and augmented MPO activity are indices of leukocyte infiltration into the brain. Enhanced ICAM-1 expression facilitates leukocyte adhesion to endothelial cells and the subsequent migration of leukocytes through the vascular wall (Danton and Dietrich, 2003). The measurement of MPO activity has been shown to be a reliable estimate of the extent of neutrophil infiltration into the brain (Matsuo et al, 1994a). The role of leukocyte infiltration in enhanced BBB permeability after ischemia–reperfusion (Gidday et al, 2005; Strbian et al, 2006; Veldhuis et al, 2003a) and in other inflammatory pathologies of the brain (Koedel et al, 2002; Veldhuis et al, 2003b) has been demonstrated in many studies. The administration of antibodies or antisense oligonucleotids against ICAM-1 or the depletion of neutrophils in the blood with antibodies has led to the protection of the brain and BBB against cerebral ischemia (Matsuo et al, 1994a, b; Vemuganti et al, 2004). In our experiments, the observed correlation between the MPO activity and the extravasated dye contents in the control ischemic animals suggests the detrimental effect of the infiltrating leukocytes on the BBB as well. PARP administration was able to decrease ICAM-1 expression at both 24 and 48 h and reduce MPO levels at 48 h. These results are consistent with other studies in a variety of organs in which PARP inhibition protects against leukocyte infiltration via the suppression of production of endothelial cell adhesion molecules and proinflammatory cytokines (Hassa and Hottiger, 2002; Mabley et al, 2001; Park et al, 2004; Haddad et al, 2006). Recently, Gidday et al (2005) have shown the importance of leukocyte-derived matrix metalloproteinases in postischemic cerebral vascular damage, which is in agreement with our findings regarding the role of neutrophil infiltration on the BBB.
Based on the beneficial effects of preischemic administration of PJ34 in the late phase of reperfusion, we tried a delayed treatment approach as well, and thus we gave the drug at the start of reperfusion, with an additional dose administered 6 h later. We focused on the 24 h reperfusion time point because this was a key time for examining BBB function. In this post-treatment protocol, the EB extravasation was lower compared with the saline treatment, but the difference was not significant. In contrast, NaF permeability was significantly decreased in the post-treated animals. At 24 h reperfusion in the PJ34 pre- and post-treated animals, the MPO activity was intermediate between the sham and the saline-treated animals, but the differences were not significant because of the large variability. Probably the most dramatic effect of post-treatment was on the preservation of occludin levels, which was greater than that seen with pre-treatment. Thus, postischemic administration of a PARP inhibitor may be effective in the clinical setting.
Although we cannot make a definitive statement, we speculate that the protection of the BBB after ischemia–reperfusion is a direct effect on cerebral endothelium. This view is based upon our findings that the attenuation of BBB permeability by PJ34 is associated with a decrease in expression of ICAM-1, as well as the preservation of occludin levels, both markers characteristic for endothelial cells. However, our experiments do not allow us to eliminate the contribution of effects on damaged neurons, glia, and immune cells.
In conclusion, inhibition of PARP reduced the disruption of the BBB and attenuated brain edema after global cerebral ischemia in rats. A primary mechanism of protection appears to be the attenuation of leukocyte infiltration and the preservation of the tight junction proteins such as occludin. The well-known role of PARP activation in inflammation, together with the wide range of cerebral diseases with inflammatory pathology, makes PARP inhibition a promising future target to protect BBB function.