
Abstract
Blood brain barrier (BBB) breakdown and neuroinflammation are key events in ischemic stroke morbidity and mortality. The present study investigated the effects of mast cell deficiency and stabilization on BBB breakdown and neutrophil infiltration in mice after transient middle cerebral artery occlusion (tMCAo). Adult male C57BL6/J wild type (WT) and mast cell-deficient (C57BL6/J KitWsh/Wsh (Wsh)) mice underwent tMCAo and BBB breakdown, brain edema and neutrophil infiltration were examined after 4 hours of reperfusion. Blood brain barrier breakdown, brain edema, and neutrophil infiltration were significantly reduced in Wsh versus WT mice (
Introduction
Ischemic stroke contributes significantly to morbidity and mortality in the aging population and despite many years of promising research there remains a paucity of treatment options available. 1
Therefore, identifying novel therapeutic targets, by improving our understanding of the progression of injury, is of paramount importance. 2 Early damaging events of the ischemic cascade including blood brain barrier (BBB) hyperpermeability, vasogenic edema, and onset of inflammation are important in determining survival and recovery of individuals suffering stroke. 3 Recent evidence from human4, 5 and animal6, 7 studies suggest that mast cells may be important in determining stroke severity.
Mast cells, derived from hematopoietic lineage, are involved in a number of normal physiologic functions such as immunity,
8
angiogenesis, and tissue remodelling,
9
as well as being implicated in multiple pathologic processes.
10
To exert these effects, mast cells store and release upon activation granules containing preformed histamine, serotonin, heparin, neutral proteases, major basic protein, acid hydrolases, peroxidase, phospholipases, and tumor necrosis factor alpha (TNF-α), among others. Additionally, mast cells synthesize
Increased mast cell degranulation was evident after stroke in the immature brain,
15
and after transient global ischemia in the adult rat
16
as well as after oxygen glucose deprivation
A number of mediators released by mast cells may have a role in stroke. Of particular interest is TNF-α as it comprises 25% of mast cell granule content, and a number of studies have highlighted the role of this cytokine in stroke. For example, TNF-α is thought to be key to enhanced BBB permeability and inhibition of TNF-α significantly reduced infarct size in mice after transient middle cerebral artery occlusion (tMCAo).19, 20 The percentage of TNF-α-containing mast cells has been shown to increase dramatically, ipsilaterally immediately after hypoxia ischemia in rat pups 21 and mast cell-derived TNF-α has been shown to increase T-cell infiltration, proliferation, function, and cytokine production, 22 which may contribute to ischemic damage.
Previous
Whereas previous studies of mast cells in MCAo have used rats; this is the first study that investigates whether mast cells promote BBB breakdown and neutrophil infiltration after focal cerebral ischemia in mice using an
Here, we test the hypothesis that mast cells and their secreted mediators potentiate BBB damage, vasogenic edema, and inflammation in the acute post-ischemic phase in a mouse model of stroke. We report on the function of mast cells in tMCAo, and reveal some of the mechanisms by which they may be exerting their effect
Materials and methods
Induction of Ischemia
The regulations, as specified by the Animals (Scientific Procedures) Act (1986), were strictly adhered to throughout and were performed under ethical approval of University of Strathclyde and the appropriate Home Office license (Project License No. PPL 60/3775; Personal license 60/11900). All procedures were performed in accordance with ARRIVE
23
and STAIR
24
guidelines where possible. All experiments were performed on adult male C57BL/6 wild type (WT,
Male WT or Wsh mice aged 10 to 12 weeks and weighing 25 to 30 g were anesthetized by inhalation of 3% isoflurane/1,000 mL per minute oxygen (O2) and maintained between 1.5% and 2% isoflurane/1,000 mL per minute O2 for the remainder of the procedure. Body temperature was monitored using a rectal probe and maintained at 37±0.5°C with an automatic heat mat (Harvard Apparatus, Kent, Edenbridge, UK). Perfusion of the MCA territory was measured in some animals throughout the surgical procedure using a laser Doppler flowmeter (Moor Instruments, Axminster, UK). Using Bregma as a reference point, the probe was glued in place approximately anterior/posterior −1 mm, medial/lateral +2.5 mm and readings were recorded at 5-minute intervals throughout the procedure.
Transient middle cerebral artery occlusion was performed by adaptation of the Longa method for mice. 27 A monofilament (20 mm length of which 9 mm is coated with silicone giving an overall diameter of 0.23±0.01 mm) (Doccol Corporation, Sharon, MA, USA) was inserted into the left external carotid artery and advanced along the internal carotid artery until resistance was met at the origin of the MCA. At this point, the filament was tied in place and remained for 20 minutes for TNF-α study or 45 minutes for all other animals before being withdrawn to allow reperfusion. The 20-minute occlusion animals were recovered for 5 minutes (time after occlusion onset 25 minutes), while the 45-minute occlusion animals recovered for 45 minutes (time after occlusion onset 90 minutes), 4 hours, or 72 hours. For animals undergoing sham operation, all procedures were followed as described, however the filament was removed immediately after insertion. A priori exclusion criteria were any animal found to be moribund due to excessive weight loss (>20% of start weight) or that exhibited no ischemic injury. To establish severity of deficit, animals recovering to 72 hours were assessed for development of general (e.g., posture and spontaneous activity) and focal (e.g., circling) neurologic deficits using the Clarks deficit scoring system at 24, 48, and 72 hours after tMCAo, where scores for both range from 0 (healthy) to 28. 28
Stereotaxic Injection of Cromoglycate
In one set of experiments, WT and Wsh mice were randomly assigned to receive either cromoglycate (Sigma, Poole, UK) (75 μg in 2 μL saline) or sterile saline (vehicle), injected into the ventricle, anterior/posterior −0.6 mm medial/lateral, +1 mm, relative to Bregma and 2.4 mm ventral, using the surface of the dura for zero reference. Stereotaxic injection was performed immediately before onset of MCAo under the same anesthesia using a 32-gauge needle attached to a Hamilton syringe. Blinding of vehicle/cromoglycate assignment before stroke induction was achieved by independent investigator. Over a 2-minute period 2 μL of sodium cromoglycate or vehicle was injected, and the needle was left in place for a further 2 minutes. The needle was then slowly retracted and the wound sutured.
Termination and Tissue Processing
The experimenter was blinded to the experimental group to which the animal belongs (mouse genotype, treatment received, stroke, or sham surgery received) in all postmortem analysis by recoding of animals by an independent investigator. At the end of each experiment, animals were killed by injection of 200 μL of sodium pentobarbital. The brains were then immediately removed and either fresh frozen in isopentane (−40°C) for histology or the hemispheres separated and homogenized in 1% protease inhibitor (in 20 mmol/L TRIS, pH 7.4) at a 5 times w/v ratio, and kept on ice. Triton X-100 was added to samples to a concentration of 1% and aliquots were stored at −80°C. Before use, samples were centrifuged at 2,000
Histologic Processing
Hematoxylin and eosin-stained 20-μm-thick coronal sections from eight distinct neuroanatomic regions representative of the forebrain
29
were analyzed for measurement of lesion and brain edema using a densitometer. For each section, the ipsilateral and contralateral hemispheric areas were measured, as well as areas of ischemic lesion represented by regions of pallor. For confirmation, ischemic damage was assessed under a light microscope through identification of regions containing pyknotic and necrotic neurons. The volumes of each hemisphere and of the lesion were calculated from area under the curve of areas measured at each of the eight coronal levels against their interaural (IA) distance, where
Toluidine blue metachromasia was used to identify mast cells in tissue. For each animal, a total of 24 coronal, 4% paraformaldehyde-fixed sections were stained, 2 from each of the 8 regions of the forebrain, with additional sections from regions 2.86 mm, 1.98 mm, 1.00 mm and 0.16 mm IA, due to a previous observation that mast cell numbers were higher in the posterior regions of the forebrain (unpublished observation). Cells were manually counted under a light microscope (× 200 magnification).
Assessment of Blood Brain Barrier Permeability
Permeability of the BBB was measured by labelling endogenous immunoglobulin G (IgG) within brain tissue, which would normally be excluded by an intact BBB. 30 Coronal acetone-fixed sections at regions of the MCA territory, at the level of the septal nucleus (3.94 mm IA) and at the level of the hypothalamus (2.86 mm IA), were stained with either FITC-conjugated goat anti-mouse IgG (prepared in 5% normal goat serum) (1:250) (Abcam Ltd, Cambridge, UK), or FITC-conjugated horse anti-mouse IgG (1:250) (Abcam Ltd) and Vectashield (Vector Laboratories, Peterborough, UK), containing the nuclear stain DAPI (4′,6-diamidino-2-phenylindole). Negative controls were incubated in the absence of antibody.
Stained sections were analyzed using an upright epifluorescent microscope (Nikon Eclipse E600, Tokyo, Japan) at × 200 magnification, at excitation wavelengths of 405 and 488 nm for DAPI and FITC, respectively, along with the Metamorph imaging software (Molecular Devices, Sunnyvale, CA, USA). A fluorescent threshold was set by reduction of the exposure time of the negative controls until no signal was detected. Thereafter, images were acquired of fluorescently-stained tissue within the ipsilateral hemisphere. Using the Image J software (National Institute of Health, Bethesda, MD, USA), areas and density of FITC fluorescence were measured.
Neutrophil Quantification
Immunofluorescent staining was performed to identify and quantify neutrophil infiltration post-MCAo in adjacent sections to BBB measurements at the level of the septal nucleus (3.94 mm IA) as well as at the level of the anterior hippocampus (1.98 mm IA). Neutrophils were labelled using SJC, a custom rabbit polyclonal antibody (1:1,000 dilution) (Gifted by Daniel Anthony, University of Oxford, UK), and secondary FITC-conjugated goat anti-rabbit (1:250) with cell nuclei stained using Vectashield. Neutrophil infiltration was identified and quantified using an upright epifluorescent microscope (Nikon Eclipse E600), at excitation wavelengths of 405 and 488 nm for DAPI and FITC, respectively. Cells fluorescently marked in both hemispheres were quantified in duplicate sections and the mean number of cells expressed.
Enzyme Linked Immunosorbent Assay
The concentration of TNF-α was measured in brain homogenates using a commercially available set, Mouse TNF-α set 1 (BD Biosciences, Oxford, UK). Brains from mice which underwent tMCAo of 20 and 45 minutes followed by 5 and 45 minutes reperfusion, respectively, were removed on termination. Brains were placed in 1% protease inhibitor (in 20 mmol/L TRIS, pH 7.4) (Merck Chemicals, Nottingham, UK), at a 5 times w/v ratio, and kept on ice. The hemispheres were separated and homogenized with a hand-held homogenizer until the sample was cloudy and contained no visible pieces of tissue. Triton X-100 (Sigma, Poole, UK) was added to samples to a concentration of 1% and aliquots were stored at −70°C. Before use, samples were centrifuged at 2,000
Protein Concentration Assay
Protein concentration in brain homogenates was determined using the Bio-Rad Protein assay (Bio-Rad Laboratories, Hemel Hempstead, UK) using the same samples as above following manufacturer's guidelines. Samples were diluted 1:100 with dye reagent and the absorbance of each sample then read at 595 nm. The protein concentration of each sample was determined from the absorbance values of the protein standards, using linear regression,
Mouse Angiogenesis Proteome Profiler
Relative expression of an array of proteins associated with angiogenesis was measured in brain tissue homogenates, using the same samples as above, using a commercially available proteome profiler kit ARY015 (R&D Systems, Abingdon, UK).
The protocol was performed following the manufacturer's guidelines. Samples containing detection antibodies were incubated on a membrane permeated with capture antibodies. Array membranes were then placed in an autoradiography cassette and exposed to X-ray film for multiple periods between 1 and 10 minutes. Five minutes exposure time was used for analysis due to uniformity of positive control.
Statistics
Data are presented as mean±standard error of mean (s.e.m.). Comparisons between two groups were performed using Students’ unpaired
Results
Cerebral Blood Flow and Neurologic Deficit Were Unaffected by Mast Cell Deficiency
Given the potential vasoactive effects of mast cell-derived mediators and potential vascular differences between the two strains of mice used, we monitored CBF within the MCA territory throughout the occlusion period by laser Doppler flowmetry in WT and Wsh mice. However, we found that blood flow was reduced to a similar level in both WT and Wsh mice during the occlusion (Figure 1A). After placement of the microvascular clip on the left internal carotid artery, blood flow dropped to around 60% of baseline levels. Perfusion was further decreased upon insertion of the filament, to 45% of baseline flow, where it remained stable throughout the occlusion period. Removal of the intraluminal thread restored blood flow to a level similar to that before insertion. These data would suggest that mast cells do not have an effect on local CBF during the ischemic period, and also suggest that Wsh mice have no inherent abnormalities that increase severity of ischemia.
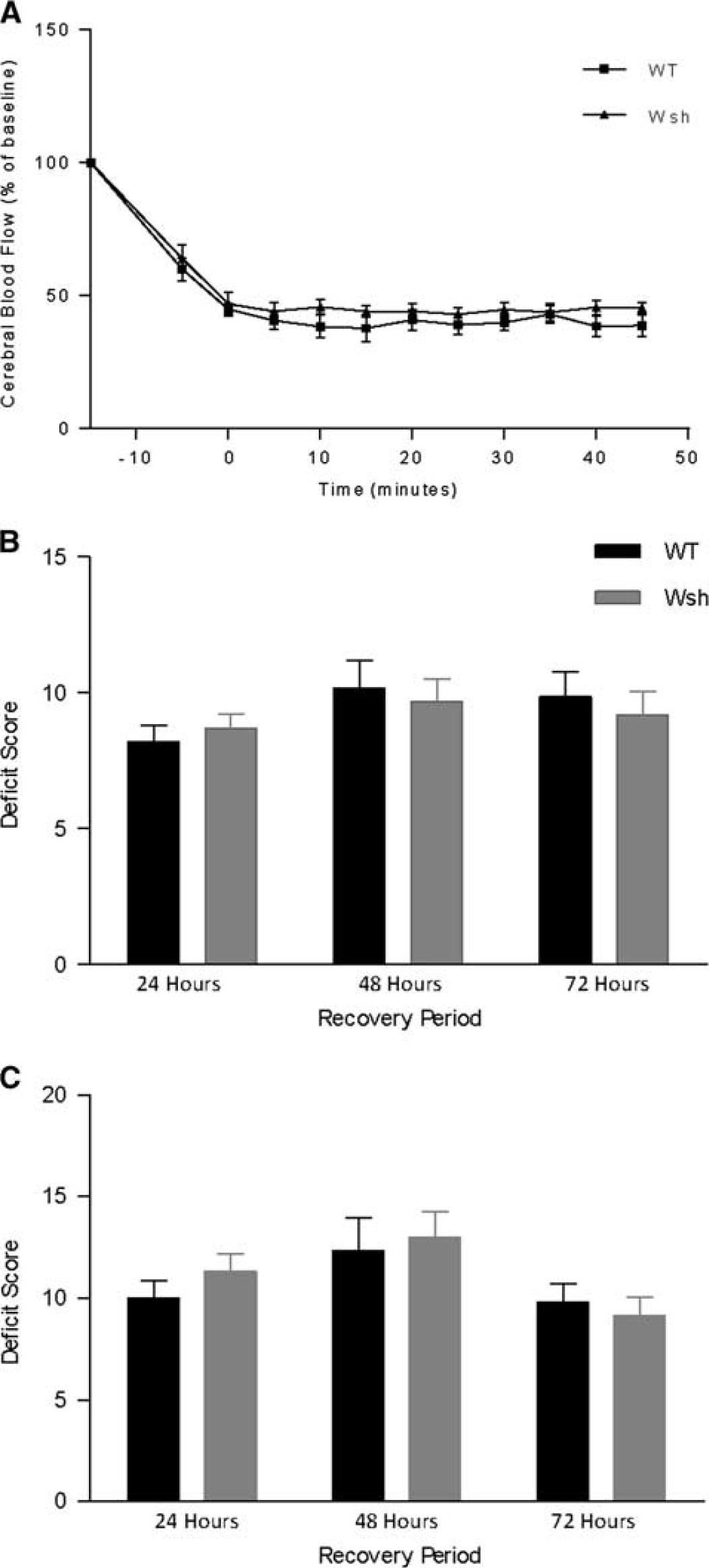
Cerebral blood flow and neurologic deficit were unaffected by mast cell deficiency. In wild-type (WT) (
There were no animals excluded from either group due to excessive weight loss, according to the above-mentioned exclusion criteria. However, there were two premature deaths in the WT group, while no mice died in the Wsh group. The two deaths in the WT group occurred during the first 24 hours and were attributed to severe brain edema after ruling out hemorrhagic transformation and on observation of an enlarged ipsilateral hemisphere. In addition, three mice were excluded (WT (
Mast Cells Increased in the Ischemic Hemisphere in Wild-Type Mice
The population of mast cells is known to increase in a number of disease states, which is indicative of active mast cell involvement in disease progression.
11
Therefore, toluidine blue-positive mast cells were counted in brains 4 hours after tMCAo. Mast cells were present within brains 4 hours after tMCAo and in sham and naïve WT animals, located predominantly perivascularly within the posterior regions of the forebrain (Figures 2A and 2B). There were no mast cells observed in Wsh mice brains and there was no difference in mast cell numbers between the two hemispheres in the sham and naïve WT animals (Figure 2D). Whereas the total brain numbers of mast cells after tMCAo did not change compared with sham (Figure 2C), mast cell numbers were increased by around 50% in the ipsilateral compared with the contralateral hemisphere after tMCAo in WT mice (
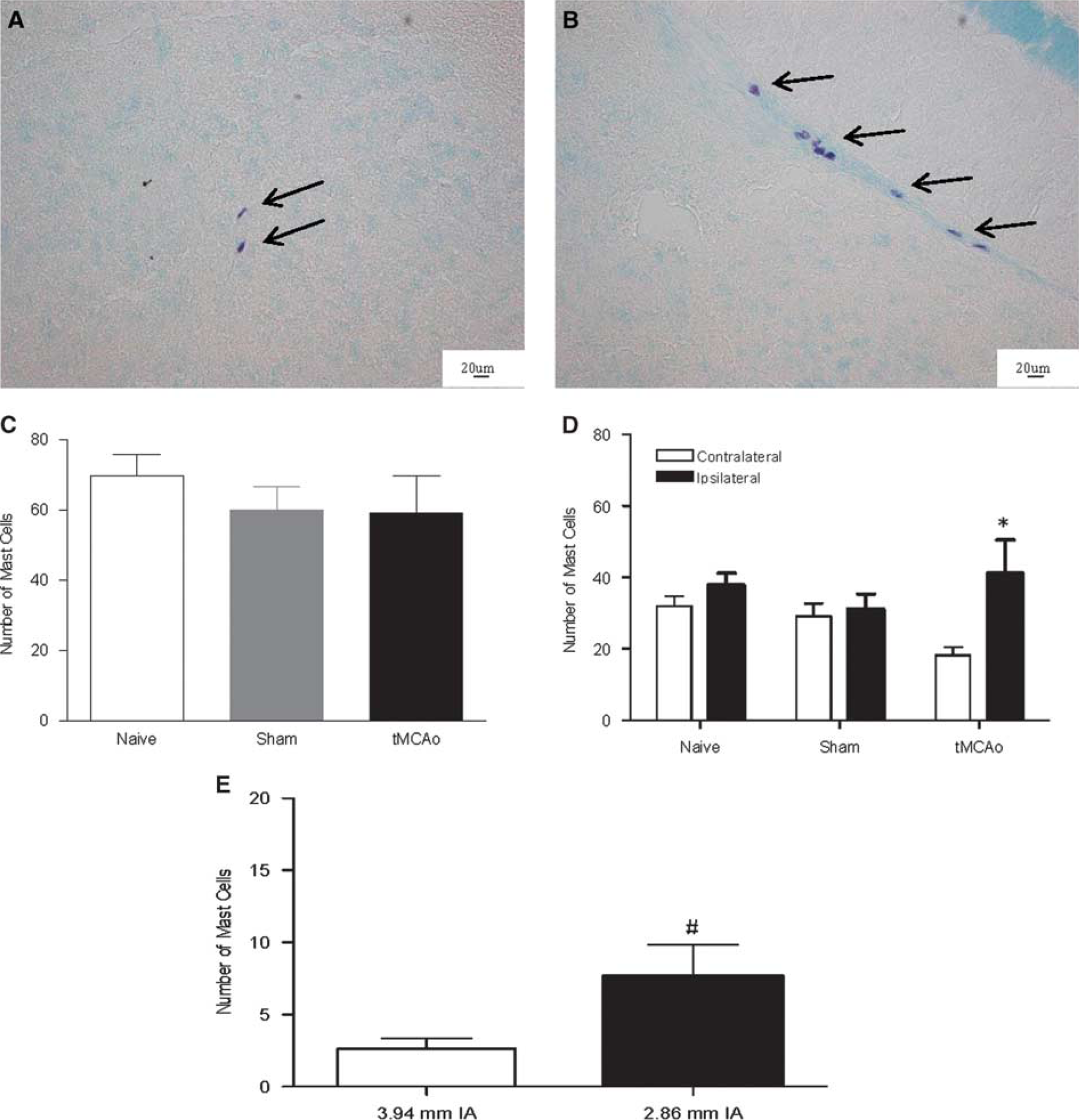
Mast cell numbers increased in the ischemic hemisphere post transient middle cerebral artery occlusion (tMCAo). Toluidine blue-stained mast cells, found within the neuropil (
Blood Brain Barrier Permeability Was Reduced by Mast Cell Stabilization and Deficiency
Mast cells have been shown previously to increase BBB permeability post-MCAo in a rat model.
6
Therefore, we measured endogenous IgG within brain tissue, which would be excluded from entering brain tissue by a healthy BBB. At 4 hours after tMCAo, cromoglycate treatment significantly reduced IgG within the tissue compared with vehicle treatment in WT mice at the level of the hypothalamus (
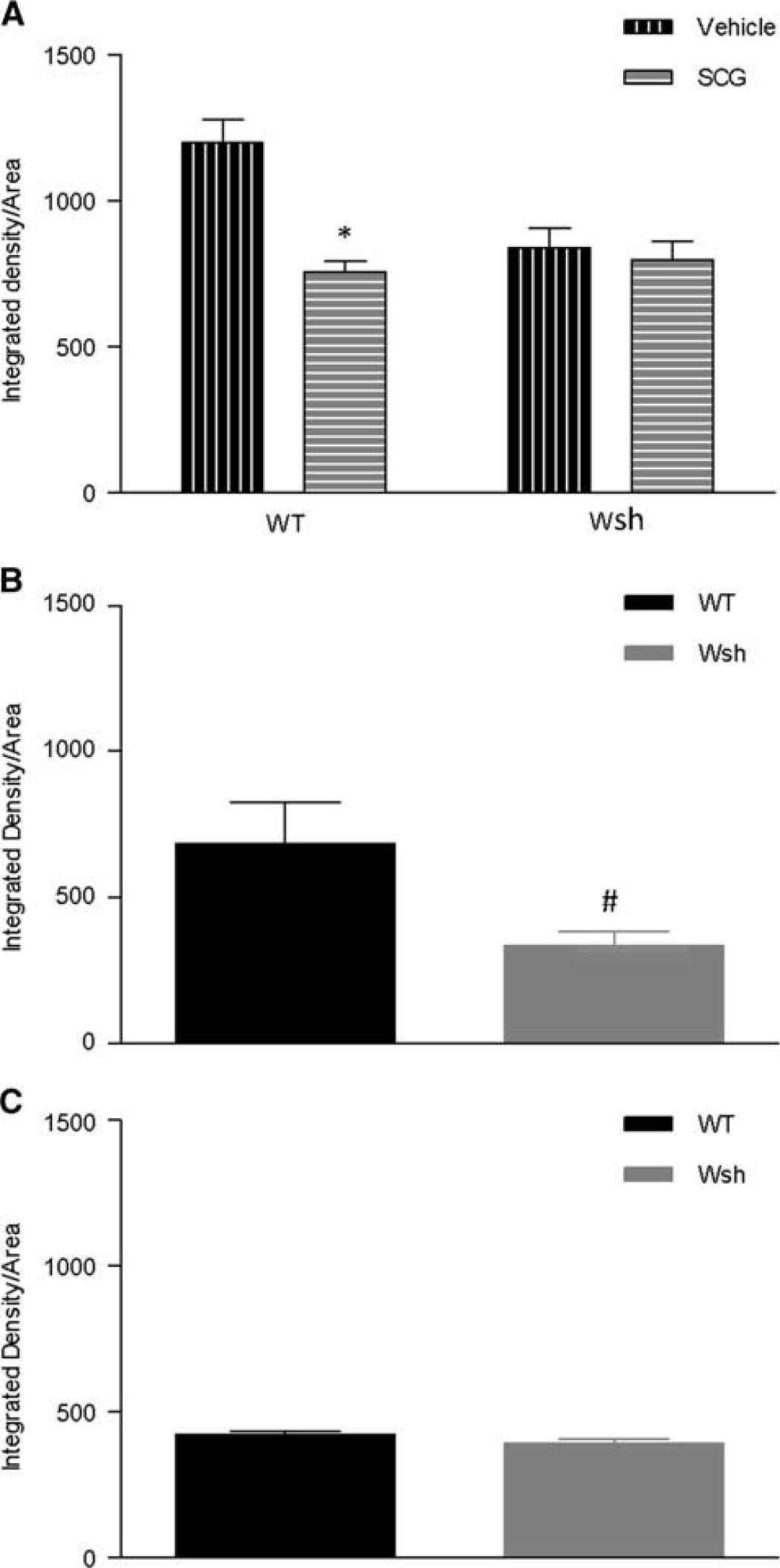
Immunoglobulin G (IgG) leakage into brain tissue was reduced by mast cell stabilization and deficiency. Endogenous IgG was detected by immunostaining of the level of the hypothalamus in wild-type (WT) and Wsh mice pretreated with cromoglycate versus vehicle (
Brain Edema Volume Was Reduced by Mast Cell Stabilization and Deficiency
Mast cells have been shown to contribute to post-MCAo brain edema,
6
and consistent with this, edema volume was significantly reduced by ∼60% after cromoglycate treatment compared with vehicle in WT mice (
Edema volume was reduced by mast cell stabilization and deficiency. The degree of swelling of the ipsilateral hemisphere compared with the contralateral hemisphere was assessed in hematoxylin and eosin stained tissues sections from the MCA territory in Wsh and wild-type (WT) mice pre-treated with cromoglycate versus vehicle (
Neutrophil Recruitment Was Attenuated by Mast Cell Stabilization/Deficiency
Neutrophils are known to contribute to post-stroke injury through release of neurotoxic proteases and neutrophil elastase, and mast cells have been shown to be involved in their recruitment in the rat.
6
Cromoglycate treatment significantly decreased neutrophil recruitment compared with vehicle treatment at the level of the septal nuclei in WT mice (
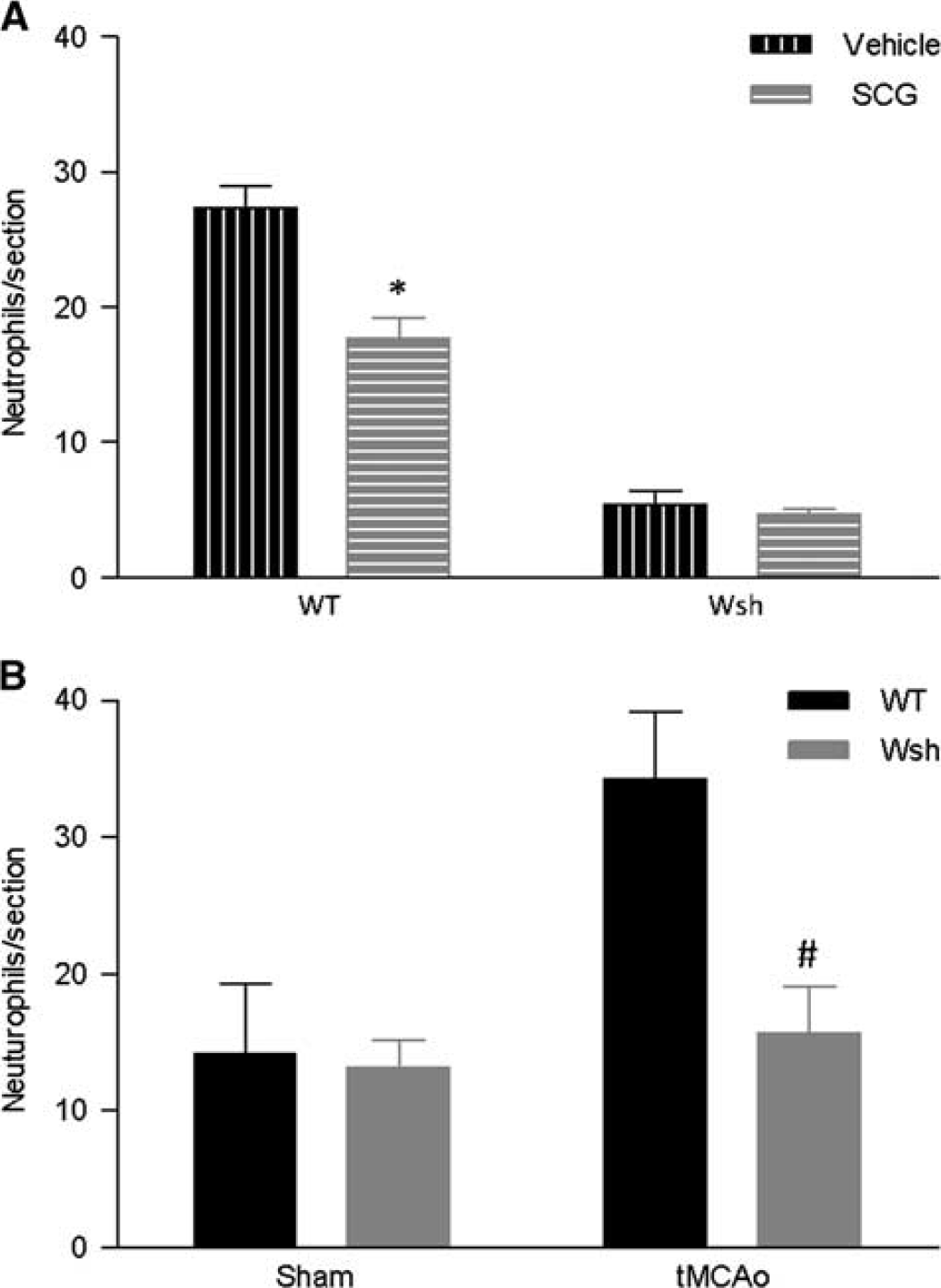
Neutrophils recruitment was attenuated by mast cell stabilization and deficiency. Immunostained neutrophil numbers were quantified in duplicate tissue sections in wild-type (WT) (
Lesion Volume Was Reduced by Mast Cell Stabilization in Wild-Type mice
To assess the contribution of mast cells to the development of the ischemic lesion, volume of injury was assessed on tissue sections at termination of the experiment. Lesion volume was significantly reduced by around 50% in WT mice pretreated with cromoglycate versus vehicle at 4 hours after tMCAo (
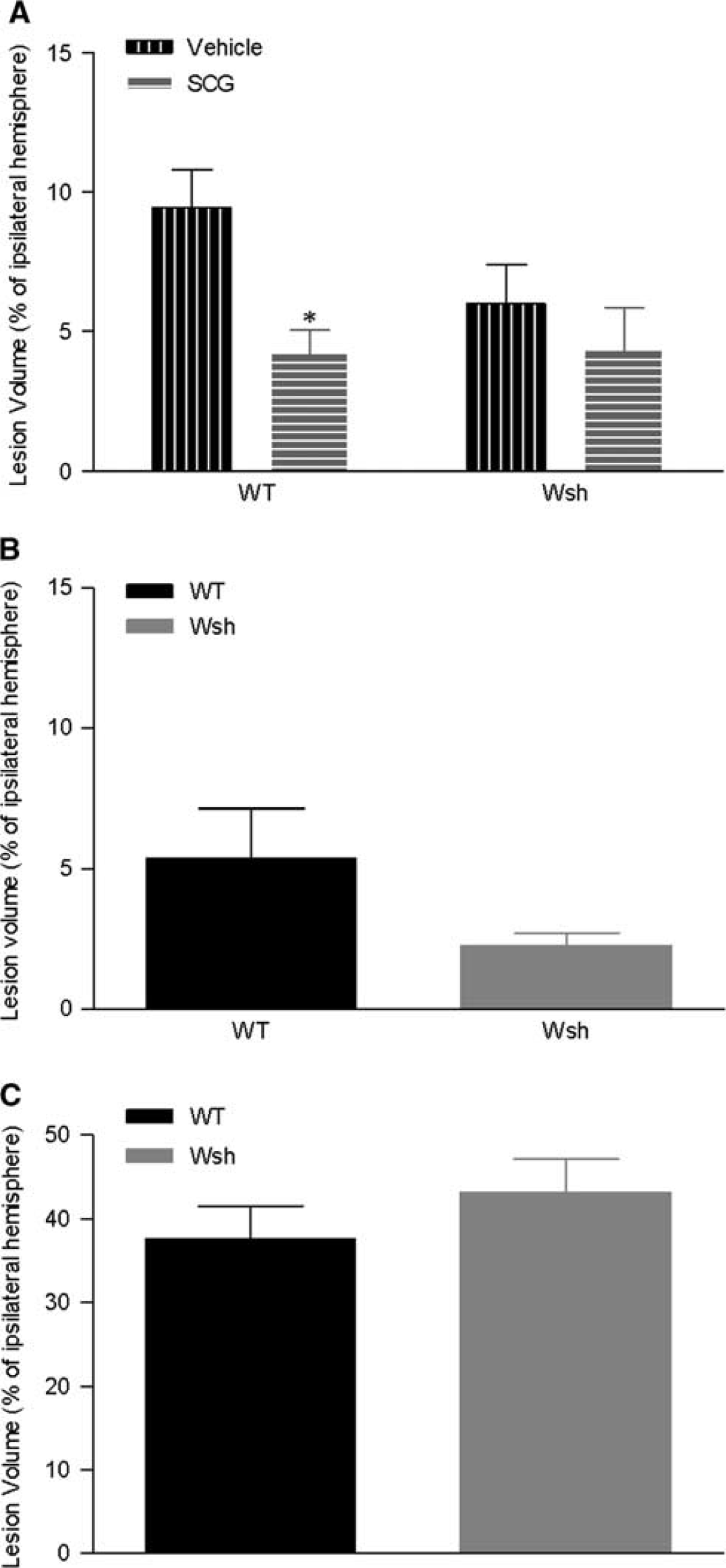
Volume of tissue damage was decreased by mast cell stabilization. Ischemic lesion volume at 4 hours after transient middle cerebral artery occlusion (tMCAo) was measured in hematoxylin and eosin stained tissue section representative of the MCA territory in wild-type (WT) and Wsh mice pretreated with cromoglycate (WT
Tumor Necrosis Factor-α Levels in Brain Tissue
A wealth of evidence indicates TNF-α to both increase ischemic injury and to protect the brain from injury under certain conditions in animal models of stroke, and that it is expressed in the acutely injured ischemic brain.19, 20 Mast cells are known to store preformed TNF-α, 21 and may be an early source of this cytokine. Tissue concentrations of TNF-α, measured by ELISA, were similar in WT and Wsh mice at 25 minutes after onset of occlusion (20 minutes of occlusion and 5 minutes of reperfusion) or in sham animals (Figure 7A). Likewise, the TNF-α concentration was similar in all WT and Wsh groups at 90 minutes after onset of occlusion (45 minutes of occlusion and 45 minutes of reperfusion) (Figure 7B). No TNF-α was detectable in serum samples at either time point analyzed (data not shown). These data do not support the idea that mast cells release a significant amount of TNF-α in the acute period post-MCAo measured in this study.
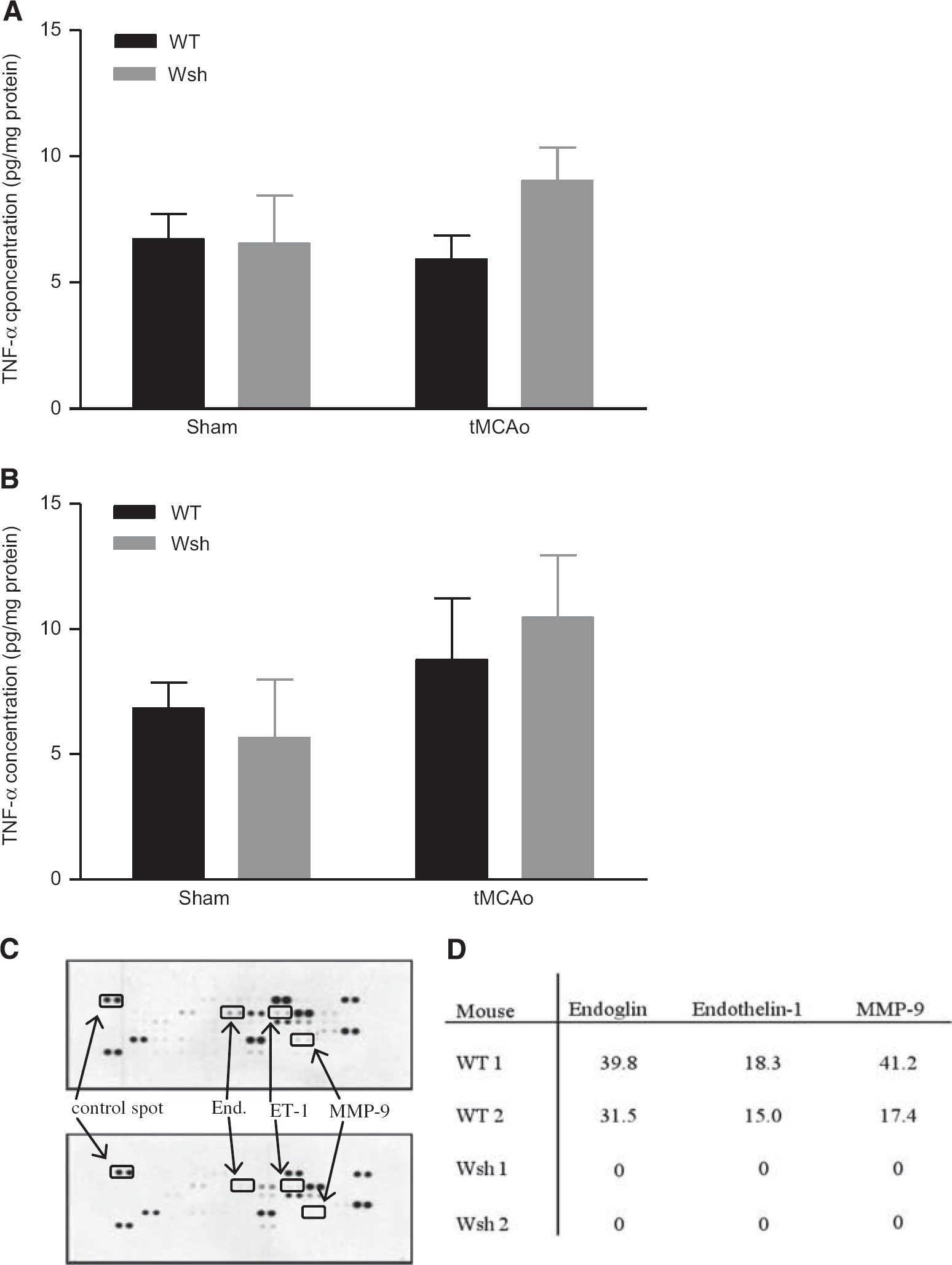
Angiogenesis proteome profile but not tumor necrosis factor alpha (TNF-α) concentration was altered by mast cell deficiency. TNF-α concentration was measured by ELISA in brain homogenates at 25 minutes after onset of occlusion in Wsh (
Angiogenesis Array of Brain Tissue
Mast cells store and release upon stimulation a broad array of vasoactive and proangiogenic mediators, 11 which may be deleterious in the ischemic brain. Therefore, we assessed the contribution of mast cells to the expression of 53 proteins known to be expressed by mast cells. At 90 minutes after onset of ischemia, the proteomic profiles between WT and Wsh mice were strikingly different for three mediators. Despite expression in WT, there was a complete deficiency of expression of endoglin, endothelin-1, and MMP-9 in Wsh mice (Figure 7C), highlighting a potential role of mast cells in the expression of these mediators posttMCAo. This result was reproduced in another set of WT and Wsh mice. In WT, endoglin was expressed with levels around 30% to 40% of the positive control spot, while MMP-9 expression was 20% to 40% of the positive control spot and endothelin-1 expression was approximately 18% of the control spot (Figure 7D).
Discussion
Being resident in the brain, in perivascular locations, and already present at onset of ischemia, mast cells are likely early initiators of neuroinflammation and BBB failure in stroke. The key findings of the present study are that mast cells increase in numbers in the ischemic hemisphere and promote neutrophil infiltration, BBB breakdown, and edema within 4 hours, but not by 72 hours, after tMCAO in mice. These mast cell-mediated effects are not likely to be due to mast cell-derived TNF-α but possibly caused by MC-derived endothelin, endoglin, and MMP-9.
In a previous study, mast cells were shown to increase after hypoxia-ischemia in the immature brain
18
but in the present study, although the total number of mast cells in the brain does not change, this is the first time mast cells have been shown to increase in localization in the ischemic hemisphere after tMCAo in adult mice. In addition, mast cells stabilization has been shown to reduce BBB breakdown and brain edema after tMCAo in the adult rat though no inflammatory response was assessed. We now extend these studies showing success of an
We have addressed our hypothesis using two strategies, namely an
Mast cells contain and release upon activation an array of mediators which can potentially decrease the integrity of the BBB leading to vasogenic edema, through degradation of extracellular matrix and tight junction proteins, such as MMP-9. 7 In the present study, we observed a lack of expression of MMP-9 in Wsh mice observed at 90 minutes after onset of ischemia. Matrix metalloproteinase-9 is capable of digesting most of the extracellular matrix components including laminin, fibronectin, vitronectin, and collagen, and also the tight junction proteins occludin and claudin. Additionally, mast cell-derived chymase, tryptase, and cathepsin G activate MMP-9, from its inactive form, and also directly degrade the BBB independent of MMP-9. 16 Alternatively, mast cells may promote neutrophil infiltration, as shown in the present study, a potent source of MMP-9. 32 Therefore, the deficiency in expression of MMP-9 in the Wsh compared with WT mice indicates a potential direct or indirect mast cell contribution to BBB degradation in focal cerebral ischemia.
Deficiencies in endothelin-1 and endoglin expression were also revealed in Wsh compared with WT mice after focal ischemia. In both experimental and human stroke, the vasoconstrictor endothelin-1 is widely regarded to increase injury severity by contributing to lesion development, BBB disruption, and edema. In mice overexpressing endothelin-1, BBB permeability was increased, as was brain edema and lesion volume compared with WT controls. 33 Also, the development of brain edema post-ischemia has been shown in animal models to be dependent on endothelin-1, which is thought to increase aquaporin4 channel expression on astrocytic end-feet, facilitating water uptake across the BBB. 33 Endoglin (CD105) is an accessory protein of the transforming growth factor-β receptor system, is expressed predominantly on endothelial cells, and is increased in expression during angiogenesis, in which it contributes to smooth muscle and endothelial proliferation, pericyte migration, and production of extracellular matrix proteins. 34 Under ischemic conditions, endoglin expression is increased on mouse endothelium, and is also found abundantly in the ischemic hemisphere 28 days after permanent MCAo in mice. 35 Therefore in the present study the absence of endoglin in the Wsh mouse brain may be indicative of a less responsive vascular endothelium, due to a reduction of injury to the BBB. The reduced expression profile of these mediators was determined at 90 minutes after onset of occlusion. Therefore, mast cells may be involved in reinforcing and maintaining the on-going expression of these mediators after induction of ischemia to contribute to BBB breakdown observed at 4 hours after tMCAo.
The present study establishes that mast cells may promote neutrophil infiltration at 4 hours after tMCAo. However given that neutrophil accumulation peaks at later points, further studies are required to establish whether there are differences in kinetics of recruitment of neutrophils after mast cell deficiency or stabilization. In either case, these results highlight a potential role of mast cells in orchestrating an early neuroinflammatory response in the ischemic brain. Indeed, a full repertoire of immune competent cells, including neutrophils, has been shown to infiltrate the ischemic hemisphere in the hours and days after tMCAo, with the potential to be either damaging or protective. 32 Since mast cells are resident in the brain and capable of responding rapidly to produce an array of chemokines and cytokines, such as eotaxin, IL-8 (in rodents), which promote recruitment, vascular adhesion, diapadesis, and activation of leukocytes, they may act as a beacon to initiate neuroinflammation during the acute ischemic period. This concept is supported by the finding that mast cells were activated before microglia in a rat model of hypoxia-ischemia, and that cromoglycate reduced both microglia and astrocyte activation in the subacute and chronically injured brain through stabilization of mast cells. 18 However, since neutrophils also release MMP-9 and have also been shown to affect BBB permeability, it is unclear in the present study whether BBB breakdown is caused by mediators derived from mast cells or neutrophils or both. In future studies, Wsh mice reconstituted with bone marrow-derived cultured mast cells from MMP-9−/− or WT mice can be used to show whether MMP-9 induced BBB breakdown is mast cell derived.
The reduction in recruitment of neutrophils, BBB permeability, and brain edema may have been expected to translate to reduced lesion size. A more stable BBB should reduce exposure to potentially toxic circulating proteins, offering neuroprotection. Additionally, alleviation of edema could reduce lesion development by preventing vascular compression, secondary ischemia, and herniation. 36 However, while mast cell stabilization by cromoglycate reduced lesion development compared with vehicle, there was no significant effect on lesion size in Wsh compared with WT mice. This discrepancy may be either due to other known abnormalities in Wsh mice, albeit these would be expected to increase not decrease infarct size, or due to additional non-specific contributing protective effects of cromoglycate, however this is unlikely given that cromoglycate did not affect lesion size in Wsh mice, indicating an effect specific to mast cells. Alternatively, this discrepancy possibly reflects the 4-hour ischemic period measured and that cromoglycate may be delaying evolution of infarct.
Despite this it appears mast cells may be important for mortality beyond 4 hours of reperfusion. The extent of brain edema, around a 10% increase in the ipsilateral hemisphere volume, was similar in WT mice after both 4 and 72 hours of reperfusion, and at the latter time point Wsh mice were affected to a similar degree. This could indicate that brain edema develops at a slower rate in the absence of mast cells. Additionally, in WT mice recovering to 72 hours there was 25% (2 animals) mortality within the first 24 hours, which was attributed to brain edema, while there was no mortality in the Wsh mice. Therefore, mast cells may be causal to this increased mortality by mediating the development of brain edema in the period of reperfusion between 4 and 24 hours.
After 72 hours of recovery, the role of mast cells seems to be less important in this model compared with the acute recovery period. It may be the case that after the initial BBB opening in the acute ischemic period that the mast cell population is exhausted from overt degranulation and is no longer capable of influencing the BBB. There is some evidence to support this idea from studies in ischemic stroke and traumatic brain injury in rats. After 1 day of recovery from traumatic brain injury, there was a dramatic decrease in the numbers of mast cells in the brain compared with uninjured controls, and the population remained low at the fourth day of recovery. Also, evaluation of BBB opening for 5 weeks after 90 minutes tMCAo in rats indicated a gradual repair of the BBB after an initial surge in opening during the first 6 hours of reperfusion. Interestingly, it took around 1 week for the mast cell population to be restored to normal levels after traumatic brain injury, while the BBB in rats who underwent tMCAo was again highly permeable after 1 week of reperfusion.37, 38 The alternative to this is that mast cells are essential for maintaining the integrity of the BBB in the period beyond the 72-hour reperfusion time point analyzed. It is known that mast cells are important modulators of angiogenesis, 9 and in their absence normal BBB repair mechanisms may be impaired. A better understanding of the temporal profile of mast cell contribution to BBB opening will increase our understanding of when any mast cell directed intervention may be appropriate.
The role of TNF-α in experimental stroke is not clear cut, as it has been shown to be protective and damaging in animal models.19, 20 The TNF receptor (TNFR) subtype appears to dictate whether TNF-α elicits protective or detrimental responses dependent on downstream adaptor proteins, with TNFR1 activation leading to cell death and TNFR2 activation promoting survival. However, in this model TNF-α does not appear to be responsible for ischemic pathology related to mast cells or otherwise as there was no detected decrease in TNF-α in brain homogenates after tMCAo in Wsh mice compared with WT or indeed sham-operated mice of either strain. This was evident at both 25 minutes after onset of occlusion, and also 90 minutes after onset of occlusion. Worthy of note is that release of preformed TNF-α from mast cell granules may be concentrated to specific regions of the brain in WT mice and any local increases in TNF-α might not be detected in the total hemispheric concentration, precluding any differences being detected.
In summary, the data presented here indicate that mast cell numbers increase in localization in the ischemic hemisphere in the acute period of reperfusion after tMCAo and are potentially initiators of neuroinflammation and BBB breakdown. The successful utilization of
Footnotes
ACKNOWLEDGMENTS
The authors thank Prof Daniel Anthony, University of Oxford, UK, for the kind gift of the antibody SJC.
The authors declare no conflict of interest.