
Abstract
Brain edema is a major and often mortal complication of brain ischemia. Vascular endothelial growth factor (VEGF) is also known as a potent vascular permeability factor and may play detrimental roles at the acute stage of brain infarction. Our goal in this study was to explore protective effects of gene transfer of soluble flt-1 (sFlt-1), a natural inhibitor of VEGF, on focal brain ischemia. Adenoviral vector encoding sFlt-1 or β-galactosidase as control was injected into the lateral ventricle 90 mins after photochemical distal middle cerebral artery occlusion in male spontaneously hypertensive rats. The transduced sFlt-1 was released to the cerebrospinal fluid from the ventricular wall and significantly increased 6 h, 1 and 7 days after sFlt-1 transfection. One day after brain ischemia, sFlt-1 gene transfer significantly reduced infarct volume (by 35%), brain edema (by 35%), and blood—brain barrier permeability (Evans blue extravasation; by 69%) with diminished phosphorylation of focal adhesion kinase (FAKtyr397 and FAKtyr861) in the ischemic vessels. Seven days after ischemia, sFlt-1 gene transfer also significantly attenuated infarct volume (by 29%) and monocyte/macrophage infiltration (by 27%), although there were no reductions in angiogenesis by sFlt-1 overexpression. These results suggest that sFlt-1 gene therapy targeting brain edema in acute stage of brain ischemia may be useful for brain infarction.
Introduction
Brain edema is a major and often mortal complication of brain ischemia, and is led by increased vascular permeability (Kimelberg, 1995). Vascular endothelial growth factor (VEGF) is a dimeric glycoprotein that is mitogenic for endothelial cells. Vascular endothelial growth factor, however, was also discovered as a vascular permeability factor (Senger et al, 1983) and is induced in the ischemic brain (Hayashi et al, 1997). While several studies reported that administration or overexpression of VEGF protects against brain ischemia (Hayashi et al, 1998; Wang et al, 2005), there are studies that revealed favorable effects of blocking VEGF before induction of brain ischemia (van Bruggen et al, 1999; Schoch et al, 2002). Thus, it is important to elucidate whether inhibition of VEGF activity protects against brain ischemia, even after induction of brain ischemia.
Gene transfer is an attractive tool for studies of neurobiology and has a potential for therapy of cerebrovascular disease (Heistad and Faraci, 1996). Recent studies have shown that gene transfer may protect against the ischemic damage when gene products were already expressed (Betz et al, 1995). To rationalize gene therapy for brain ischemia, it is important to show efficacy of gene transfer even when genes are administered after induction of brain ischemia. We have recently shown that gene transfer is successfully accomplished after induction of brain ischemia either to brain parenchyma or ventricular walls (Ooboshi et al, 2001; Takada et al, 2003; Kumai et al, 2003) and that postischemic gene therapy may protect against cerebral ischemia (Kumai et al, 2004; Takada et al, 2005; Ooboshi et al, 2005).
We have also reported usefulness of gene transfer of a soluble form of the VEGF receptor-1 (soluble flt-1 (sFlt-1)) for inhibition of VEGF activity (Ohtani et al, 2004). Soluble flt-1, which is expressed endogenously by vascular endothelial cells, can directly sequester VEGF and function as a dominant-negative inhibitor by heterodimerizing with the extracellular ligand-binding region of the membrane spanning VEGF receptor-1 (Flt-1) and VEGF receptor-2 (KDR/Flk-1) (Kendall and Thomas, 1993; Kendall et al, 1996). In this study, we delivered the sFlt-1 gene into the cerebral ventricle 90 mins after focal brain ischemia, and examined the effect of sFlt-1 overexpression on vascular permeability and whether sFlt-1 gene therapy would protect against focal brain ischemia.
Materials and methods
Adenoviral Vectors
We used replication-deficient recombinant adenoviral vectors expressing β-galactosidase (AdlacZ) or sFlt-1 (AdsFlt-1). The 3.3-kb mouse sFlt-1 gene was obtained from a mouse lung cDNA library and cloned into BamHI (5′) and NotI (3′) sites of the pcDNA3 expression vector (Ohtani et al, 2004). The DNA constructs of vectors comprised of a full-length copy of the adenovirus genome of approximately 36 kb, from which the early region 1 gene (E1) was replaced by the CAG (cytomegarovirus enhancer, chicken β-actin enhancer—promoter, and rabbit β-globin poly-A signal) promoter and a cDNA for lacZ or sFlt-1. After purification, the virus was suspended in phosphate-buffered saline with 3% sucrose, and was kept at -80°C until used.
Animals
All animal procedures were approved by the Animal Care and Use Review Committee at the Kyushu University (12-053-0). Fifty-nine male spontaneously hypertensive rats (SHR), aged 5 to 8 months and weighed 340 to 435 g, were used to study transgene expression (n = 9), ischemia-induced VEGF releases (n = 10), or neuroprotection by gene therapy (n = 40).
Analysis of Transgene Expression (β-Galactosidase and Soluble flt-1)
Three male SHR were analyzed for transgene expression of β-galactosidase as described previously (Kumai et al, 2004). Briefly, rats were anesthetized with pentobarbital (65 mg/kg, intraperitoneally) and mounted on a stereotaxic head holder in the prone position. After a burr hole was made with a dental drill, a 27 G needle on a Hamilton syringe was stereotaxically inserted into the right lateral ventricle (4.5 mm in depth), and 30 μL of AdlacZ (6.8 × 109 plaque-forming units/mL) was injected over 10 mins. One day after injection, the rats were anesthetized and perfused transcardially with 2% paraformaldehyde in phosphate-buffered saline. The brain was cut into coronal sections at intervals of 2 mm, incubated in 5-bromo-4-chloro-3-indolyl-β-
Six male SHR were analyzed for transgene expression of sFlt-1. The same surgical procedure with transgene expression of β-galactosidase was performed. A 30 μL of viral suspension of AdlacZ (6.8 × 109 plaque-forming units/mL: n = 3) or AdsFlt-1 (6.8 × 109 plaque-forming units/mL: n = 3) was injected into the lateral ventricle over 10 mins. One day after injection, the rats were deeply anesthetized and the brain was removed. Tissue samples at the periventricular area in the injected hemisphere were dissected out from the 2 mm thick coronal slice at the caudate—putamen level, weighted, and homogenized in the lysis buffer, followed by centrifugation at 10,000 g for 10 mins as described previously (Kumai et al, 2003). The supernatants were used to assay by the sandwich enzyme-linked immunosorbent assay for sFlt-1 (R&D Systems, Minneapolis, MN, USA).
Brain Ischemia and Gene Transfer
Forty male SHR were used for brain ischemia study as described previously (Kumai et al, 2004; Ooboshi et al, 2005). Briefly, rats were anesthetized and the left femoral artery and vein were cannulated. Mean arterial pressure was continuously monitored. The rat was mounted on a stereotaxic head holder in the prone position, and the distal segment of middle cerebral artery (MCA) above the rhinal fissure was revealed with the dura left intact. Cerebral blood flow before and during ischemia at the parietal cortex was measured by laser Doppler flowmetry (ALF21D, Advance Co., Ltd, Tokyo, Japan). Brain ischemia was produced by photochemical occlusion of the distal MCA of SHR as described previously (Kumai et al, 2004; Ooboshi et al, 2005). Physiologic variables were determined before and 1 h after the distal MCA occlusion.
For the injection of adenoviral vectors into the lateral ventricle ipsilateral to the ischemic side, a small burr hole was made in the parietal region as described in the above experiments. Ninety minutes after induction of ischemia, 30 μL of viral suspension of AdlacZ (6.8 × 109 plaque-forming units/mL: n = 20) or AdsFlt-1 (6.8 × 109 plaque-forming units/mL: n = 20) was injected into the lateral ventricle over 10 mins. Two hours after the distal MCA occlusion, the rats were returned to the home cage after regaining the ability to breathe independently. After the injection of vectors, the rats were housed for 6 h (n = 8), 1 day (n = 14), or 7 days (n = 18).
Measurement of Soluble flt-1 and Vascular Endothelial Growth Factor in Cerebrospinal Fluid and Blood
Cerebrospinal fluid (CSF) and blood samples were collected (Ooboshi et al, 1995) for the rats 6 h (n = 8), 1 day (n = 14), or 7 days (n = 18) after ischemia and gene therapy and the control rats (nonischemia and nontransfection, n = 6). The CSF and blood samples were analyzed to measure mouse sFlt-1 by the enzyme-linked immunosorbent assay. Rat VEGF in the CSF was measured for the rats 6 h (n = 8), 1 day (n = 14), or 7 days (n = 18) after ischemia and transfection, for the rats 1 day after ischemia alone (nontransfection, n = 4), and for the control rats (n = 6) using an enzyme-linked immunosorbent assay (RayBiotech Inc., Norcross, GA, USA).
Evaluation of Blood—Brain Barrier Permeability
To evaluate the effect of sFlt-1 overexpression on blood—brain barrier (BBB) permeability, Evans blue dye was used as a marker of albumin extravasation. Briefly, Evans blue dye (Sigma-Aldrich Inc., St Louis, MO, USA, 2% in saline, 3 mL/kg) was injected via the femoral vein under pentobarbital anesthesia (65 mg/kg, intraperitoneally) 1 day after ischemic insult (Shimamura et al, 2004). One hour after the injection, the rats were anesthetized with pentobarbital (overdose, intraperitoneally) and perfused transcardially with physiologic saline. The brain was rapidly removed, cooled in ice-cold saline, and cut into 2 mm coronal sections. The posterior surface of each section was photographed, and the area of dye extravasation, regarded as BBB disruption, was measured with National Institutes of Health Image software (version 1.63) using the following formula: BBB disruption (%) = [LT–(RT–RB)]/LT × 100, where LT is the left hemisphere, RT the right hemisphere, and RB the area stained blue.
Quantification of Brain Infarction and Edema
After evaluation of BBB permeability, brain slices were stained with 2,3,5-triphenyletetrazolium chloride (Wako Pure Chemical, Osaka, Japan) at 37°C for 30 mins in the dark. The crosssectional area of infarct was measured with National Institutes of Health Image software (version 1.63) and infarct volume of each rat was calculated by the indirect method to exclude the influence of edema (Swanson et al, 1990). Brain edema was evaluated as a percentage of the net difference in ischemic cortical volume over the contralateral cortical volume (Kitayama et al, 2001). The brain was then processed for paraffin embedding.
Seven days after ischemic insult, rats were anesthetized with pentobarbital and perfused transcardially with physiologic saline. The brain was rapidly removed, cooled in ice-cold saline, and cut into 2 mm coronal sections followed by postfixation with 4% formaldehyde. The fixed tissue was then processed for paraffin embedding, and sections (5 μm thick) were cut from the block with microtomes for cresyl violet. Infarct volume of each rat was calculated by the indirect method.
Immunohistochemistry
For immunohistochemistry, paraffin slices 5 μm thick at the caudate—putamen levels were preincubated with 3% skim milk to decrease nonspecific binding. The sections were incubated overnight at 4°C with the rabbit anti-rat antibody for phosphorylated focal adhesion kinase (nonreceptor tyrosine kinase linked to permeability) at tyrosine 861 (pFAKtyr861) or 397 (pFAKtyr397) site (BioSource International Inc., Camarillo, CA, USA) or for von Willebrand factor (vWF, Novocastra Laboratories Ltd, Newcastle, UK), the mouse anti-rat antibody for macrophage/monocyte (ED1, Serotec Inc., Oxford, UK), Ki67 (Novocastra Laboratories Ltd, Newcastle, UK) or proliferating cell nuclear antigen (PCNA) (DAKO, Glostrup, Denmark), or nonimmune rabbit or mouse immunoglobulin G (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) as negative control. The slides were washed and incubated with biotinylated, affinity-purified anti-rabbit or mouse immunoglobulin G (Nichirei Corporation, Tokyo, Japan) as the secondary antibody. After avidin—biotin amplification, the slides were incubated with 3′,3′-diaminobenzidine. The slides of Ki67 or PCNA, markers of proliferated cells, were incubated overnight at 4°C for double staining after immunohistochemistry of vWF, a marker of endothelium. The slides were washed and incubated with simple stain AP (Nichirei Corporation, Tokyo, Japan) and incubated with new fuchsin chromogen. For quantification of macrophage/monocyte infiltration, numbers of ED1-positive cells at ischemic hemisphere were analyzed with National Institutes of Health Image software version 1.63 (Kumai et al, 2004). For quantification of vascular density, vWF-positive area in the ischemic area was analyzed as well. Ki67-vWF and PCNA-vWF double-positive cells were regarded as angiogenetic cells, and those angiogenetic cells and pFAKtyr861- or pFAKtyr397-positive cells in vessels were counted within the ischemic area. The numbers of immunohistochemically positive cells and area were divided by ischemic area and was expressed as cells/cm2 or %. The slides, except Ki67-vWF and PCNA-vWF double staining, were counterstained with hematoxylin for nuclear staining.
Statistical Analysis
Data are presented as mean ± s.e.m. Differences between AdlacZ and AdsFlt-1 groups were analyzed by unpaired t-test. P < 0.05 was regarded as statistically significant. Differences in the amount of sFlt-1 were analyzed with one-way analysis of variance followed by Bonferroni's post hoc t-test.
Results
Transgene Expression and Vascular Endothelial Growth Factor Release
Expression of the reporter gene (β-galactosidase) was detected at the periventricular areas 1 day after gene transfer (Figure 1A). The expression was not observed in the cortex, as reported previously (Kumai et al, 2004). Expression of sFlt-1 at the periventricular area was also detected 1 day after gene transfer (Figure 1B).
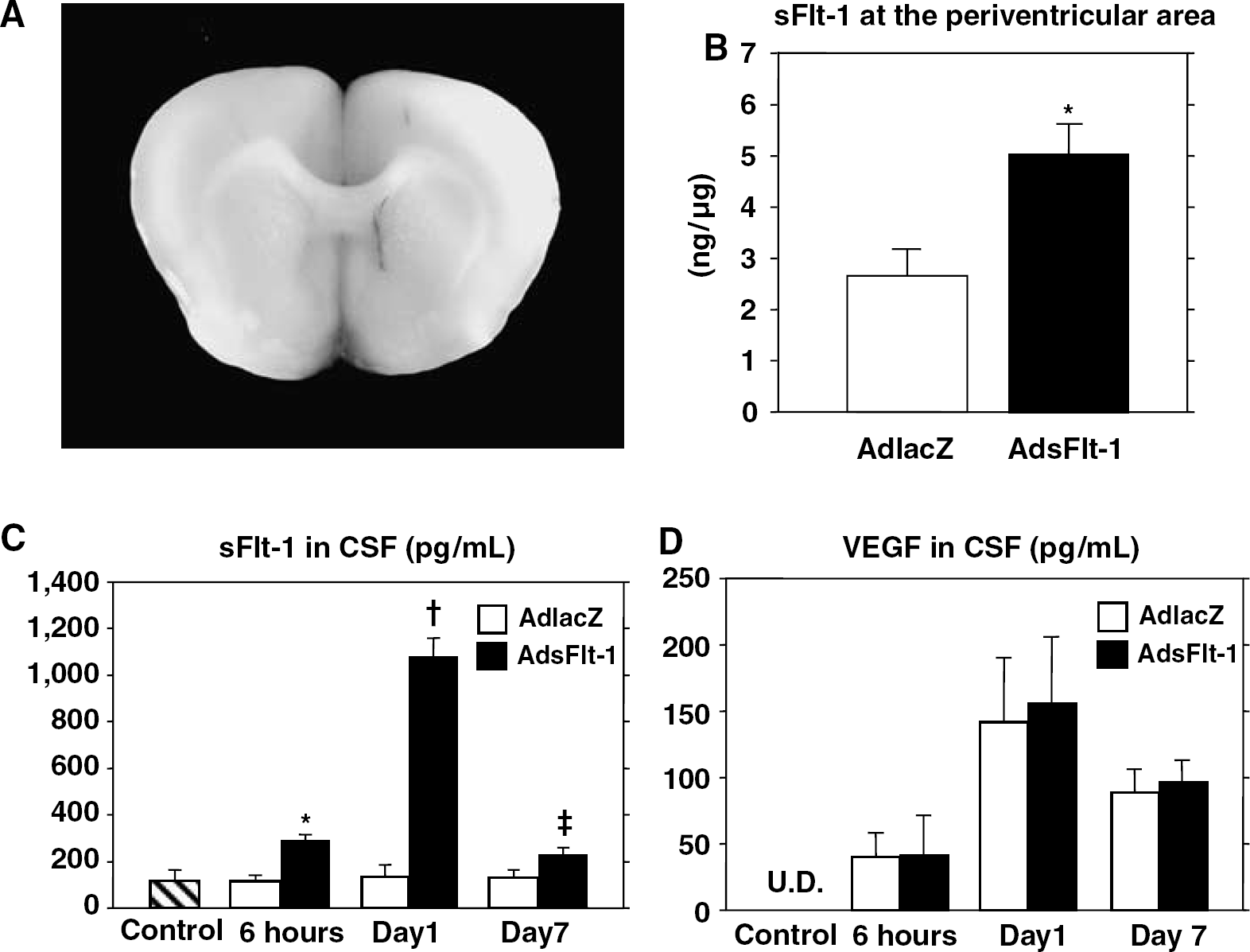
Transgene expression and VEGF release. (
Secreted sFlt-1 in CSF after ischemia and sFlt-1 transfection was detected at 6 h (289 ± 19 pg/mL) and markedly increased (1,072 ± 81 pg/mL) at day 1 and remained higher at day 7 (225 ± 26 pg/mL) as compared with control (117 ± 42 pg/mL) and β-galactosidase transfection (6 h: 115 ± 14 pg/mL; day 1: 134 ± 43 pg/mL; day 7: 130 ± 34 pg/mL; Figure 1C). There was no difference in serum sFlt-1 levels among the control (198 ± 84 pg/mL), AdlacZ (day 1: 212 ± 49 pg/mL; day 7: 181 ± 31 pg/mL) and AdsFlt-1 (day 1: 140 ± 65 pg/mL; day 7: 163 ± 31 pg/mL) groups at each phase after ischemia and sFlt-1 transfection. Although the CSF VEGF was undetectable in the control rats (n = 6), the ischemia increased the VEGF concentrations 1 day after the insult (56.1 ± 26.1 pg/mL; n = 4). The CSF VEGF after ischemia and gene transfer was detected as early as 6 h (AdlacZ: 40.2 ± 18.3 pg/mL; AdsFlt-1: 40.8 ± 30.5 pg/mL), increased at day 1 (AdlacZ: 141 ± 49 pg/mL; AdsFlt-1: 156 ± 50 pg/mL) and decreased at day 7 (AdlacZ: 88.9 ± 17.7 pg/mL; AdsFlt-1: 96.6 ± 16.8 pg/mL; Figure 1D). There was no difference in CSF VEGF levels between AdlacZ and AdsFlt-1 groups at each phase.
Physiologic Variables Before and After Ischemia
There were no significant differences in physiologic variables before and after ischemia between the two groups (Table 1). Blood flow to the cortex on the occlusion side began to decrease within 10 mins after focal ischemia and lasted for more than 60 mins. Cerebral blood flow reductions in AdlacZ and AdsFlt-1 group were -66 ± 2% and -66 ± 2%, respectively, at 60 mins (Figure 2). Changes in cerebral blood flow were not different between the two groups.
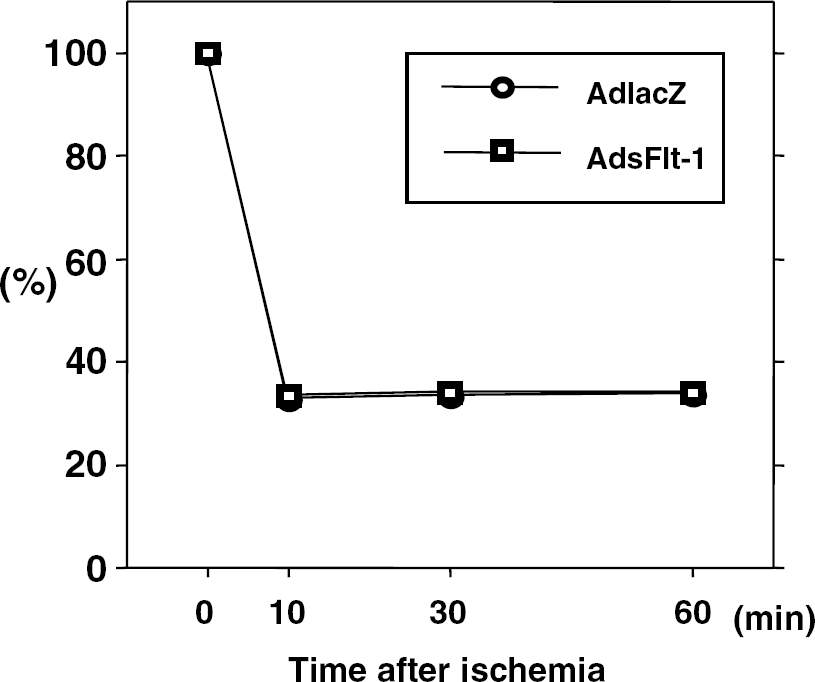
Changes in cerebral blood flow during distal MCA occlusion. Blood flow to the parietal cortex on the occlusion side began to decrease within 10 mins after focal ischemia and lasted for more than 60 mins. Changes in cerebral blood flow were not significantly different between AdlacZ (n = 16) and AdsFlt-1 (n = 16) groups.
Physiological variables
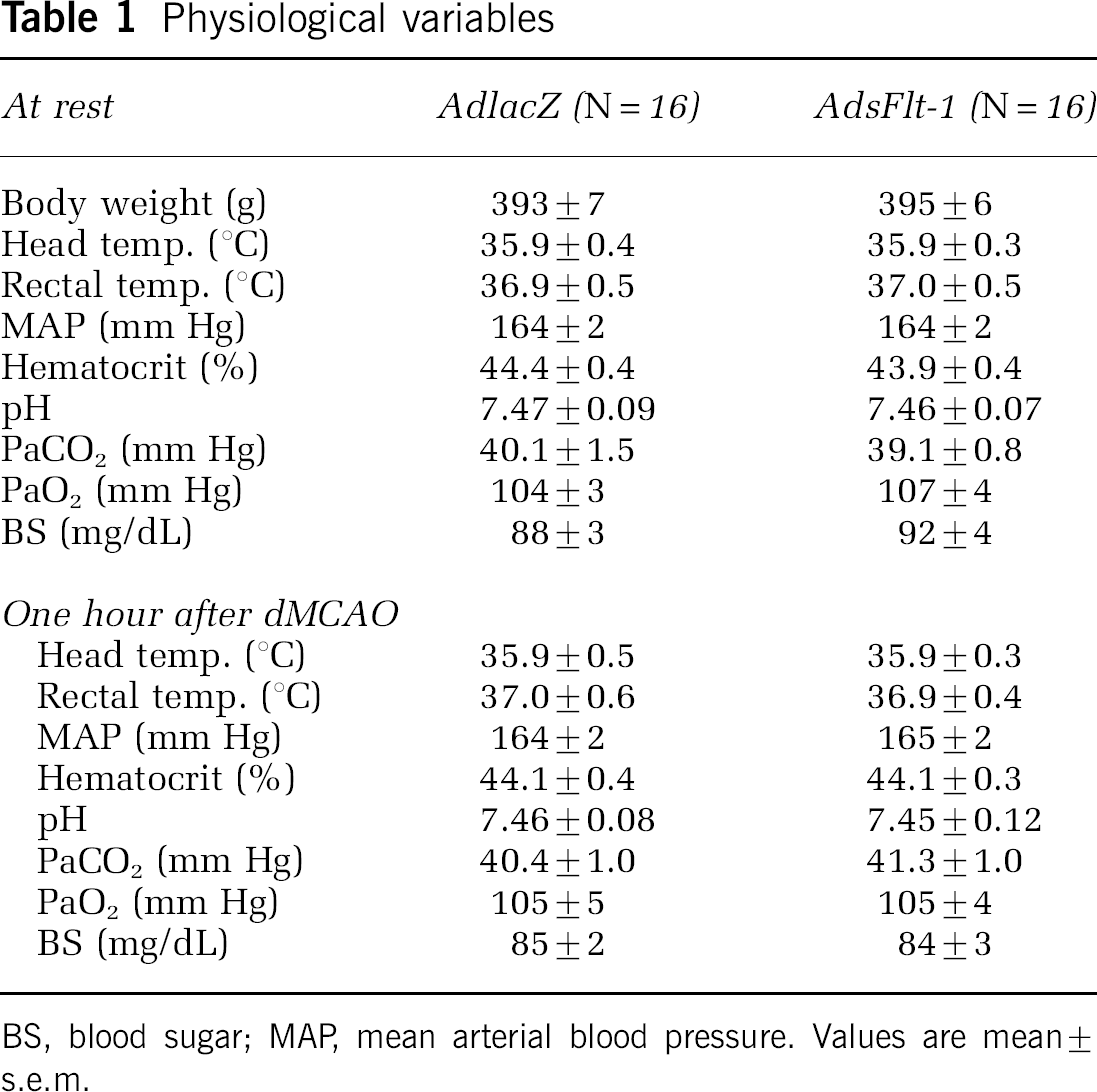
BS, blood sugar; MAP, mean arterial blood pressure. Values are mean ± s.e.m.
Infarct Volume, Brain Edema, and Blood—Brain Barrier Permeability
Infarct volume of AdsFlt-1 group (46 ± 9 mm3; n = 7) was significantly smaller (by 35%) than that of AdlacZ group (71 ± 6 mm3; n = 7, P < 0.05) 1 day after ischemic insult (Figure 3A and Figure 3B). Brain edema of AdsFlt-1 group (8.8% ± 1.5% of contralateral hemisphere) was significantly smaller (by 35%) than that of AdlacZ group (13.6% ± 0.8%; P < 0.05; Figure 3B). Blood—brain barrier disruption evaluated by Evans blue extravasation was markedly attenuated (by 69%) in AdsFlt-1 group (0.93% ± 0.25% of hemisphere) as compared with that in AdlacZ group (3.01% ± 0.83%; P < 0.05; Figure 3C and Figure 3D). Seven days after ischemic insult, infarct volume of AdsFlt-1 group (46 ± 4 mm3; n = 9) remained to be smaller (by 29%) than that of AdlacZ group (65 ± 5 mm3; n = 9, P < 0.01; Figure 3E and Figure 3F).
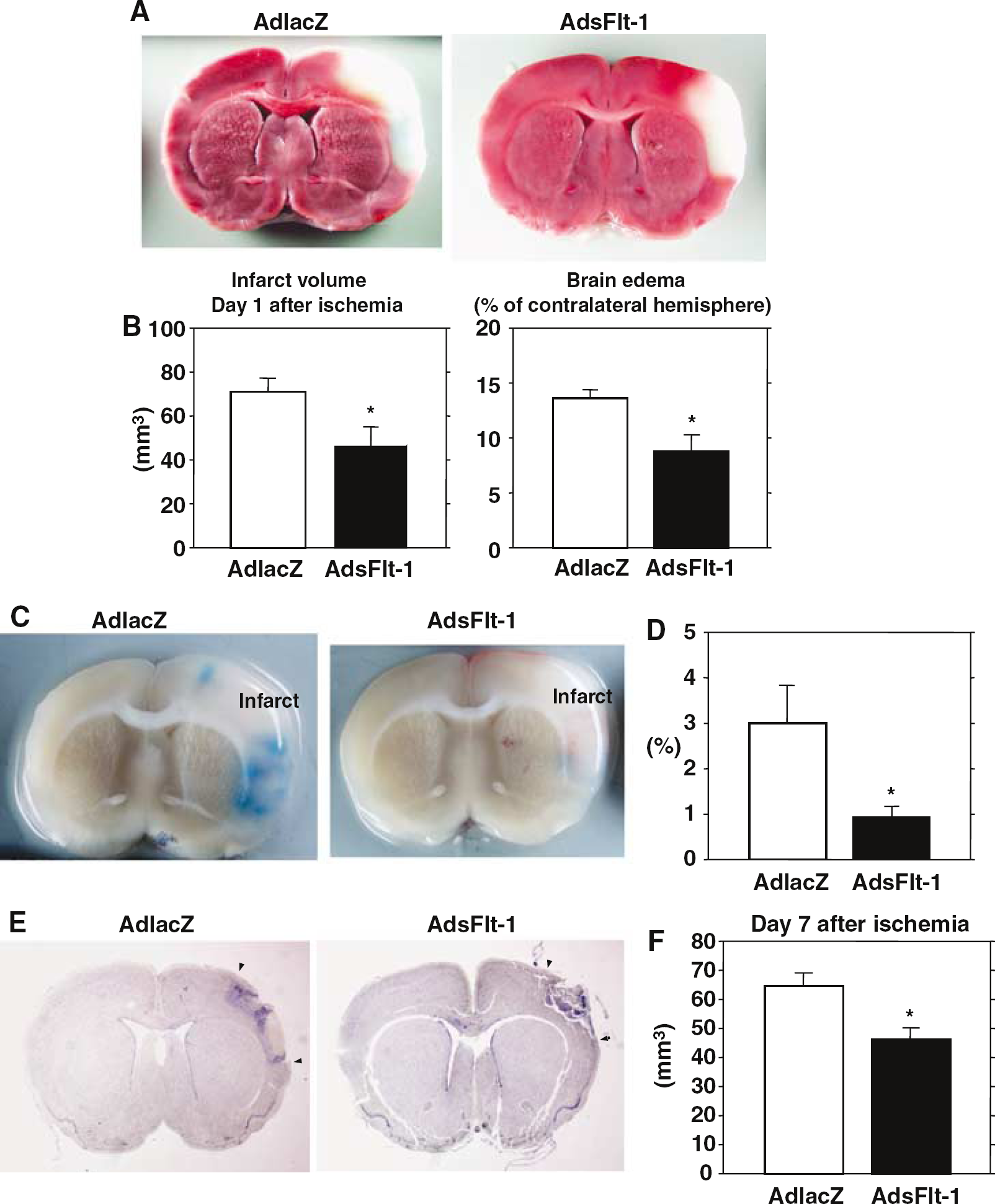
Brain infarction, brain edema, and BBB permeability after gene transfer. (
Phosphorylated Focal Adhesion Kinase of Vessels at the Ischemic Area
One day after brain ischemia, numbers of pFAK-tyr397-positive cells in the vessels at the ischemic area were 593 ± 117 cells/cm2 in AdlacZ group, and were significantly attenuated (by 53%) in AdsFlt-1 group (276 ± 29 cells/mm2; P < 0.05; Figure 4A and 4B). Phosphorylation of vascular FAKtyr861 in the ischemic area were 64 ± 12 cells/cm2 in AdlacZ group and were also halved in AdsFlt-1 group (32 ± 7 cells/cm2; P < 0.05; Figures 4C and Figure 4D).
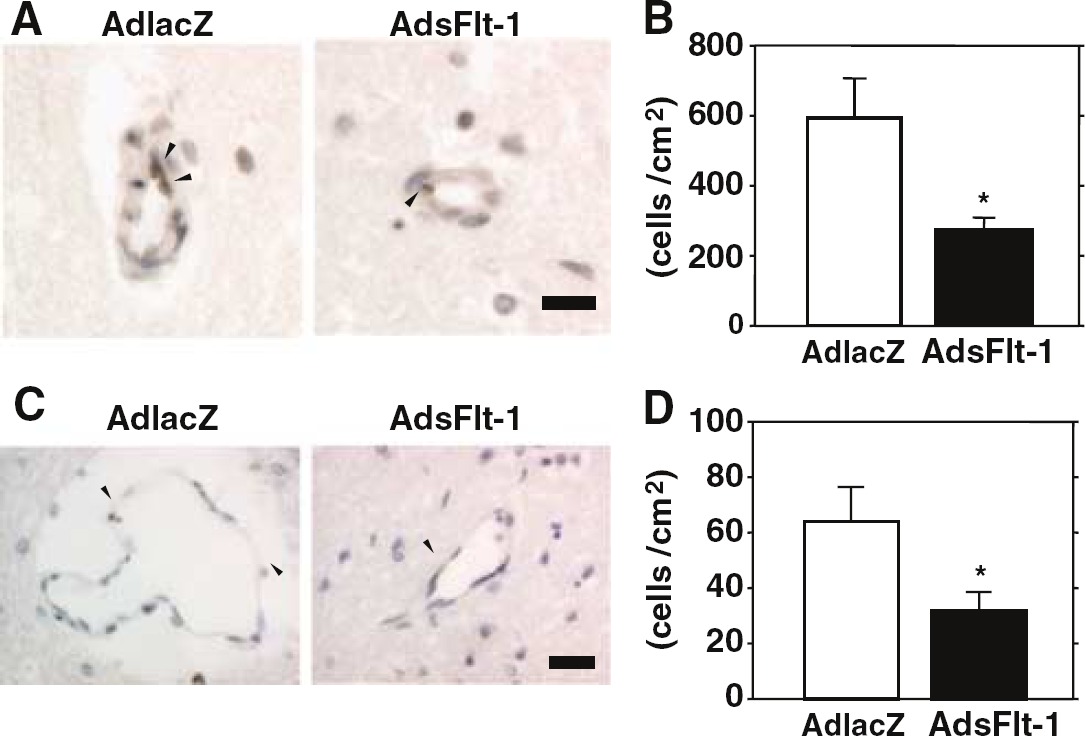
Phosphorylated FAK in vessels at the ischemic area. (
Macrophage Infiltration
Infiltrated macrophages/monocytes were mainly located in the border of the ischemic area (data not shown) as reported previously (Kumai et al, 2004). One day after brain ischemia, numbers of macrophages/monocytes in the ischemic hemisphere of AdsFlt-1 group were 422 ± 66 cells/cm2, which was significantly less than that of AdlacZ group (847 ± 180 cells/cm2; P < 0.05). Seven days after brain ischemia, infiltrations of macrophage/monocyte markedly increased in AdlacZ group (30,509 ± 3,937 cells/cm2) and were significantly less in AdsFlt-1 group (22,334 ± 1,302 cells/cm2; P < 0.05; Figures 5A and Figure 5B).
Angiogenesis After Brain Ischemia and Gene Transfer
von Willebrand factor-positive cells were mainly observed in the ischemic area (Figure 6A). There was no significant difference in the vascular density within the ischemic area between AdlacZ (0.06% ± 0.01%) and AdsFlt-1 (0.10% ± 0.03%; P = 0.188) groups 1 day after brain ischemia. Even 7 days after brain ischemia, the vascular area and numbers of vWF-Ki67 or vWF-PCNA double-positive cells were not significantly different between the AdlacZ (0.32% ± 0.06%, 288 ± 43 cells/cm2, 868 ± 111 cells/cm2) and AdsFlt-1 (0.52% ± 0.10%, P = 0.104; 409 ± 55 cells/cm2, P = 0.292; 1,037 ± 108 cells/cm2, P = 0.101, respectively), suggesting that gene transfer of sFlt-1 did not inhibit angiogenesis (Figures 6B–Figure 6E).
Discussion
In this study, we first showed that gene transfer of sFlt-1 into the lateral ventricle provided marked releases of transgene products into the CSF but not in the serum. Secondly, gene transfer of sFlt-1 significantly reduced infarct volume and brain edema even when vectors were delivered after induction of focal brain ischemia. The reduction of infarct volume was associated with marked attenuations of both BBB permeability and macrophage/monocyte infiltration. We also revealed that the sFlt-1 gene transfer inhibited phosphorylation of FAK, which is an important downstream signal of VEGF receptors. Gene transfer of sFlt-1, however, did not inhibit angiogenesis at the acute stage of focal brain ischemia. These results suggest that the sFlt-1 gene therapy is protective against focal brain ischemia, even after induction of brain ischemia.
Several studies reported beneficial effects of VEGF on brain ischemia in vivo (Hayashi et al, 1998; Wang et al, 2005), and in vitro (Jin et al, 2000). However, there are several concerns over the VEGF therapy for acute brain ischemia because VEGF promotes vascular permeability. While the administration of a low-dose VEGF after brain ischemia lead to neuroprotection, higher doses did not show the protection, suggesting that the protective effect on brain ischemia is dependent on concentrations of VEGF (Manoonkitiwongsa et al, 2004). Although administration of VEGF 48 h after brain ischemia reduced infarct volume and BBB disruption, the treatment 1 h after ischemia exacerbated them (Zhang et al, 2000). Furthermore the delivery of VEGF 2 h after brain ischemia aggravated hemorrhagic transformation at the ischemic area (Abumiya et al, 2005). These lines of evidence suggest that acute administration of VEGF in brain ischemia may exacerbate BBB disruption. Brain edema formation is a major life-threatening complication of brain ischemia in the early phase, and the protection of acute brain edema appears to be an important function of anti-VEGF therapy. Because we and others showed protective effects of acute blockage of VEGF on brain ischemia or hypoxia (van Bruggen et al, 1999; Schoch et al, 2002; Kimura et al, 2005), inhibition of VEGF in the early phase would be useful for the treatment of brain ischemia.
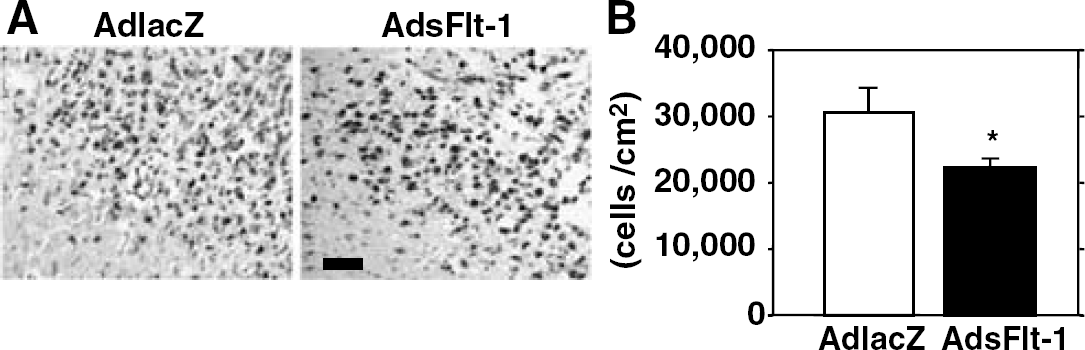
Macrophage infiltration after brain ischemia and gene transfer. (
Vascular endothelial growth factor-mediated vascular permeability is dependent on a subset of Src-family kinases (Eliceiri et al, 1999). Src deficiency or blockade of Src activity prevents VEGF-mediated vascular permeability, thereby reducing neuronal damage after brain ischemia (Paul et al, 2001). Tyrosine-397 is a major phosphorylation site of FAK and its phosphorylation also creates a high-affinity binding site recognized by the Src homology 2 domains of Src-family kinases (Schaller et al, 1994). The phosphorylation may influence the complexity of integrin-associated protein, by mediating baseline levels of FAK/integrin interactions (Wennerberg et al, 2000). Tyrosine-861 is also a major Src site in the carboxyl-terminal domain of FAK, and the phosphorylation of FAKtyr861 may function in additional interactions between FAK and Src homology 2 domains (Calalb et al, 1996). Vascular endothelial growth factor induces robust phosphorylation of FAKtyr861 and FAKtyr397, and promotes Src-mediated phosphorylation of FAKtyr861, which contributes to the formation of a FAK/αvβ5 signaling complex, suggesting that phosphorylation of FAKtyr861 is associated with VEGF-mediated vascular permeability (Eliceiri et al, 2002). However, the association between phosphorylation of FAKtyr397 and VEGF-mediated vascular permeability remains to be established. Although we showed that blockage of VEGF by sFlt-1 gene transfer significantly attenuated phosphorylation of FAKtyr397 as well as FAKtyr861 in the ischemic vessels, further studies are necessary to clarify the role of FAKtyr397 phosphorylation in BBB permeability.
Vascular endothelial growth factor has been reported to be involved in the recruitment of monocyte in several experimental models, including brain inflammation (Clauss et al, 1990; Proescholdt et al, 1999). Flt-1 is expressed by monocyte/macrophage, and VEGF can induce their migration across collagen membranes and endothelial cell monolayers (Clauss et al, 1990; Barleon et al, 1996). We showed that sFlt-1 gene transfer significantly attenuated infiltration of monocyte/macrophage both 1 and 7 days after stroke, which were associated with reduction in infarct size. The beneficial effect of sFlt-1 gene transfer may be partly attributable to inhibition of monocyte/macrophage recruitment and activation.
While sFlt-1 gene transfer may be effective for antiangiogenic strategies in the experimental tumor models, the relation between sFlt-1 and angiogenesis in the brain ischemia was unclear. Interestingly, sFlt-1 gene therapy did neither reduce vascular area nor angiogenesis in the ischemic area 1 and 7 days after ischemic insult. Although stimulation of VEGF receptors on either luminal or abluminal side by different administration routes may lead to differences in signaling pathways in angiogenesis, intravenous (Zhang et al, 2000), intra-arterial (Abumiya et al, 2005), or intraventricular (Sun et al, 2003) administration of VEGF promotes angiogenesis in ischemic brain, suggesting that the delivery route of VEGF is not essential for angiogenesis. In the above studies, late administrations of VEGF (a couple days after ischemia) provoked augmentation of angiogenesis (Zhang et al, 2000; Sun et al, 2003). Because the gene transfer of sFlt-1 provides higher expression at the earlier phase than later phase in our study, the temporary change in transgene expression may lead to discrepancy of effects on vascular permeability and angiogenesis. The amount of sFlt-1 in our study may also explain diverse effects on the permeability and angiogenesis. To elucidate the effect of sFlt-1 gene therapy on angiogenesis in brain ischemia, further examinations are inevitable. Nevertheless, the preserved angiogenesis in this study appears to contribute to the neuronal protection by sFlt-1 gene therapy.
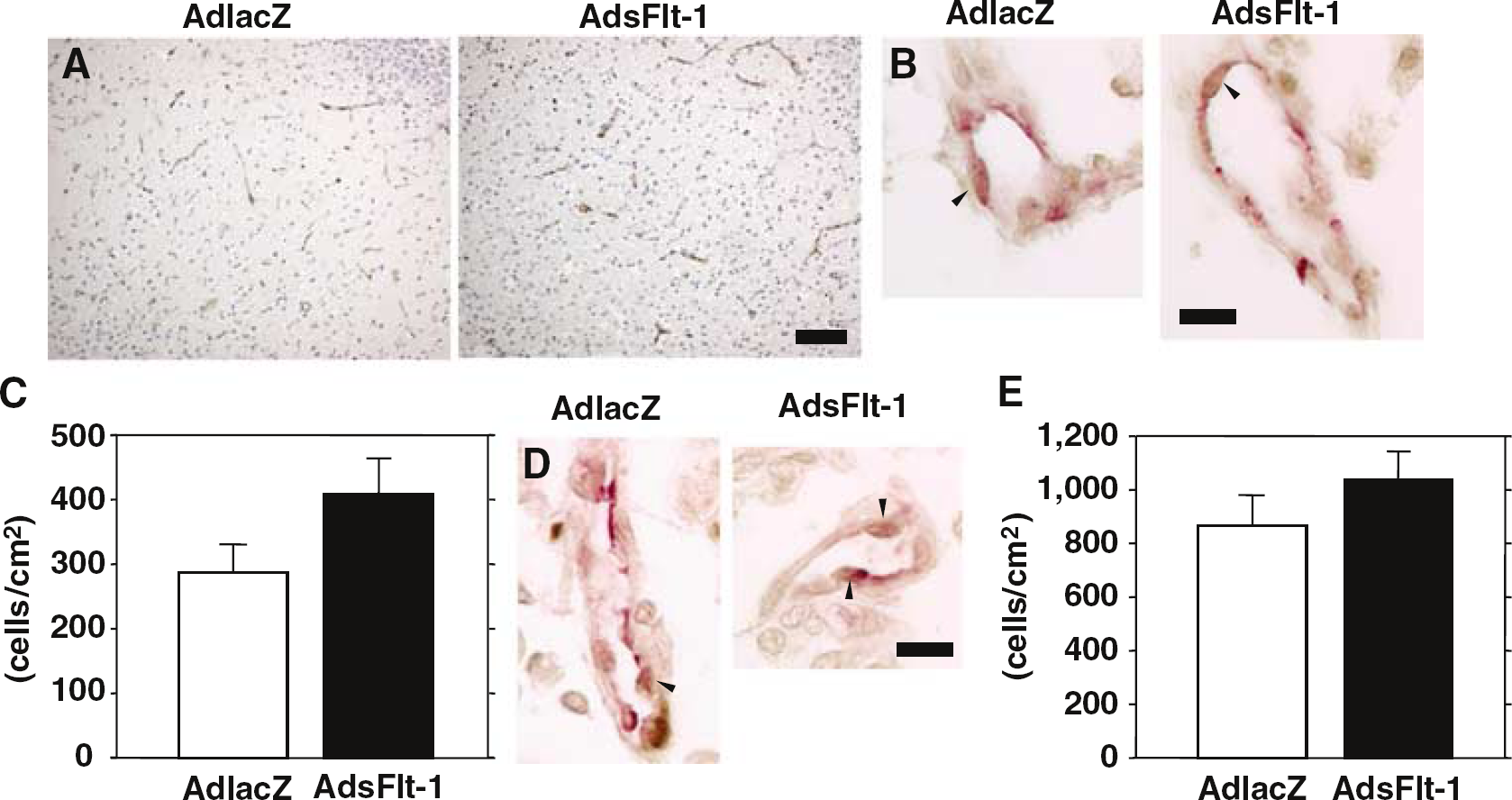
Angiogenesis after brain ischemia and gene transfer. (
In conclusion, gene transfer of sFlt-1 in acute stage of brain ischemia markedly attenuated the vascular permeability and infarct volume, which was accompanied by the reduction of FAK phosphorylation. The beneficial effect of sFlt-1 gene transfer may be also attributable to the inhibition of monocyte/macrophage infiltration and the preservation of angiogenesis. Thus, the sFlt-1 gene therapy may be a promising approach for the treatment of brain ischemia.